HIV Infection Drives Foam Cell Formation via NLRP3 Inflammasome Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
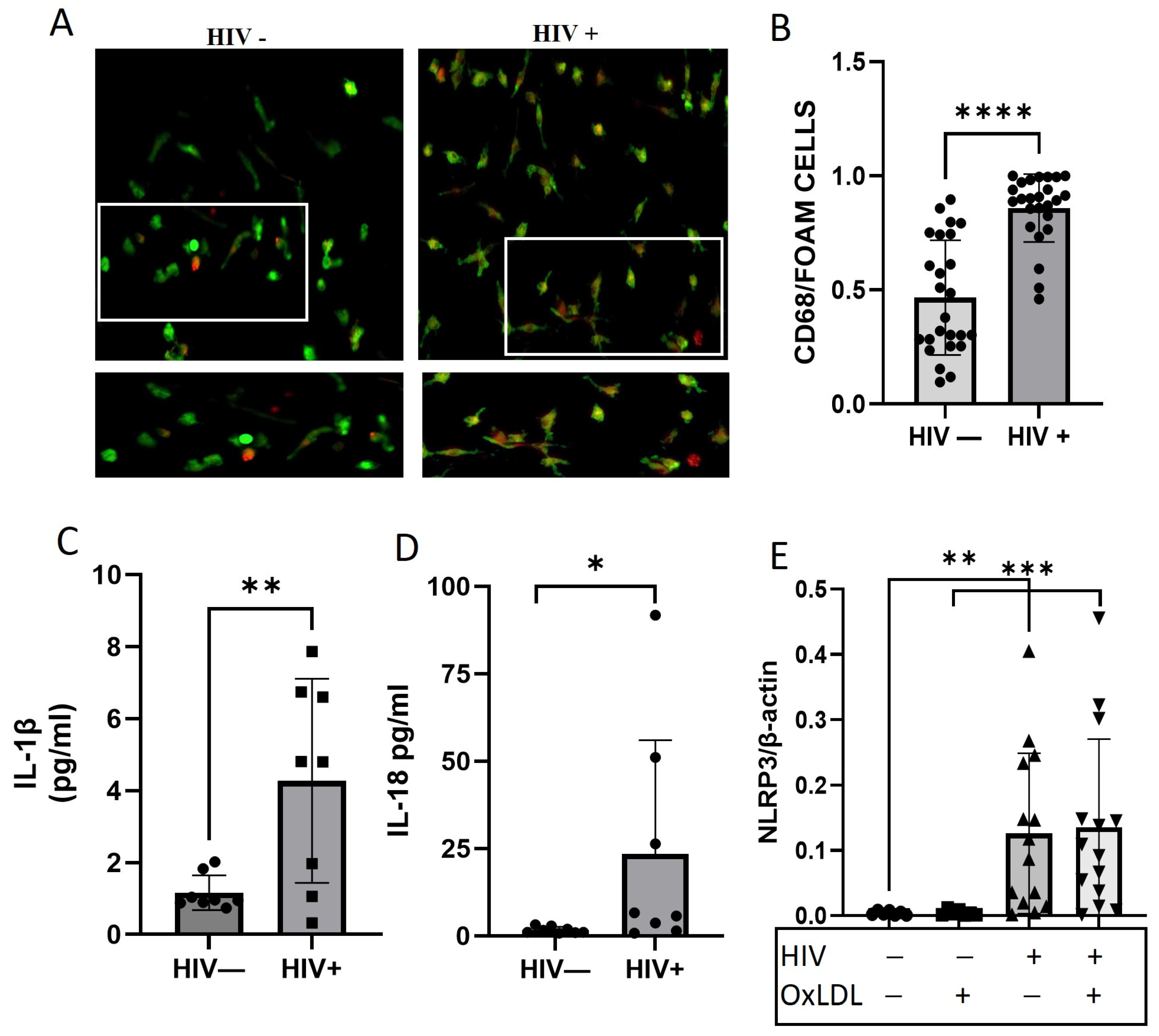
2.1. HIV Increases Foam Cell Formation and NLRP3 Downstream Cytokines
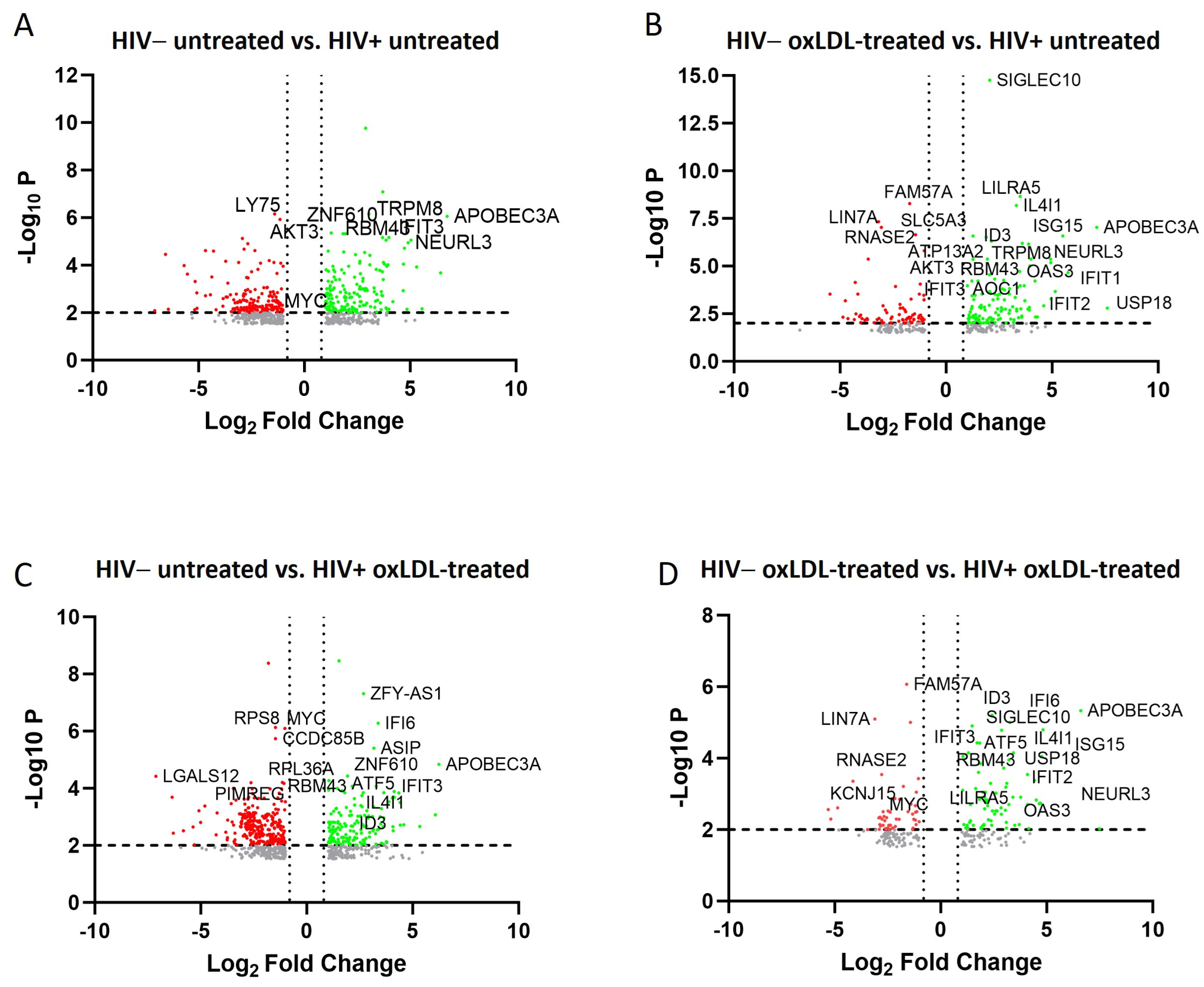
2.2. Transcriptomic and Pathway Changes in Macrophages after HIV Infection and oxLDL Treatment
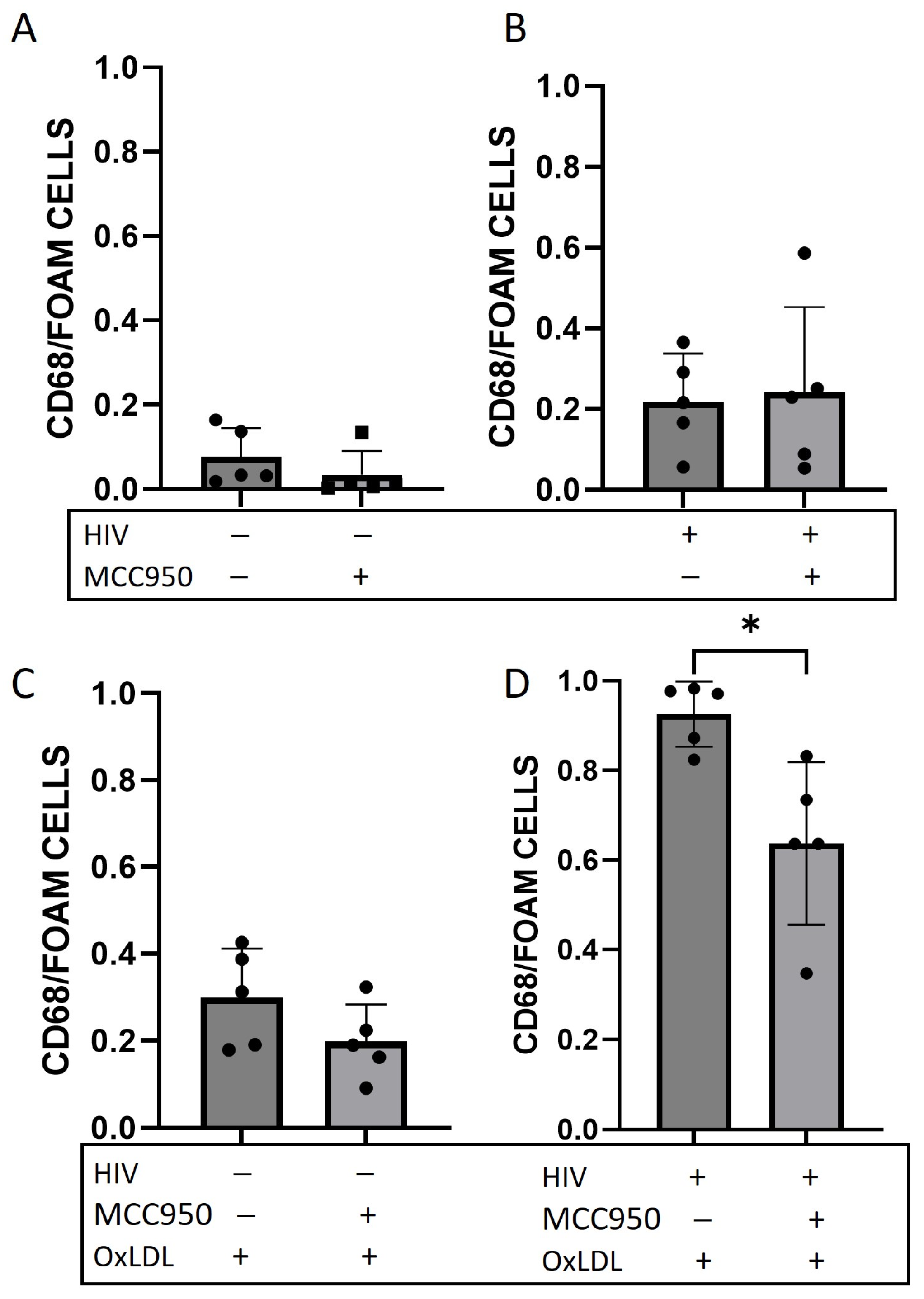
2.3. Foam Cell Formation Is Reduced after HIV Infection and NLRP3 Inhibition
2.4. ART and HIV Increase Foam Cell Formation
2.5. PWH Show an Increased Foam Cell Formation and NLRP3 Expression
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. ELISA
4.3. FLICA-FAM Caspase-1 Staining
4.4. Foam Cell Formation Assay
4.5. qRT-PCR
4.6. RNA Extraction, Library Preparation, Sequencing, and Analysis
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoffmann, U.; Lu, M.T.; Foldyna, B.; Zanni, M.V.; Karady, J.; Taron, J.; Zhai, B.K.; Burdo, T.; Fitch, K.V.; Kileel, E.M.; et al. Assessment of Coronary Artery Disease with Computed Tomography Angiography and Inflammatory and Immune Activation Biomarkers Among Adults with HIV Eligible for Primary Cardiovascular Prevention. JAMA Netw. Open 2021, 4, e2114923. [Google Scholar] [CrossRef] [PubMed]
- Sackoff, J.E.; Hanna, D.B.; Pfeiffer, M.R.; Torian, L.V. Causes of Death among Persons with AIDS in the Era of Highly Active Antiretroviral Therapy: New York City. Ann. Intern. Med. 2006, 145, 397. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Tsakiris, D.A.; Weber, R.; Erb, P.; Battegay, M. Antiretroviral Therapy Reduces Markers of Endothelial and Coagulation Activation in Patients Infected with Human Immunodeficiency Virus Type 1. J. Infect. Dis. 2002, 185, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Cotter, B.R.; Torriani, F.J. Cardiovascular Effects of Antiretroviral Therapy and Noninvasive Assessments of Cardiovascular Disease in HIV Infection. Cardiovasc. Toxicol. 2004, 4, 281–286. [Google Scholar] [CrossRef]
- Kearns, A.C.; Liu, F.; Dai, S.; Robinson, J.A.; Kiernan, E.; Tesfaye Cheru, L.; Peng, X.; Gordon, J.; Morgello, S.; Abuova, A.; et al. Caspase-1 Activation Is Related with HIV-Associated Atherosclerosis in an HIV Transgenic Mouse Model and HIV Patient Cohort. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1762–1775. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.I.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-omics analyses reveal that HIV-1 alters CD4+ T cell immunometabolism to fuel virus replication. Nat. Immunol. 2021, 22, 423–433. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008, 1, 23–30. [Google Scholar] [CrossRef]
- Dennis, E.A.; Smythies, L.E.; Grabski, R.; Li, M.; Ballestas, M.E.; Shimamura, M.; Sun, J.J.; Grams, J.; Stahl, R.; Niederweis, M.E.; et al. Cytomegalovirus promotes intestinal macrophage-mediated mucosal inflammation through induction of Smad7. Mucosal Immunol. 2018, 11, 1694–1704. [Google Scholar] [CrossRef]
- Schnittman, S.R.; Kitch, D.W.; Swartz, T.H.; Burdo, T.H.; Fitch, K.V.; McCallum, S.; Flynn, J.M.; Fulda, E.S.; Diggs, M.R.; Stapleton, J.T.; et al. Coronary Artery Plaque Composition and Severity Relate to the Inflammasome in People with Treated Human Immunodeficiency Virus. Open Forum Infect. Dis. 2023, 10, ofad106. [Google Scholar] [CrossRef]
- Toribio, M.; Burdo, T.H.; Fulda, E.S.; Cetlin, M.; Chu, S.M.; Feldpausch, M.N.; Robbins, G.K.; Nelian, T.G.; Melbourne, K.; Grinspoon, S.K.; et al. Effects of Integrase Inhibitor–Based ART on the NLRP3 Inflammasome Among ART-Naïve People with HIV. Open Forum Infect. Dis. 2020, 7, ofaa459. [Google Scholar] [CrossRef]
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340. [Google Scholar] [CrossRef]
- Grinspoon, S.K.; Fitch, K.V.; Zanni, M.V.; Fichtenbaum, C.J.; Umbleja, T.; Aberg, J.A.; Edgar, E.T.; Malvestutto, C.D.; Bloomfield, G.S.; Currier, J.S.; et al. Pitavastatin to Prevent Cardiovascular Disease in HIV Infection. N. Engl. J. Med. 2023, 389, 687–699. [Google Scholar] [CrossRef]
- Alam, M.A.; Caocci, M.; Ren, M.; Chen, Z.; Liu, F.; Khatun, M.S.; Kolls, J.K.; Qin, C.; Burdo, T.H. Deficiency of Caspase-1 Attenuates HIV-1-Associated Atherogenesis in Mice. Int. J. Mol. Sci. 2023, 24, 12871. [Google Scholar] [CrossRef]
- Angelovich, T.A.; Trevillyan, J.M.; Hoy, J.F.; Wong, M.E.; Agius, P.A.; Hearps, A.C.; Jaworowski, A. Monocytes from men living with HIV exhibit heightened atherogenic potential despite long-term viral suppression with antiretroviral therapy. AIDS 2020, 34, 513–518. [Google Scholar] [CrossRef]
- Albert, M.; Vázquez, J.; Falcón-Pérez, J.M.; Balboa, M.A.; Liesa, M.; Balsinde, J.; Guerra, S. ISG15 Is a Novel Regulator of Lipid Metabolism during Vaccinia Virus Infection. Martinez MA, editor. Microbiol. Spectr. 2022, 10, e03893-22. [Google Scholar] [CrossRef]
- Qi, F.; Zhang, X.; Wang, L.; Ren, C.; Zhao, X.; Luo, J.; Lu, D. E3 ubiquitin ligase NEURL3 promotes innate antiviral response through catalyzing K63 -linked ubiquitination of IRF7. FASEB J. 2022, 36, e22409. Available online: https://onlinelibrary.wiley.com/doi/10.1096/fj.202200316R (accessed on 28 November 2023). [CrossRef]
- Gao, J.; Wang, S.; Liu, S. The involvement of protein TNFSF18 in promoting P-STAT1 phosphorylation to induce coronary microcirculation disturbance in atherosclerotic mouse model. Drug Dev. Res. 2021, 82, 115–122. [Google Scholar] [CrossRef]
- Hornsby, E.; King, H.W.; Peiris, M.; Buccafusca, R.; Lee WY, J.; Wing, E.S.; Blackshaw, L.A.; Lindsay, J.O.; Stagg, A.J. The cation channel TRPM8 influences the differentiation and function of human monocytes. J. Leukoc. Biol. 2022, 112, 365–381. [Google Scholar] [CrossRef]
- Doran, A.C.; Lipinski, M.J.; Oldham, S.N.; Garmey, J.C.; Campbell, K.A.; Skaflen, M.D.; Cutchins, A.; Lee, D.J.; Glover, D.K.; Kelly, K.A.; et al. B-Cell Aortic Homing and Atheroprotection Depend on Id3. Circ. Res. 2012, 110, e1–e12. Available online: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.111.256438 (accessed on 28 November 2023). [CrossRef]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1–Associated Atherosclerosis. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef]
- The Data Collection on Adverse Events of Anti-HIV Drugs (DAD) Study Group. Combination Antiretroviral Therapy and the Risk of Myocardial Infarction. N. Engl. J. Med. 2003, 349, 1993–2003. [Google Scholar] [CrossRef]
- Riddler, S.A.; Li, X.; Otvos, J.; Post, W.; Palella, F.; Kingsley, L.; Visscher, B.; Jacobson, L.; Sharrett, A.R. Antiretroviral Therapy is Associated with an Atherogenic Lipoprotein Phenotype Among HIV-1-Infected Men in the Multicenter AIDS Cohort Study. JAIDS J. Acquir. Immune Defic. Syndr. 2008, 48, 281–288. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Dong, R.; Jiang, G.; Tian, Y.; Shi, X. Identification of immune-related biomarkers and construction of regulatory network in patients with atherosclerosis. BMC Med. Genom. 2022, 15, 245. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caocci, M.; Niu, M.; Fox, H.S.; Burdo, T.H. HIV Infection Drives Foam Cell Formation via NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2024, 25, 2367. https://doi.org/10.3390/ijms25042367
Caocci M, Niu M, Fox HS, Burdo TH. HIV Infection Drives Foam Cell Formation via NLRP3 Inflammasome Activation. International Journal of Molecular Sciences. 2024; 25(4):2367. https://doi.org/10.3390/ijms25042367
Chicago/Turabian StyleCaocci, Maurizio, Meng Niu, Howard S. Fox, and Tricia H. Burdo. 2024. "HIV Infection Drives Foam Cell Formation via NLRP3 Inflammasome Activation" International Journal of Molecular Sciences 25, no. 4: 2367. https://doi.org/10.3390/ijms25042367