
ELK1/MTOR/S6K1 Pathway Contributes to Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
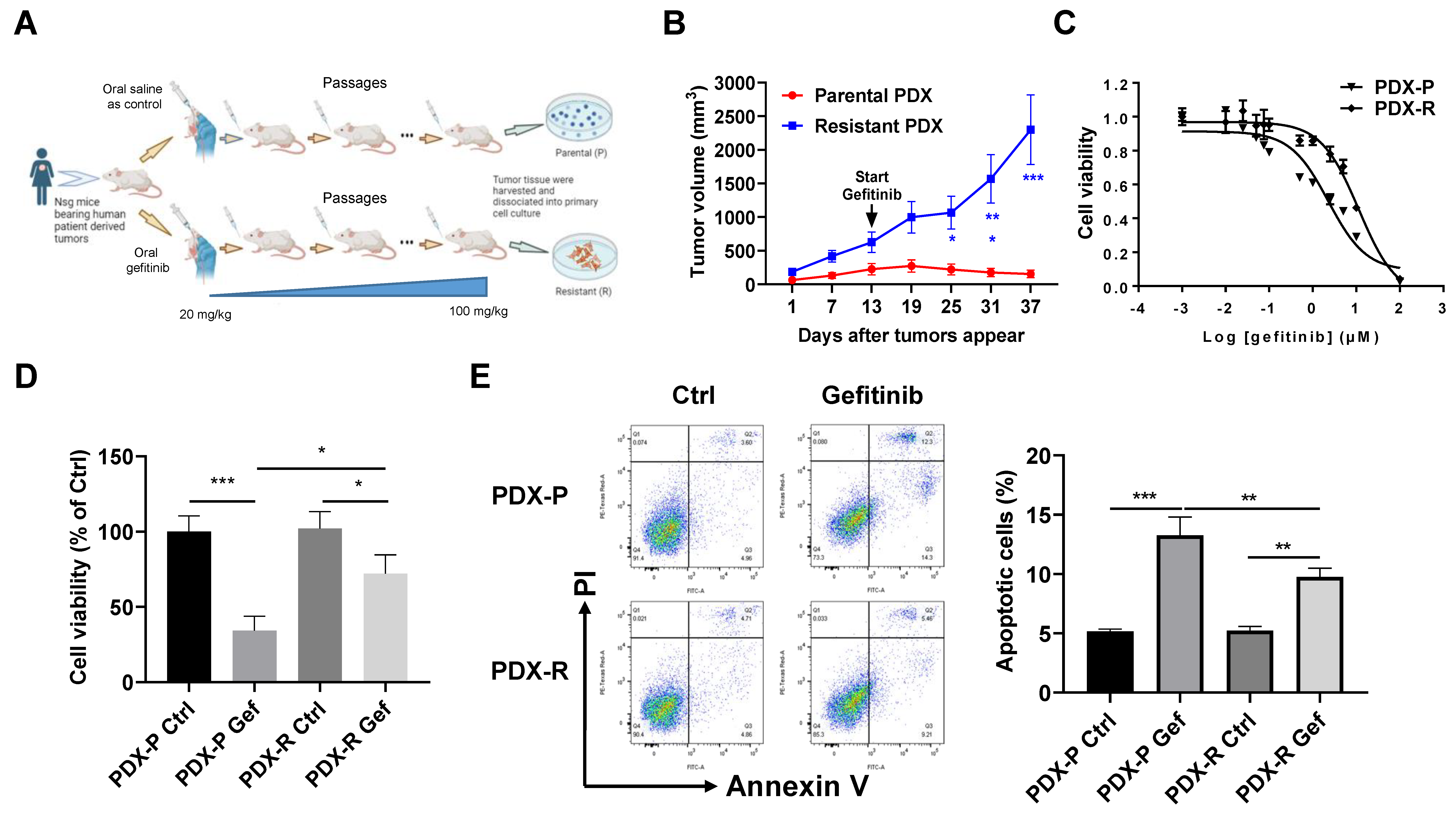
2.1. Development of Gefitinib-Resistant PDX Model In Vivo
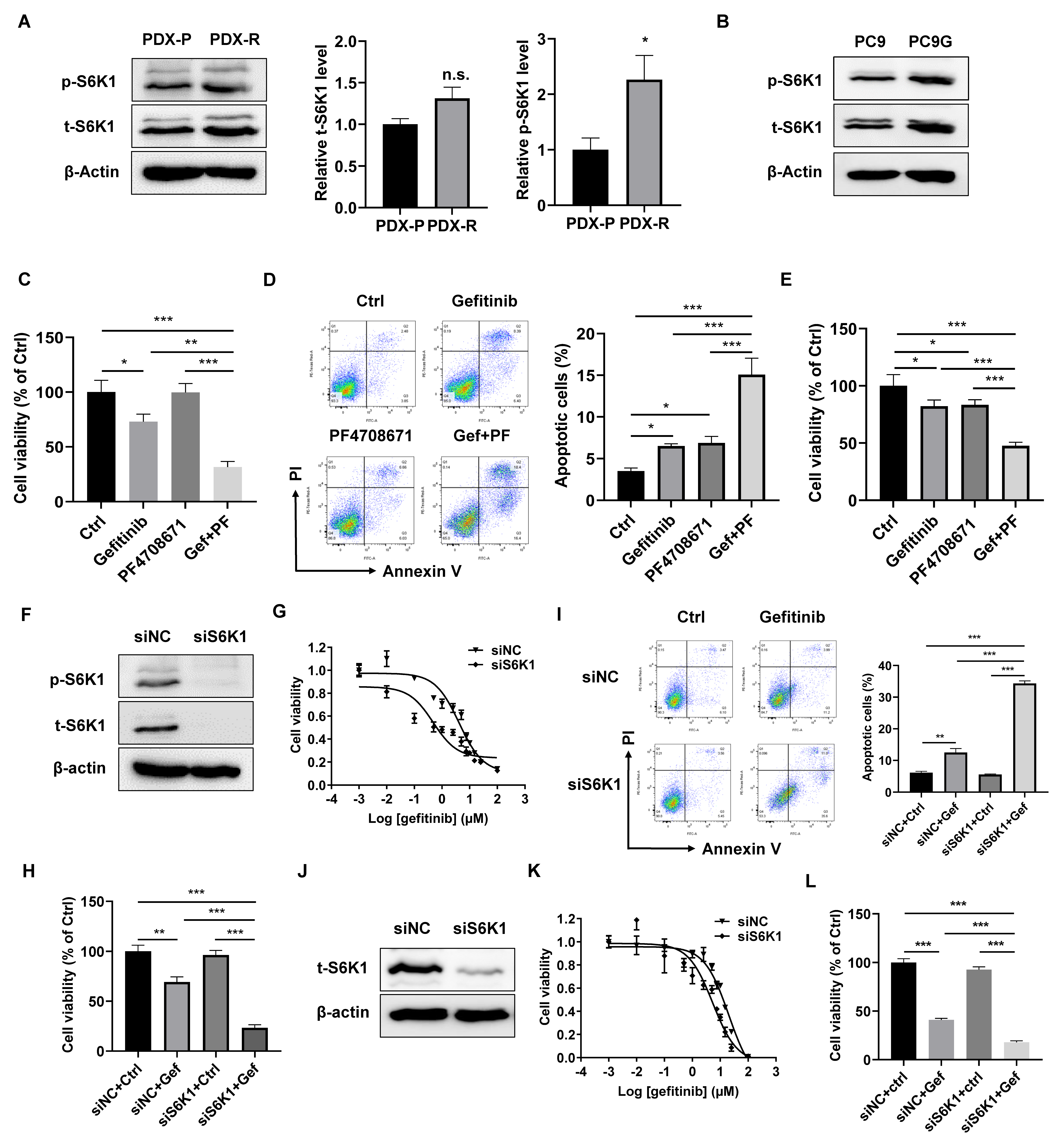
2.2. Inhibition of S6K1 Improved the Efficacy of Gefitinib in PDX-R Cells
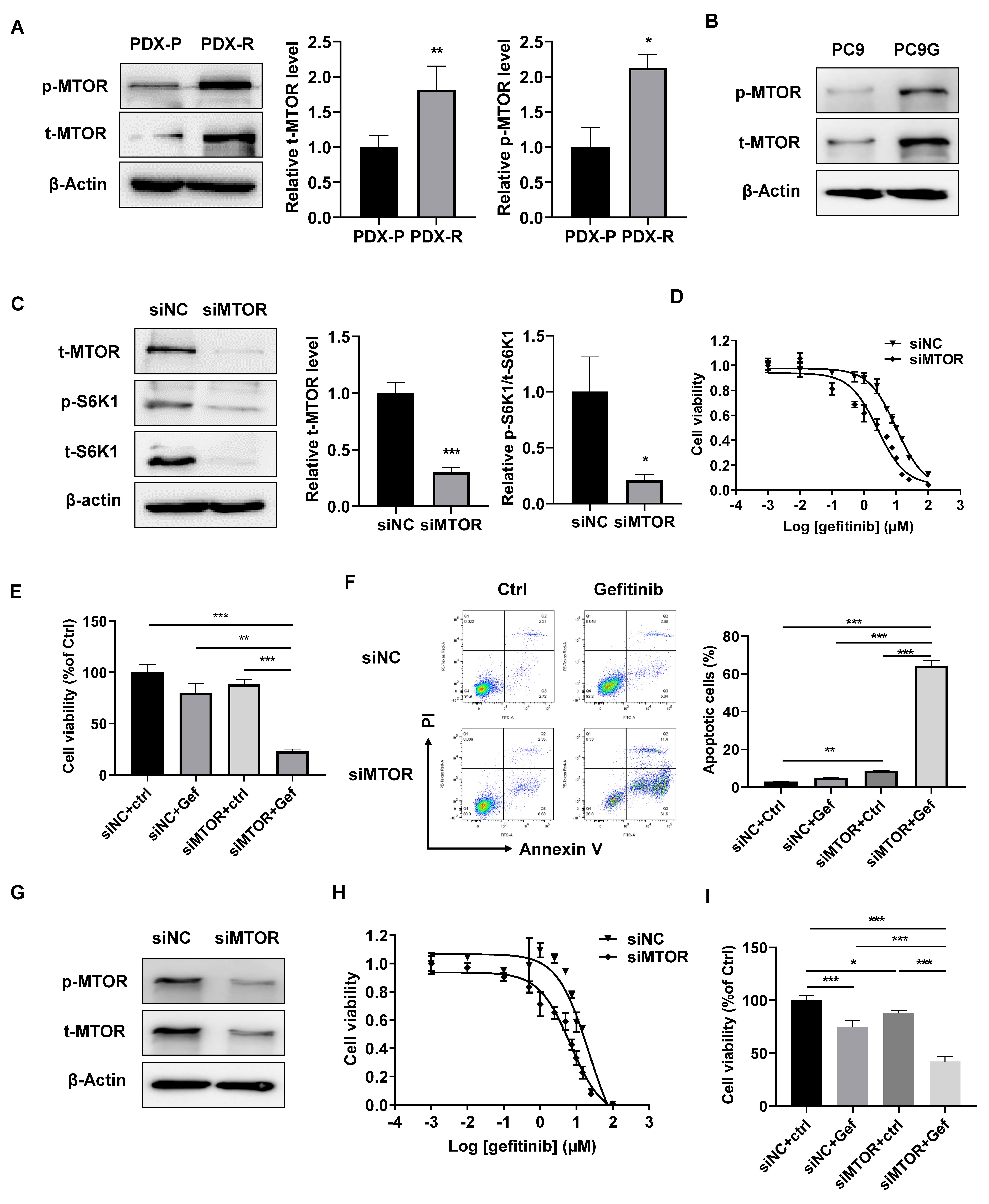
2.3. Increased MTOR Activity Contributes to S6K1-Mediated Gefitinib Resistance
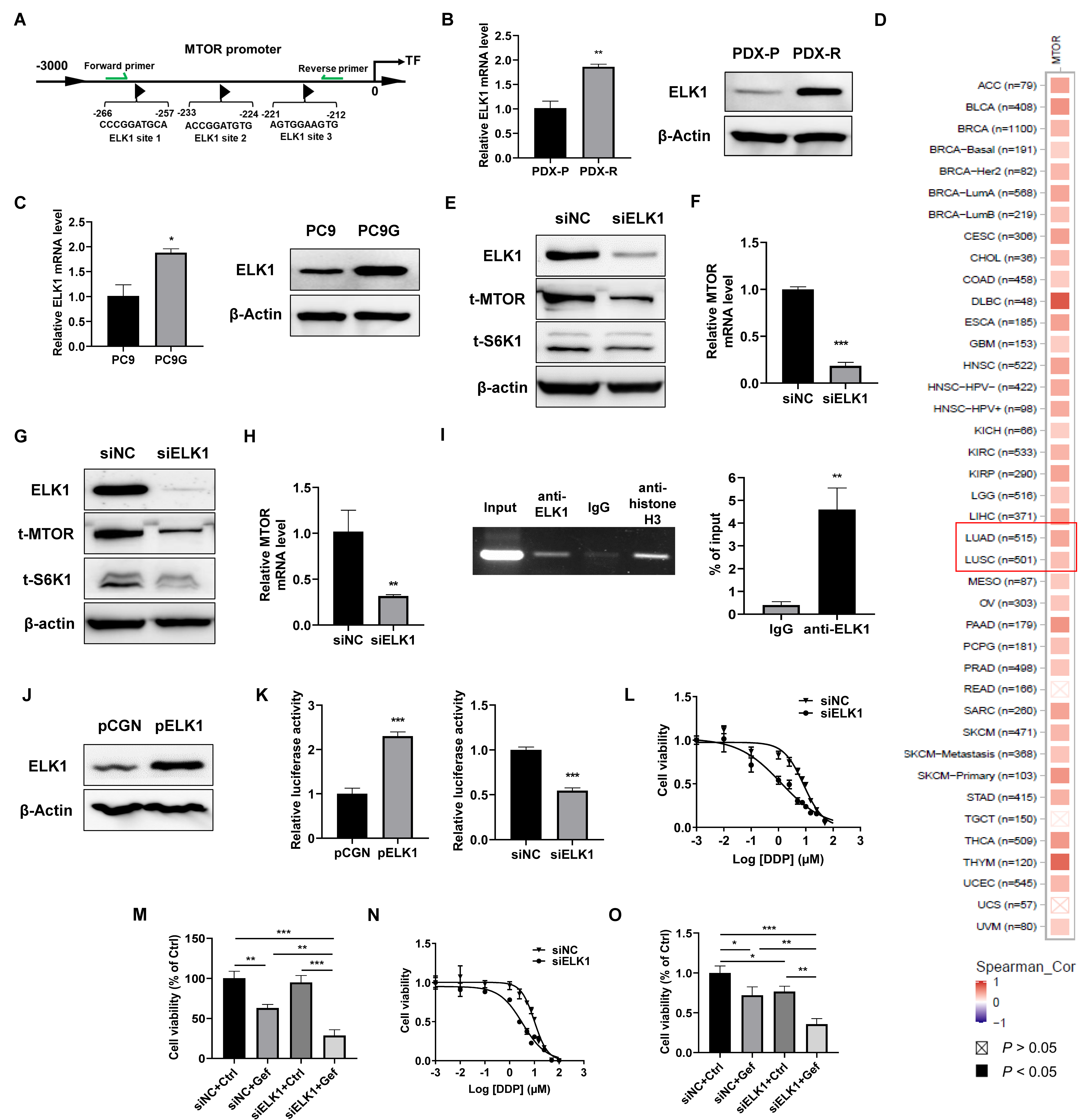
2.4. Upregulation of ELK1 Mediates Gefitinib Resistance through MTOR at the Transcriptional Level
3. Discussion
4. Materials and Methods
4.1. Ethics of the Animal Studies
4.2. Establishment of the NSCLC PDX Model
4.3. Development of the Gefitinib-Resistant NSCLC PDX Model In Vivo
4.4. Dissociation of Primary Cells from the PDX Tumor
4.5. Cell Culture
4.6. Transfection of the siRNA Oligos
4.7. Measurement of the IC50 of Gefitinib in Cells
4.8. Cell Apoptosis Assay
4.9. Real-Time PCR (RT-qPCR)
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Western Blotting
4.12. Dual-Luciferase Reporter Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Pless, M.; Stupp, R.; Ris, H.-B.; A Stahel, R.; Weder, W.; Thierstein, S.; Gerard, M.-A.; Xyrafas, A.; Früh, M.; Cathomas, R.; et al. Induction chemoradiation in stage IIIA/N2 non-small-cell lung cancer: A phase 3 randomised trial. Lancet 2015, 386, 1049–1056. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Chen, R.; Manochakian, R.; James, L.; Azzouqa, A.-G.; Shi, H.; Zhang, Y.; Zhao, Y.; Zhou, K.; Lou, Y. Emerging therapeutic agents for advanced non-small cell lung cancer. J. Hematol. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef]
- Nan, X.; Xie, C.; Yu, X.; Liu, J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget 2017, 8, 75712–75726. [Google Scholar] [CrossRef]
- Wu, L.; Ke, L.; Zhang, Z.; Yu, J.; Meng, X. Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 602762. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2011, 441, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bostner, J.; Karlsson, E.; Eding, C.B.; Perez-Tenorio, G.; Franzén, H.; Konstantinell, A.; Fornander, T.; Nordenskjöld, B.; Stål, O. S6 kinase signaling: Tamoxifen response and prognostic indication in two breast cancer cohorts. Endocrine-Related Cancer 2015, 22, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, J.; Yu, G.; Guo, T.; Hu, C.; Ren, P. Expression and clinical significance of mammalian target of rapamycin/P70 ribosomal protein S6 kinase signaling pathway in human colorectal carcinoma tissue. Oncol. Lett. 2015, 10, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Artemenko, M.; Zhong, S.S.; To, S.K.; Wong, A.S. p70 S6 kinase as a therapeutic target in cancers: More than just an mTOR effector. Cancer Lett. 2022, 535, 215593. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H.; Yi, S.A.; Nam, G.; Noh, J.S.; Park, J.W.; Lee, M.G.; Park, J.H.; Oh, H.; Lee, J.; Lee, K.R.; et al. Identification of a novel S6K1 inhibitor, rosmarinic acid methyl ester, for treating cisplatin-resistant cervical cancer. BMC Cancer 2019, 19, 773. [Google Scholar] [CrossRef] [PubMed]
- Grasso, S.; Tristante, E.; Saceda, M.; Carbonell, P.; Mayor-López, L.; Carballo-Santana, M.; Carrasco-García, E.; Rocamora-Reverte, L.; García-Morales, P.; Carballo, F.; et al. Resistance to Selumetinib (AZD6244) in Colorectal Cancer Cell Lines is Mediated by p70S6K and RPS6 Activation. Neoplasia 2014, 16, 845–860. [Google Scholar] [CrossRef]
- Shen, H.; Wang, G.-C.; Li, X.; Ge, X.; Wang, M.; Shi, Z.-M.; Bhardwaj, V.; Wang, Z.-X.; Zinner, R.G.; Peiper, S.C.; et al. S6K1 blockade overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Oncogene 2020, 39, 7181–7195. [Google Scholar] [CrossRef]
- Park, J.S.; Kang, D.H.; Lee, D.H.; Bae, S.H. PF-4708671, a specific inhibitor of p70 ribosomal S6 kinase 1, activates Nrf2 by promoting p62-dependent autophagic degradation of Keap1. Biochem. Biophys. Res. Commun. 2015, 466, 499–504. [Google Scholar] [CrossRef]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, Y.; Ding, Y.; Zhang, Z.; Feng, J.; Hu, J.; Chen, J.; Lian, Z.; Chen, Y.; Hu, K.; et al. BCL6 is regulated by the MAPK/ELK1 axis and promotes KRAS-driven lung cancer. J. Clin. Investig. 2022, 132, e161308. [Google Scholar] [CrossRef]
- Xie, W.; Li, S.; Guo, H.; Zhang, J.; Tu, M.; Wang, R.; Lin, B.; Wu, Y.; Wang, X. Androgen receptor knockdown enhances prostate cancer chemosensitivity by down-regulating FEN1 through the ERK/ELK1 signalling pathway. Cancer Med. 2023, 12, 15317–15336. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Xie, F.; Wang, F.; Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J. Hematol. Oncol. 2022, 15, 173. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Ren, S.; Li, A.; Xu, J.; Xu, Q.; Su, C.; Guo, J.; Deng, Q.; Zhou, C. Epidermal growth factor receptor-tyrosine kinase inhibitor therapy is effective as first-line treatment of advanced non-small-cell lung cancer with mutated EGFR: A meta-analysis from six phase III randomized controlled trials. Int. J. Cancer 2011, 131, E822–E829. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Yang, H.; Ding, G.; Jin, B.; Lou, Y.; Zhang, Y.; Wang, H.; Han, B. Comparison of outcomes of tyrosine kinase inhibitor in first- or second-line therapy for advanced non-small-cell lung cancer patients with sensitive EGFR mutations. Oncotarget 2016, 7, 68442–68448. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Lung Cancer: Distinct Natural History of Patients with Tumors Harboring the T790M Mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef]
- Qiu, Z.-X.; Sun, R.-F.; Mo, X.-M.; Li, W.-M. The p70S6K Specific Inhibitor PF-4708671 Impedes Non-Small Cell Lung Cancer Growth. PLoS ONE 2016, 11, e0147185. [Google Scholar] [CrossRef]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Aghdaei, H.A.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Han, L.-L.; Du, F.; Liu, X.-M.; Li, J.; Wang, H.-H.; Song, M.-H.; Li, Z.; Li, G.-Y. FGFR1 Induces Acquired Resistance Against Gefitinib By Activating AKT/mTOR Pathway In NSCLC. OncoTargets Ther. 2019, 12, 9809–9816. [Google Scholar] [CrossRef]
- Yamazaki, S.; Higuchi, Y.; Ishibashi, M.; Hashimoto, H.; Yasunaga, M.; Matsumura, Y.; Tsuchihara, K.; Tsuboi, M.; Goto, K.; Ochiai, A.; et al. Collagen type I induces EGFR-TKI resistance in EGFR-mutated cancer cells by mTOR activation through Akt-independent pathway. Cancer Sci. 2018, 109, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Bai, J.; Chang, J.Y.; Yuan, Z.; Wang, P. MTOR inhibition reversed drug resistance after combination radiation with erlotinib in lung adenocarcinoma. Oncotarget 2016, 7, 84688–84694. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, D.; Takeuchi, S.; Nakagawa, T.; Sano, T.; Nakade, J.; Nanjo, S.; Yamada, T.; Ebi, H.; Zhao, L.; Yasumoto, K.; et al. mTOR Inhibitors Control the Growth of EGFR Mutant Lung Cancer Even after Acquiring Resistance by HGF. PLoS ONE 2013, 8, e62104. [Google Scholar] [CrossRef]
- Kawahara, T.; Shareef, H.K.; Aljarah, A.K.; Ide, H.; Li, Y.; Kashiwagi, E.; Netto, G.J.; Zheng, Y.; Miyamoto, H. ELK1 is up-regulated by androgen in bladder cancer cells and promotes tumor progression. Oncotarget 2015, 6, 29860–29876. [Google Scholar] [CrossRef]
- Hu, R.; Zhu, Z. ELK1-activated GPC3-AS1/GPC3 axis promotes the proliferation and migration of cervical cancer cells. J. Gene Med. 2019, 21, e3099. [Google Scholar] [CrossRef]
- Yano, S.; Wu, S.; Sakao, K.; Hou, D.-X. Involvement of ERK1/2-mediated ELK1/CHOP/DR5 pathway in 6-(methylsulfinyl)hexyl isothiocyanate-induced apoptosis of colorectal cancer cells. Biosci. Biotechnol. Biochem. 2019, 83, 960–969. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, J.; Zhang, X.; Cao, L.; Wu, Y.; Miao, X. Transcription factor ELK1 accelerates aerobic glycolysis to enhance osteosarcoma chemoresistance through miR-134/PTBP1 signaling cascade. Aging 2021, 13, 6804–6819. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Ide, H.; Kashiwagi, E.; Patterson, J.D.; Inoue, S.; Shareef, H.K.; Aljarah, A.K.; Zheng, Y.; Baras, A.S.; Miyamoto, H. Silodosin inhibits the growth of bladder cancer cells and enhances the cytotoxic activity of cisplatin via ELK1 inactivation. Am. J. Cancer Res. 2015, 5, 2959–2968. [Google Scholar]
- Kawahara, T.; Aljarah, A.K.; Shareef, H.K.; Inoue, S.; Ide, H.; Patterson, J.D.; Kashiwagi, E.; Han, B.; Li, Y.; Zheng, Y.; et al. Silodosin inhibits prostate cancer cell growth via ELK1 inactivation and enhances the cytotoxic activity of gemcitabine. Prostate 2016, 76, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Pang, C.; Chang, N.; Zhang, J.; Liu, W. Overexpression of PAD4 suppresses drug resistance of NSCLC cell lines to gefitinib through inhibiting Elk1-mediated epithelial-mesenchymal transition. Oncol. Rep. 2016, 36, 551–558. [Google Scholar] [CrossRef]
- Canbaz, D.; Kırımtay, K.; Karaca, E.; Karabay, A. SPG4 gene promoter regulation via Elk1 transcription factor. J. Neurochem. 2011, 117, 724–734. [Google Scholar] [CrossRef]
- Yang, B.; Wang, H.; Xiao, J.; Chen, W.; Chen, W. ELK1/KIFC1 axis promotes breast cancer cell proliferation by regulating glutathione metabolism. J. Obstet. Gynaecol. Res. 2023, 49, 2175–2184. [Google Scholar] [CrossRef]
- Li, T.; Kuang, T.; Yang, Z.; Zhang, Q.; Zhang, W.; Fan, Y. Co-treatment With Everolimus, an mTOR-Specific Antagonist, or Downregulation of ELK1 Enhances the Sensitivity of Pancreatic Cancer Cells to Genistein. Front. Cell Dev. Biol. 2021, 9, 633035. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Lehmann, K.; Jefferies, H.B.; McMahon, M.; Downward, J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001, 15, 981–994. [Google Scholar] [CrossRef]
- Park, J.; Cho, Y.-H.; Shin, W.-J.; Lee, S.-K.; Lee, J.; Kim, T.; Cha, P.-H.; Yang, J.S.; Cho, J.; Min, D.S.; et al. A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci. Rep. 2019, 9, 648. [Google Scholar] [CrossRef]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and Activation of p70 s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.-P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Hahn, K.; Miranda, M.; Francis, V.A.; Vendrell, J.; Zorzano, A.; Teleman, A.A. PP2A Regulatory Subunit PP2A-B′ Counteracts S6K Phosphorylation. Cell Metab. 2010, 11, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Moose, D.L.; Krog, B.L.; Kim, T.-H.; Zhao, L.; Williams-Perez, S.; Burke, G.; Rhodes, L.; Vanneste, M.; Breheny, P.; Milhem, M.; et al. Cancer Cells Resist Mechanical Destruction in Circulation via RhoA/Actomyosin-Dependent Mechano-Adaptation. Cell Rep. 2020, 30, 3864–3874.e6. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Wang, Y.; Sun, X.; Zhang, X.; Simone, N.; He, J. ELK1/MTOR/S6K1 Pathway Contributes to Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 2382. https://doi.org/10.3390/ijms25042382
Zhao L, Wang Y, Sun X, Zhang X, Simone N, He J. ELK1/MTOR/S6K1 Pathway Contributes to Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2024; 25(4):2382. https://doi.org/10.3390/ijms25042382
Chicago/Turabian StyleZhao, Lei, Yifang Wang, Xin Sun, Xiujuan Zhang, Nicole Simone, and Jun He. 2024. "ELK1/MTOR/S6K1 Pathway Contributes to Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 25, no. 4: 2382. https://doi.org/10.3390/ijms25042382
APA StyleZhao, L., Wang, Y., Sun, X., Zhang, X., Simone, N., & He, J. (2024). ELK1/MTOR/S6K1 Pathway Contributes to Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 25(4), 2382. https://doi.org/10.3390/ijms25042382