
Epidemiology of Wilson’s Disease and Pathogenic Variants of the ATP7B Gene Leading to Diversified Protein Disfunctions
 ,
,
Abstract
:1. Introduction
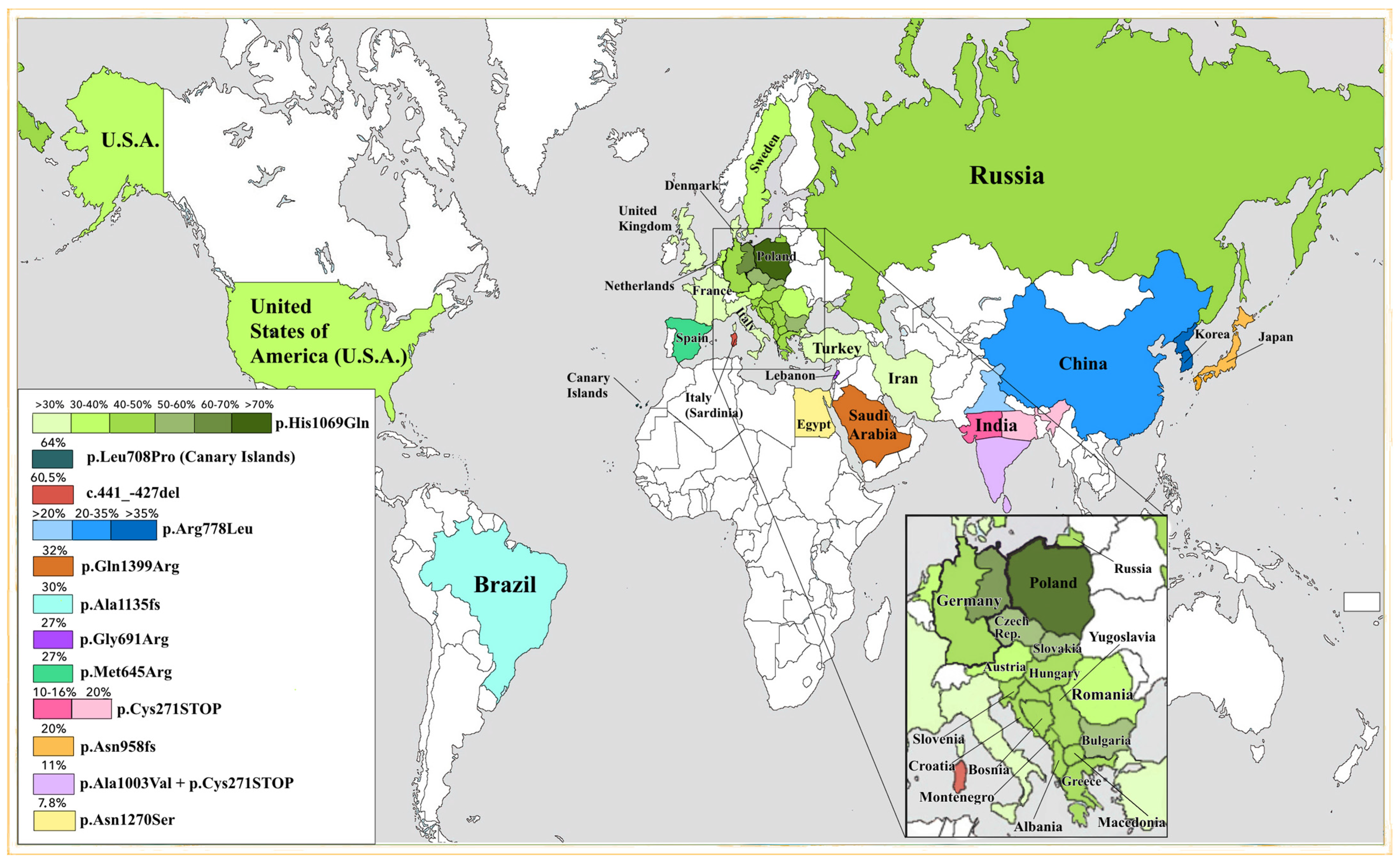
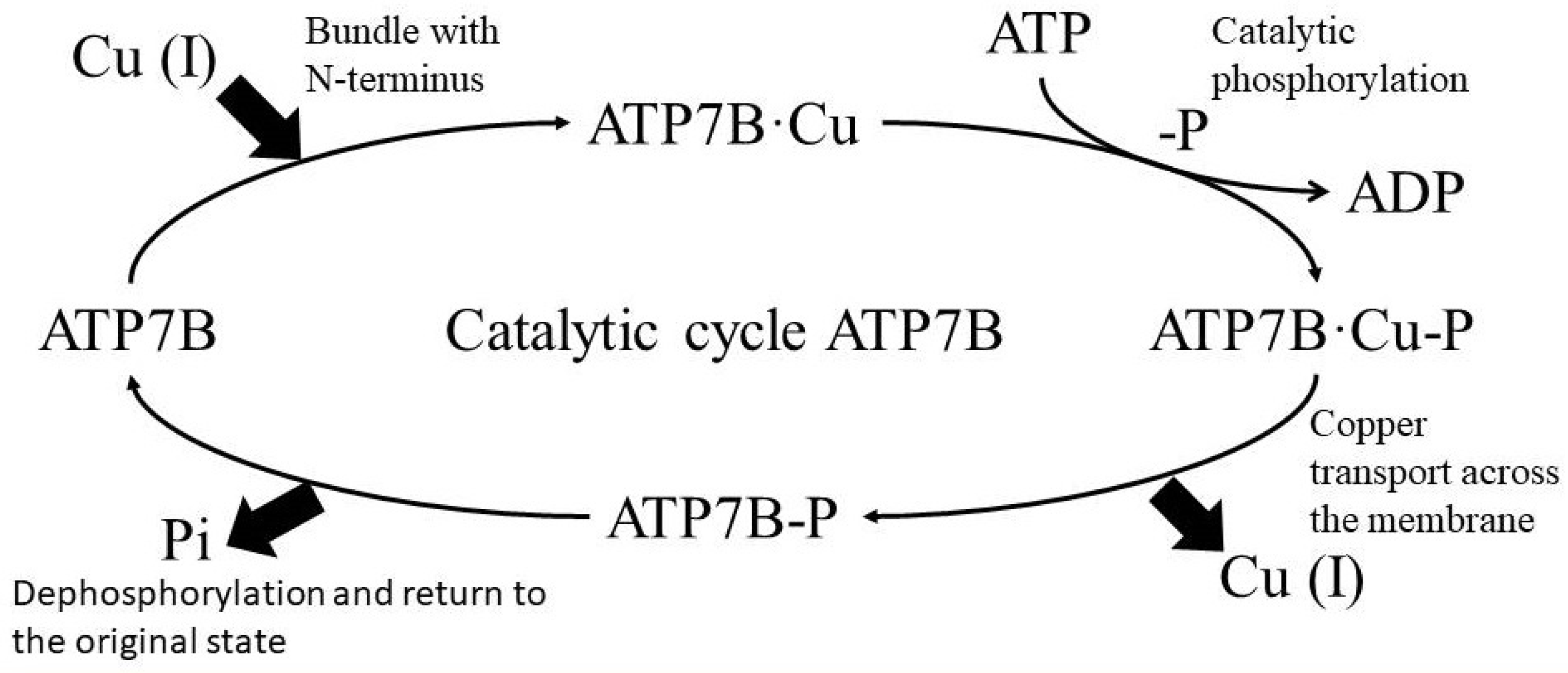
2. Discussion
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poujois, A.; Woimant, F. Wilson’s disease: A 2017 update. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 512–520. [Google Scholar] [CrossRef]
- Albertovich, S.A.; Nikolaevich, D.V.; Yuryevna, K.E. Providing Medical Care and Drug Supply to Patients Suffering from Life-Threatening and Chronic Progressive Rare Diseases. Wilson’s Disease (Hepatolenticular Degeneration); Problems of Standardization in Healthcare: Moscow, Russia, 2015; pp. 30–35. (In Russian) [Google Scholar]
- Asanov, A.Y.; Sokolov, A.A.; Volgina, S.Y.; Goryacheva, L.G.; Gustov, A.V.; Ivanova-Smolenskaya, I.A. Federal Clinical Guidelines for the Diagnosis and Treatment of Wilson-Konovalov Disease (Hepatolenticular Degeneration); Ministry of Health of Russia: Moscow, Russia, 2013; 71p. (In Russian) [Google Scholar]
- Ferenci, P.; Stremmel, W.; Członkowska, A.; Szalay, F.; Viveiros, A.; Stättermayer, A.F.; Bruha, R.; Houwen, R.; Pop, T.L.; Stauber, R.; et al. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology 2019, 69, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Sukezaki, A.; Chu, P.-S.; Shinoda, M.; Hibi, T.; Taniki, N.; Yoshida, A.; Kawaida, M.; Hori, S.; Morikawa, R.; Kurokouchi, A.; et al. Late-onset acute liver failure due to Wilson’s disease managed by plasmapheresis and hemodiafiltration successfully serving as a bridge for deceased donor liver transplantation: A case report and literature review. Clin. J. Gastroenterol. 2020, 13, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Žigrai, M.; Vyskočil, M.; Tóthová, A.; Vereš, P.; Bluska, P.; Valkovič, P. Late-Onset Wilson’s Disease. Front. Med. 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Socha, P.; Janczyk, W.; Dhawan, A.; Baumann, U.; D’Antiga, L.; Tanner, S.; Iorio, R.; Vajro, P.; Houwen, R.; Fischler, B.; et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, W.; Li, X.; Pei, P.; Dong, T.; Yang, Y.; Zhang, J. Clinical and genetic characterization of a large cohort of patients with Wilson’s disease in China. Transl. Neurodegener. 2022, 11, 13. [Google Scholar] [CrossRef]
- Nilles, C.; Obadia, M.A.; Sobesky, R.; Dumortier, J.; Guillaud, O.; Laurencin, C.; Moreau, C.; Vanlemmens, C.; Ory-Magne, F.; de Ledinghen, V.; et al. Diagnosis and Outcomes of Late-Onset Wilson’s Disease: A National Registry-Based Study. Mov. Disord. 2023, 38, 321–332. [Google Scholar] [CrossRef]
- Socha, P.; Czlonkowska, A.; Janczyk, W.; Litwin, T. Wilson’s disease- management and long term outcomes. Best Pract. Res. Clin. Gastroenterol. 2022, 56–57, 101768. [Google Scholar] [CrossRef]
- Li, M.; Ma, J.; Wang, W.; Yang, X.; Luo, K. Pathogenic variant analysis of the ATP7B gene and genotype-phenotype correlation in Chinese patients with Wilson disease. BMC Gastroenterol. 2021, 21, 339. [Google Scholar] [CrossRef]
- Møller, L.B.; Horn, N.; Jeppesen, T.D.; Vissing, J.; Wibrand, F.; Jennum, P.; Ott, P. Clinical presentation and pathogenic variants in Danish patients with Wilson disease. Eur. J. Hum. Genet. 2011, 19, 935–941. [Google Scholar] [CrossRef]
- Gialluisi, A.; Incollu, S.; Pippucci, T.; Lepori, M.B.; Zappu, A.; Loudianos, G.; Romeo, G. The homozygosity index (HI) approach reveals high allele frequency for Wilson disease in the Sardinian population. Eur. J. Hum. Genet. 2013, 21, 1308–1311, Erratum in Eur. J. Hum. Genet. 2014, 22, 295. [Google Scholar] [CrossRef]
- Zali, N.; Mohebbi, S.R.; Esteghamat, S.; Chiani, M.; Haghighi, M.M.; Hosseini-Asl, S.M.; Derakhshan, F.; Mohammad-Alizadeh, A.H.; Malek-Hosseini, S.A.; Zali, M.R. Prevalence of ATP7B Gene Pathogenic variants in Iranian Patients with Wilson Disease. Hepat. Mon. 2011, 11, 890–894. [Google Scholar] [CrossRef]
- O’Brien, M.; Kinsella, K.; Reilly, M.; Sweeney, B.J.; Walsh, C.; Hutchinson, M. Wilson’s Disease in Ireland: Increasing Prevalence Over 40 Years. J. Neurol. Neurosurg. Psychiatry 2012, 83, A13. [Google Scholar] [CrossRef]
- Rootsi, S.; Behar, D.M.; Järve, M.; Lin, A.A.; Myres, N.M.; Passarelli, B.; Poznik, G.D.; Tzur, S.; Sahakyan, H.; Pathak, A.K.; et al. Phylogenetic applications of whole Y-chromosome sequences and the Near Eastern origin of Ashkenazi Levites. Nat. Commun. 2013, 4, 2928. [Google Scholar] [CrossRef]
- Vyalova, N.V.; Proskokova, T.N.; Bayazutdinova, G.M.; Ten, N.N.; Nam, S.I.; Khelimsky, A.M. Molecular genetic analysis in patients with hepatolenticular degeneration in the Khabarovsk Territory. Far East. Med. J. 2016, 2, 72–75. (In Russian) [Google Scholar]
- Simashkova, N.V.; Ivanov, M.V.; Makushkin, E.V.; Sharlay, I.A.; Klyushnik, T.P.; Kozlovskaya, G.V. Skrining riska vozniknoveniya narushenii psikhicheskogo razvitiya u detei rannego vozrasta (dannye po 9 regionam Rossii v 2017–2019 gg.) [Screening of the risk of mental and developmental disorders in children of early age in the Russian population (2017–2019)]. Z. Nevrol. Psikhiatrii Im. S.S. Korsakova 2020, 120, 79–86. (In Russian) [Google Scholar] [CrossRef]
- Gulyaeva, S.E.; Ovchinnikov, A.V. Molecular defect of hepatolenticular degeneration and its clinical reflection. Pac. J. Med. 2013, 4, 98–102. (In Russian) [Google Scholar]
- Park, R.H.; McCabe, P.; Fell, G.S.; Russell, R.I. Wilson’s disease in Scotland. Gut 1991, 32, 1541–1545. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013, 136 Pt 5, 1476–1487. [Google Scholar] [CrossRef]
- Couchonnal, E.; Bouchard, S.; Sandahl, T.D.; Pagan, C.; Lion-François, L.; Guillaud, O.; Habes, D.; Debray, D.; Lamireau, T.; Broué, P.; et al. ATP7B variant spectrum in a French pediatric Wilson disease cohort. Eur. J. Med. Genet. 2021, 64, 104305, Erratum in Eur. J. Med. Genet. 2021, 64, 104341. Erratum in Eur. J. Med. Genet. 2022, 65, 104453. [Google Scholar] [CrossRef]
- Margarit, E.; Bach, V.; Gómez, D.; Bruguera, M.; Jara, P.; Queralt, R.; Ballesta, F. Pathogenic variant analysis of Wilson disease in the Spanish population—Identification of a prevalent substitution and eight novel pathogenic variants in the ATP7B gene. Clin. Genet. 2005, 68, 61–68. [Google Scholar] [CrossRef]
- Sipilä, J.O.T.; Hietala, M.; Kytö, V.; Kaasinen, V. Wilson’s Disease in Finland: A Nationwide Population-Based Study. Mov. Disord. 2020, 35, 2323–2327. [Google Scholar] [CrossRef]
- Otto, P.A.; Deguti, M.M.; Araújo, T.F.; Barbosa, E.R.; Bem, R.S.D.; Araujo, F.C.; Cançado, E.L.R. Estimation of Allele Frequencies and Population Incidence of Wilson Disease in Brazil. Prensa Medica Argent. 2017, 102, 1–4. Available online: https://www.researchgate.net/publication/313867352_Estimation_of_Allele_Frequencies_and_Population_Incidence_of_Wilson_Disease_in_Brazil (accessed on 10 October 2023).
- Olivarez, L.; Caggana, M.; Pass, K.A.; Ferguson, P.; Brewer, G.J. Estimate of the frequency of Wilson’s disease in the US Caucasian population: A pathogenic variant analysis approach. Ann. Hum. Genet. 2001, 65 Pt 5, 459–463. [Google Scholar] [CrossRef]
- Choe, E.J.; Choi, J.W.; Kang, M.; Lee, Y.K.; Jeon, H.H.; Park, B.K.; Won, S.Y.; Cho, Y.S.; Seo, J.H.; Lee, C.K.; et al. A population-based epidemiology of Wilson’s disease in South Korea between 2010 and 2016. Sci. Rep. 2020, 10, 14041. [Google Scholar] [CrossRef]
- Lorente-Arencibia, P.; García-Villarreal, L.; González-Montelongo, R.; Rubio-Rodríguez, L.A.; Flores, C.; Garay-Sánchez, P.; delaCruz, T.; Santana-Verano, M.; Rodríguez-Esparragón, F.; Benitez-Reyes, J.N.; et al. Wilson Disease Prevalence: Discrepancy Between Clinical Records, Registries and Mutation Carrier Frequency. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 192–199. [Google Scholar] [CrossRef]
- Gomes, A.; Dedoussis, G.V. Geographic distribution of ATP7B pathogenic variants in Wilson disease. Ann. Hum. Biol. 2016, 43, 1–8. [Google Scholar] [CrossRef]
- Karabanov, A.V.; Ovchinnikov, I.V.; Illarioshkin, S.N.; Polishchuk, V.V. Analysis of pathogenic variants in the ATP7B gene and experience of direct DNA diagnostics in hepatolenticular degeneration. J. Neurol. Psychiatry 2001, 101, 44–47, 76. (In Russian) [Google Scholar]
- Gromadzka, G.; Schmidt, H.H.; Genschel, J.; Bochow, B.; Rodo, M.; Tarnacka, B.; Litwin, T.; Chabik, G.; Członkowska, A. Frameshift and nonsense pathogenic variants in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin. Genet. 2005, 68, 524–532. [Google Scholar] [CrossRef]
- Tsivkovskii, R.; Purnat, T.; Lutsenko, S. Copper-Transporting ATPases: Key Regulators of Intracellular Copper Concentration. In Handbook of ATPases; Wiley: Hoboken, NJ, USA, 2005; pp. 99–158. [Google Scholar] [CrossRef]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337, Erratum in Nat. Genet. 1994, 6, 214. [Google Scholar] [CrossRef]
- Gul, B.; Firasat, S.; Tehreem, R.; Shan, T.; Afshan, K. Analysis of Wilson disease mutations in copper binding domain of ATP7B gene. PLoS ONE 2022, 17, e0269833. [Google Scholar] [CrossRef]
- Jayakanthan, S.; Braiterman, L.T.; Hasan, N.M.; Unger, V.M.; Lutsenko, S. Human copper transporter ATP7B (Wilson disease protein) forms stable dimers in vitro and in cells. J. Biol. Chem. 2017, 292, 18760–18774. [Google Scholar] [CrossRef]
- Huster, D.; Kühne, A.; Bhattacharjee, A.; Raines, L.; Jantsch, V.; Noe, J.; Schirrmeister, W.; Sommerer, I.; Sabri, O.; Berr, F.; et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 2012, 142, 947–956.e5. [Google Scholar] [CrossRef]
- Dusek, P.; Roos, P.M.; Litwin, T.; Schneider, S.A.; Flaten, T.P.; Aaseth, J. The neurotoxicity of iron, copper and manganese in Parkinson’s and Wilson’s diseases. J. Trace Elem. Med. Biol. 2015, 31, 193–203. [Google Scholar] [CrossRef]
- Dmitriev, O.Y.; Bhattacharjee, A.; Nokhrin, S.; Uhlemann, E.M.; Lutsenko, S. Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and HIS1069GLN mutants of copper-transporting ATPase ATP7B. J. Biol. Chem. 2011, 286, 16355–16362. [Google Scholar] [CrossRef]
- Stapelbroek, J.M.; Bollen, C.W.; van Amstel, J.K.; van Erpecum, K.J.; van Hattum, J.; van den Berg, L.H.; Klomp, L.W.; Houwen, R.H. The HIS1069GLN pathogenic variant in ATP7B is associated with late and neurologic presentation in Wilson disease: Results of a meta-analysis. J. Hepatol. 2004, 41, 758–763. [Google Scholar] [CrossRef]
- Kalita, J.; Somarajan, B.I.; Misra, U.K.; Mittal, B. R778L, HIS1069GLN, and I1102T pathogenic variant study in neurologic Wilson disease. Neurol. India 2010, 58, 627–630. [Google Scholar] [CrossRef]
- Folhoffer, A.; Ferenci, P.; Csak, T.; Horvath, A.; Hegedus, D.; Firneisz, G.; Osztovits, J.; Kosa, J.P.; Willheim-Polli, C.; Szonyi, L.; et al. Novel pathogenic variants of the ATP7B gene among 109 Hungarian patients with Wilson’s disease. Eur. J. Gastroenterol. Hepatol. 2007, 19, 105–111. [Google Scholar] [CrossRef]
- Gupta, A.; Bhattacharjee, A.; Dmitriev, O.Y.; Nokhrin, S.; Braiterman, L.; Hubbard, A.L.; Lutsenko, S. Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein. Proc. Natl. Acad. Sci. USA 2011, 108, 5390–5395. [Google Scholar] [CrossRef]
- Braiterman, L.T.; Murthy, A.; Jayakanthan, S.; Nyasae, L.; Tzeng, E.; Gromadzka, G.; Woolf, T.B.; Lutsenko, S.; Hubbard, A.L. Distinct phenotype of a Wilson disease pathogenic variant reveals a novel trafficking determinant in the copper transporter ATP7B. Proc. Natl. Acad. Sci. USA 2014, 111, E1364–E1373. [Google Scholar] [CrossRef]
- Lalioti, V.; Hernandez-Tiedra, S.; Sandoval, I.V. DKWSLLL, a versatile DXXXLL-type signal with distinct roles in the Cu(+)-regulated trafficking of ATP7B. Traffic 2014, 15, 839–860. [Google Scholar] [CrossRef]
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense pathogenic variants in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352. [Google Scholar] [CrossRef]
- Hodgkinson, V.L.; Dale, J.M.; Garcia, M.L.; Weisman, G.A.; Lee, J.; Gitlin, J.D.; Petris, M.J. X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron. J. Pathol. 2015, 236, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Donoso, M.; Cancino, J.; Lee, J.; van Kerkhof, P.; Retamal, C.; Bu, G.; Gonzalez, A.; Cáceres, A.; Marzolo, M.P. Polarized traffic of LRP1 involves AP1B and SNX17 operating on Y-dependent sorting motifs in different pathways. Mol. Biol Cell. 2009, 20, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Lozoya, J.C.; Tuma, S.; Gotthardt, D.; Reichert, J.; Ehehalt, R.; Stremmel, W.; Füllekrug, J. Copper-induced translocation of the Wilson disease protein ATP7B independent of Murr1/COMMD1 and Rab7. Am. J. Pathol. 2008, 173, 1783–1794. [Google Scholar] [CrossRef]
- Simon, I.; Schaefer, M.; Reichert, J.; Stremmel, W. Analysis of the human Atox 1 homologue in Wilson patients. World J. Gastroenterol. 2008, 14, 2383–2387. [Google Scholar] [CrossRef]
- Weiss, K.H.; Runz, H.; Noe, B.; Gotthardt, D.N.; Merle, U.; Ferenci, P.; Stremmel, W.; Füllekrug, J. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S233–S240. [Google Scholar] [CrossRef] [PubMed]
- Stättermayer, A.F.; Traussnigg, S.; Dienes, H.P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease—Role of copper and PNPLA3 pathogenic variants. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef]
- Przybyłkowski, A.; Gromadzka, G.; Członkowska, A. Polymorphisms of metal transporter genes DMT1 and ATP7A in Wilson’s disease. J. Trace Elem. Med. Biol. 2014, 28, 8–12. [Google Scholar] [CrossRef]
- Seo, J.K. Diagnosis of Wilson disease in young children: Molecular genetic testing and a paradigm shift from the laboratory diagnosis. Pediatr. Gastroenterol. Hepatol. Nutr. 2012, 15, 197–209. [Google Scholar] [CrossRef]
- Lutsenko, S. Modifying factors and phenotypic diversity in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 56–63. [Google Scholar] [CrossRef]
- Bayazutdinova, G.M.; Shchagina, O.A.; Karunas, A.S.; Vyalova, N.V.; Sokolov, A.A.; Polyakov, A.V. Spectrum of pathogenic variants in the ATP7B gene in Russian patients with Wilson–Konovalov disease. Genetics 2019, 55, 1433–1441. (In Russian) [Google Scholar] [CrossRef]
- Todorov, T.; Savov, A.; Jelev, H.; Panteleeva, E.; Konstantinova, D.; Krustev, Z.; Mihaylova, V.; Tournev, I.; Tankova, L.; Tzolova, N.; et al. Spectrum of mutations in the Wilson disease gene (ATP7B) in the Bulgarian population. Clin. Genet. 2005, 68, 474–476. [Google Scholar] [CrossRef]
- Firneisz, G.; Szonyi, L.; Ferenci, P.; Willheim, C.; Horvath, A.; Folhoffer, A.; Tulassay, Z.; Szalay, F. The other pathogenic variant is found: Follow-up of an exceptional family with Wilson disease. Am. J. Gastroenterol. 2004, 99, 2504–2505. [Google Scholar] [CrossRef]
- Caca, K.; Ferenci, P.; Kühn, H.J.; Polli, C.; Willgerodt, H.; Kunath, B.; Hermann, W.; Mössner, J.; Berr, F. High prevalence of the H1069Q mutation in East German patients with Wilson disease: Rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J. Hepatol. 2001, 35, 575–581. [Google Scholar] [CrossRef]
- Telinius, N.; Ott, P.; Sandahl, T.; Hjortdal, J. Scheimpflug Imaging of the Danish Cohort of Patients With Wilson Disease. Cornea 2019, 38, 998–1002. [Google Scholar] [CrossRef]
- Iacob, R.; Iacob, S.; Nastase, A.; Vagu, C.; Ene, A.M.; Constantinescu, A.; Anghel, D.; Banica, C.; Paslaru, L.; Coriu, D.; et al. The His1069Gln mutation in the ATP7B gene in Romanian patients with Wilson’s disease referred to a tertiary gastroenterology center. J. Gastrointestin. Liver Dis. 2012, 21, 181–185. [Google Scholar]
- Tomić, A.; Dobricić, V.; Novaković, I.; Svetel, M.; Pekmezović, T.; Kresojević, N.; Potrebić, A.; Kostić, V.S. Pathogenic variantal analysis of ATP7B gene and the genotype-phenotype correlation in patients with Wilson’s disease in Serbia. Vojnosanit. Pregl. 2013, 70, 457–462. [Google Scholar] [CrossRef]
- Vrabelova, S.; Letocha, O.; Borsky, M.; Kozak, L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol. Genet. Metab. 2005, 86, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Karunas, A.S.; Mersiianova, I.V.; Poliakov, A.V.; Evgrafov, O.V.; Khusnutdinova, E.K. Analiz mutatsiĭ i gaplotipov polimorfnykh markerov u patsientov s bolezn’iu Vil’sona-Konovalova iz Bashkirii [Analysis of mutations and haplotypes of polymorphic markers in patients with Wilson-Konovalov disease from Bashkir]. Genetika 2000, 36, 972–979. (In Russian) [Google Scholar] [PubMed]
- Voloshin-Gaponov, I.K. Molecular genetic aspects of Wilson-Konovalov disease. Ukr. Bull. Psychoneurol. 2014, 22, 23–27. (In Russian) [Google Scholar]
- Baiazutdinova, G.M.; Shchagina, O.A.; Poliakov, A.V. The study of common pathogenic variant p.HIS1069GLN in АТР7В gene in Russian WD-patients. Med. Genet. 2018, 17, 25–30. (In Russian) [Google Scholar]
- Zinchenko, R.A.; El’chinova, G.I.; Baryshnikova, N.V.; Polyakov, A.V.; Ginter, E.K. Prevalences of hereditary diseases in different populations of Russia. Russ. J. Genet. 2007, 43, 1038–1045. [Google Scholar] [CrossRef]
- Matveeva, M.; Zaklyazminskaya, E.; Polyakov, A. The molecular–genetic analysis of ATP7B gene at the Russian patients with Wilson disease. Eur. J. Hum. Genet. 2008, 16, 54. [Google Scholar]
- Gerner, E.A.; Nazarov, V.D.; Fedorova, T.F.; Lapin, S.V.; Pavlova, T.A.; Novikov, S.A.; Alekseeva, T.M.; Emanuel, V.L.; Panina, E.B. Clinical laboratory and molecular genetic diagnosis of Wilson-Konovalov disease. Russ. Neurol. J. (Ross. Nevrol. Z.) 2019, 24, 10–18. [Google Scholar] [CrossRef]
- García-Villarreal, L.; Hernández-Ortega, A.; Sánchez-Monteagudo, A.; Peña-Quintana, L.; Ramírez-Lorenzo, T.; Riaño, M.; Moreno-Pérez, R.; Monescillo, A.; González-Santana, D.; Quiñones, I.; et al. Wilson disease: Revision of diagnostic criteria in a clinical series with great genetic homogeneity. J. Gastroenterol. 2021, 56, 78–89. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Nagase, H.; Tokumoto, S.; Tomioka, K.; Nishiyama, M.; Takeda, H.; Ninchoji, T.; Nagano, C.; Iijima, K.; Nozu, K. Prevalence of Wilson disease based on genome databases in Japan. Pediatr. Int. 2021, 63, 918–922. [Google Scholar] [CrossRef]
- Chang, I.J.; Hahn, S.H. The genetics of Wilson disease. Handb. Clin. Neurol. 2017, 142, 19–34. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Rio, M.J.; Moreno, S.; Ramírez-Gomez, L.; Correa, G.; Lopera, F. New pathogenic variant (T1232P) of the ATP-7B gene associated with neurologic and neuropsychiatric dominance onset of Wilson’s disease in three unrelated Colombian kindred. Neurosci. Lett. 2004, 367, 360–364. [Google Scholar] [CrossRef]
- Panagiotakaki, E.; Tzetis, M.; Manolaki, N.; Loudianos, G.; Papatheodorou, A.; Manesis, E.; Nousia-Arvanitakis, S.; Syriopoulou, V.; Kanavakis, E. Genotype-phenotype correlations for a wide spectrum of pathogenic variants in the Wilson disease gene (ATP7B). Am. J. Med. Genet. A 2004, 131, 168–173. [Google Scholar] [CrossRef]
- Lv, T.; Li, X.; Zhang, W.; Zhao, X.; Ou, X.; Huang, J. Recent advance in the molecular genetics of Wilson disease and hereditary hemochromatosis. Eur. J. Med. Genet. 2016, 59, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Nagral, A.; Mallakmir, S.; Garg, N.; Tiwari, K.; Masih, S.; Nagral, N.; Unavane, O.; Jhaveri, A.; Phadke, S.; ArunKumar, G.; et al. Genomic Variations in ATP7B Gene in Indian Patients with Wilson Disease. Indian J. Pediatr. 2023, 90, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Naveed, A.K.; Majeed, A.; Mansoor, S. Spectrum of ATP7B gene pathogenic variants in Pakistani Wilson Disease patients: A Novel Pathogenic variant is Associated with Severe Hepatic and Neurological Complication. Int. J. Biol. 2010, 2, 117–122. [Google Scholar] [CrossRef]
- Hofer, H.; Willheim-Polli, C.; Knoflach, P.; Gabriel, C.; Vogel, W.; Trauner, M.; Müller, T.; Ferenci, P. Identification of a novel Wilson disease gene pathogenic variant frequent in Upper Austria: A genetic and clinical study. J. Hum. Genet. 2012, 57, 564–567, Erratum in J. Hum. Genet. 2021, 66, 1199. [Google Scholar] [CrossRef] [PubMed]
- Åberg, F.; Shang, Y.; Strandberg, R.; Wester, A.; Widman, L.; Hagström, H. Four-fold increased mortality rate in patients with Wilson’s disease: A population-based cohort study of 151 patients. United Eur. Gastroenterol. J. 2023, 11, 852–860. [Google Scholar] [CrossRef]
- Simsek Papur, O.; Akman, S.A.; Cakmur, R.; Terzioglu, O. Mutation analysis of ATP7B gene in Turkish Wilson disease patients: Identification of five novel mutations. Eur. J. Med. Genet. 2013, 56, 175–179. [Google Scholar] [CrossRef]
- Kumar, S.S.; Kurian, G.; Eapen, C.E.; Roberts, E.A. Genetics of Wilson’s disease: A clinical perspective. Indian J. Gastroenterol. 2012, 31, 285–293. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Stezin, A.; Kamble, N.; Mohammed Shereef, P.M.; Kashyap, B.; Pal, P.K. Retinal Degeneration in Patients with Wilson’s Disease: An OCT Study in Asian Indian Population. Ann. Indian Acad. Neurol. 2022, 25, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, I.; De Freitas, L.; Arias, S. Most frequent mutation c.3402delC (p.Ala1135GlnfsX13) among Wilson disease patients in Venezuela has a wide distribution and two old origins. Eur. J. Med. Genet. 2015, 58, 59–65. [Google Scholar] [CrossRef]
- Naorniakowska, M.; Dądalski, M.; Kamińska, D.; Jańczyk, W.; Lebensztejn, D.; Fyderek, K.; Wysocki, J.; Socha, P. Clinical presentations of Wilson disease among Polish children. Dev. Period Med. 2016, 20, 216–221. [Google Scholar] [PubMed]
- Garbuz, M.M.; Ovchinnikova, A.A.; Kumeiko, V.V. Design, Optimization and Validation of the ARMS PCR Protocol for the Rapid Diagnosis of Wilson’s Disease Using a Panel of 14 Common Pathogenic variants for the European Population. Genes 2022, 13, 1940. [Google Scholar] [CrossRef] [PubMed]
- Woimant, F.; Poujois, A.; Bloch, A.; Jordi, T.; Laplanche, J.L.; Morel, H.; Collet, C. A novel deep intronic variant in ATP7B in five unrelated families affected by Wilson disease. Mol. Genet. Genom. Med. 2020, 8, e1428. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Country | Year of Research | WD Prevalence | Source |
|---|---|---|---|
| Russia | 2014 | 0.23–2.6:100,000 (0.4:100,000) | [3] |
| RF: Khabarovsk Territory | 2018 | 1.5:100,000 | [17] |
| RF: Udmurt region | 2018 | 4:100,000 | [18] |
| RF: Primorsky Krai | 2013 | 5.6–6.5:100,000 | [19] |
| Scotland | 1989 | [20] | |
| Ireland | 1971 | 0.67:100,000 | [15] |
| 2011 | 9.0:100,000 | ||
| Great Britain | 2013 | 14.2:100,000 | [21] |
| France | 2013 | 1.5–1.6:100,000 for men, 1.44:100,000 for women | [22] |
| Spain (Gran Canaria) | 2011–2013 | 8.08:100,000 | [23] |
| Finland | 2016 | 1.44:100,000 | [24] |
| Italy (Sardinia) | 1983 | 2.9:100,000 | [25] |
| 2013 | 36.6:100,000 | [13] | |
| Brazil | 2016 | 4.1:100,000 | [25] |
| USA | 2001 | 2:100,000 | [26] |
| 2006–2011 | 6.4:100,000 | [1] | |
| Korea | 2017 | 13:100,000 | [27] |
| China (Hong Kong) | 2000–2016 | 1.793:100,000 | [28] |
| Country | % | Source |
|---|---|---|
| Russian Federation | 35–50 | [55] |
| Austria | 34 | [4] |
| Bulgaria | 58.8 | [56] |
| Great Britain | 19 | [21] |
| Hungary | 42.9 | [57] |
| East Germany | 63 | [58] |
| Denmark | 18 | [59] |
| Italy | 17.5 | [25] |
| Poland | 72 | [31] |
| Romania | 38.1 | [60] |
| Serbia | 38.4 | [61] |
| USA | 40.3 | [26] |
| France | 15 | [22] |
| Czech | 57 | [62] |
| Country | % | Pathogenic Variant | Source |
|---|---|---|---|
| Russia | 35–50 | p.His1069Gln (c.3207C<A) | [17,55] |
| Austria | 34.1 | p.His1069Gln (c.3207C>A) | [4] |
| 6.4 | p.Gly710Ser (c.2128G>A) | ||
| 3.4 | p.Met769fs (c.2298_2299insC) | ||
| Bulgaria | 58.8 | p.His1069Gln (c.3207C>A) | [56] |
| Canary Islands | 64 | p.Leu708Pro c.2123 (T>C) | [23] |
| Czech | 57 | p.His1069Gln (c.3207C>A) | [62] |
| Denmark | 18 | p.His1069Gln c.3207C>A | [59] |
| 16 | p.Trp779STOP (c.2336G>A) | ||
| France | 15 | p.His1069Gln (c.3207C>A) | [22] |
| West Germany | 47.9 | p.His1069Gln (c.3207C>A) | [4] |
| East Germany | 63 | p.His1069Gln (c.3207C>A) | [58] |
| Greece | 35 | p.His1069Gln (c.3207C>A) | [29] |
| 12 | p.Arg969Gln (c.2906G>A) | ||
| Hungary | 42.9 | p.His1069Gln (c.3207C>A) | [57] |
| Iceland | 100 | p.Tyr670STOP (c.2007_2013del) | [77] |
| Italy (continental) | 17.5 | p.His1069Gln (c.3207C>A) | [74] |
| 9 | c.2530delA p.Val845fs | ||
| Netherlands | 33 | p.His1069Gln (c.3207C>A) | [39] |
| Poland | 72 | p.His1069Gln (c.3207C>A) | [31] |
| 7.3 | p.Ala1135GlnfsTer13 (c.3402delC) | ||
| 3.7 | p.Gln1351STOP (c.4051C>T) | ||
| Romania | 38.1 | p.His1069Gln (c.3207C>A) | [60] |
| Sardinia | 92 | c.-441_-427del p.Met822fs (c. 2463delC) | [13,73] |
| Spain | 27 | p.Met645Arg (c.1934 T>G) | [23] |
| Sweden | 38 | p.His1069Gln (c.3207C>A) | [78] |
| Turkey | 17.4 | p.His1069Gln (c.3207C>A) | [79] |
| 5.3 | p.Gly710Ser (c.2128G>A) | ||
| Great Britain | 19 | p.His1069Gln (c.3207C>A) | [21] |
| 8 | p.Met769Val (c.2305A>G) | ||
| China | 31 | p.Arg778Leu (c.2332C>T) | [28] |
| 10 | p.Pro992Leu (c.2975C>T) | [70] | |
| 29 | p.Arg778Leu (c.2332C>T) | [8] | |
| Northern India | 12 | p.Ile1102Thr (c.3305 T>C) | [42] |
| 9 | p.Pro992His (c.2975C>T) | ||
| South India | 11 | p.Cys271STOP (c.813C>A) | [80] |
| 9 | p.Pro768Leu (c.2303C>T) | [81] | |
| Japan | 17.95 | p.Asn958fs (c.2871delC) | [26] |
| 16.7 | p.Arg778Leu (c.2332C>T) | [70] | |
| Korea | 37.9 | p.Arg778Leu (c.2332C>T) | [27] |
| 12.1 | p.Asn1270Ser (c.3809A>G) | ||
| Saudi Arabia | 32 | p.Gln1399Arg (c.4196A>G) | [75] |
| 16 | p.Ser774Arg (c.2230 T>C) | ||
| Iran | 19 | p.His1069Gln (c.3207C>A) | [14] |
| USA | 40.3 | p.His1069Gln (c.3207C>A) | [26] |
| 1.9 | p.Asn1270Ser (c.3809A>G) | [26] | |
| 1.9 | p.Gly1266Arg (c.3796G>A) | ||
| Brazil | 37.1 | p.His1069Gln (c.3207C>A) | [25] |
| 11.4 | p.Ala1135GlnfsTer13 (c.3402delC) | ||
| Venezuela | 26.9 | p.Ala1135GlnfsTer13 (c.3402delC) | [82] |
| 9.6 | p.Gly691Arg (c.2071G>A) |
| Country | Actual Prevalence of WD | Old Prevalence of WD | Carriage Status of WD According to Whole Exome Sequencing Data |
|---|---|---|---|
| Great Britain | 1:30,000 | 1:7026 | 1:25 |
| France | 1:67,000 | 1:9000 | 1:31 |
| Korea | 1:53 | ||
| Russia | 1:167,000 | 1:10,000 | 1:43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovchinnikova, E.V.; Garbuz, M.M.; Ovchinnikova, A.A.; Kumeiko, V.V. Epidemiology of Wilson’s Disease and Pathogenic Variants of the ATP7B Gene Leading to Diversified Protein Disfunctions. Int. J. Mol. Sci. 2024, 25, 2402. https://doi.org/10.3390/ijms25042402
Ovchinnikova EV, Garbuz MM, Ovchinnikova AA, Kumeiko VV. Epidemiology of Wilson’s Disease and Pathogenic Variants of the ATP7B Gene Leading to Diversified Protein Disfunctions. International Journal of Molecular Sciences. 2024; 25(4):2402. https://doi.org/10.3390/ijms25042402
Chicago/Turabian StyleOvchinnikova, Elena Vasilievna, Mikhail Maksimovich Garbuz, Anna Aleksandrovna Ovchinnikova, and Vadim Vladimirovich Kumeiko. 2024. "Epidemiology of Wilson’s Disease and Pathogenic Variants of the ATP7B Gene Leading to Diversified Protein Disfunctions" International Journal of Molecular Sciences 25, no. 4: 2402. https://doi.org/10.3390/ijms25042402