
Comparative Mitogenomics Analysis Revealed Evolutionary Divergence among Neopestalotiopsis Species Complex (Fungi: Xylariales)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
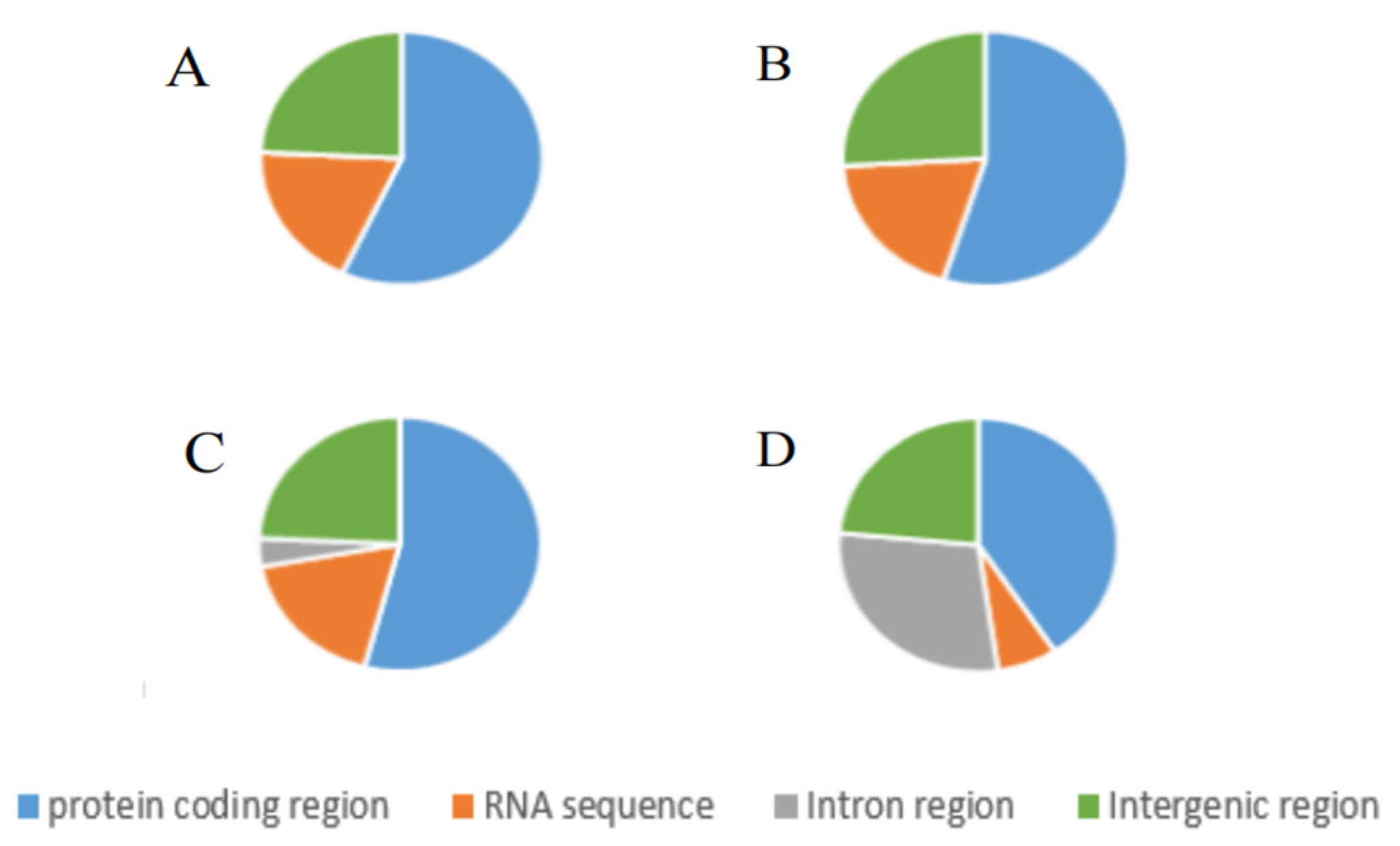
2.1. Structure and Organisation of the Mitochondrial Genome of N. cubana
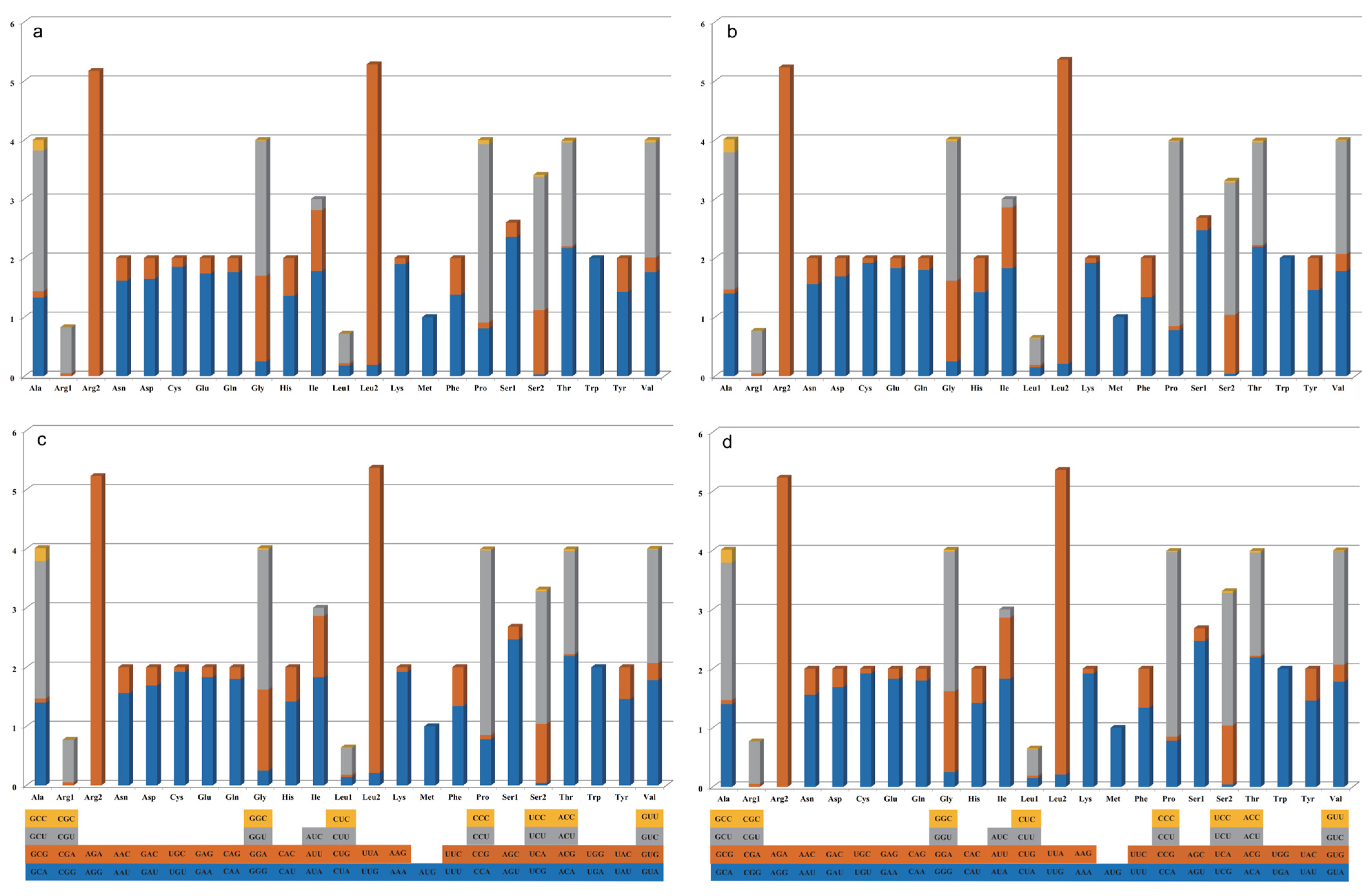
2.2. Codon Usage Analysis
2.3. RNA Genes in the N. cubana Mitogenomes
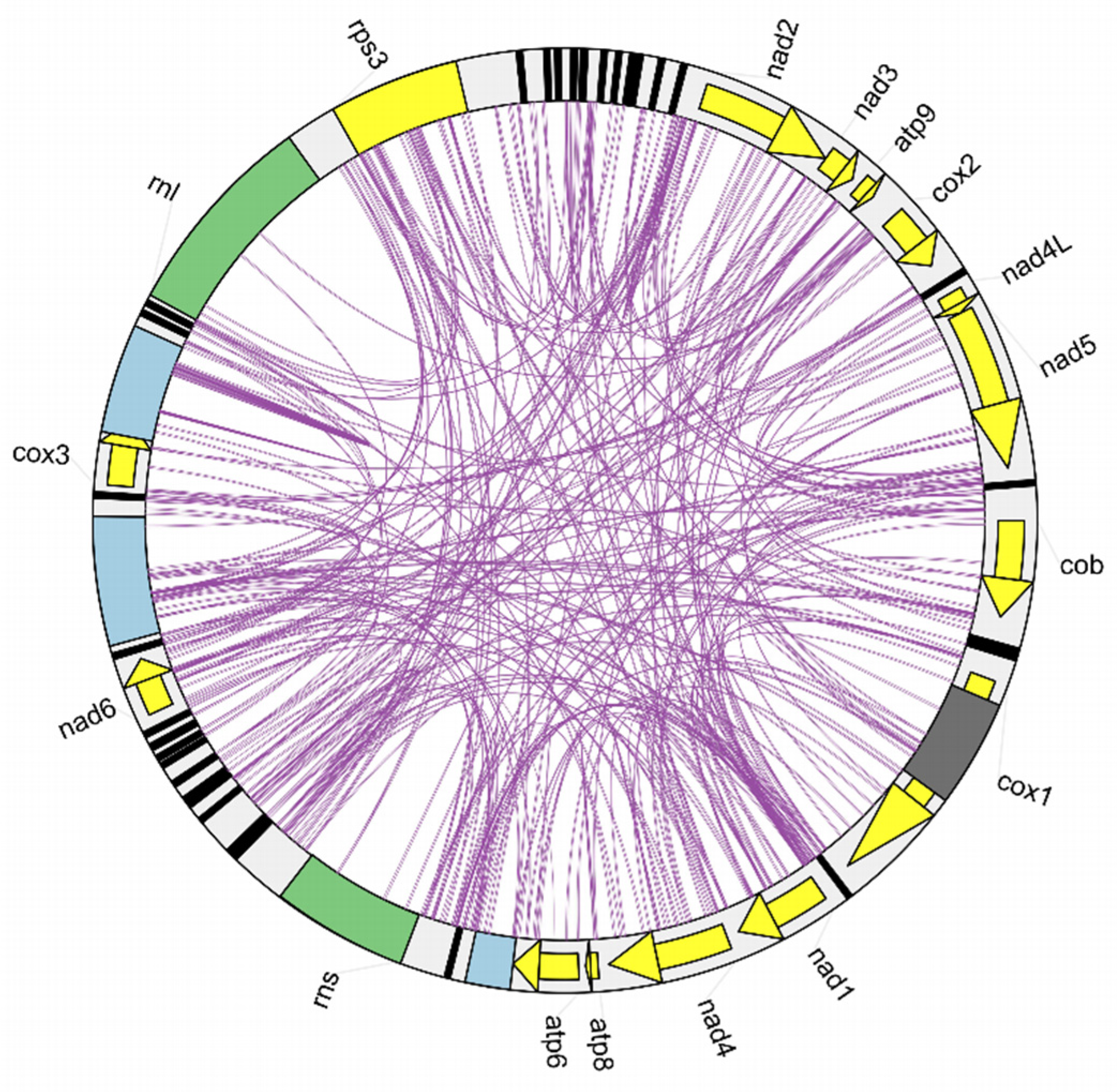
2.4. Repetitive Sequences in the Mitogenome
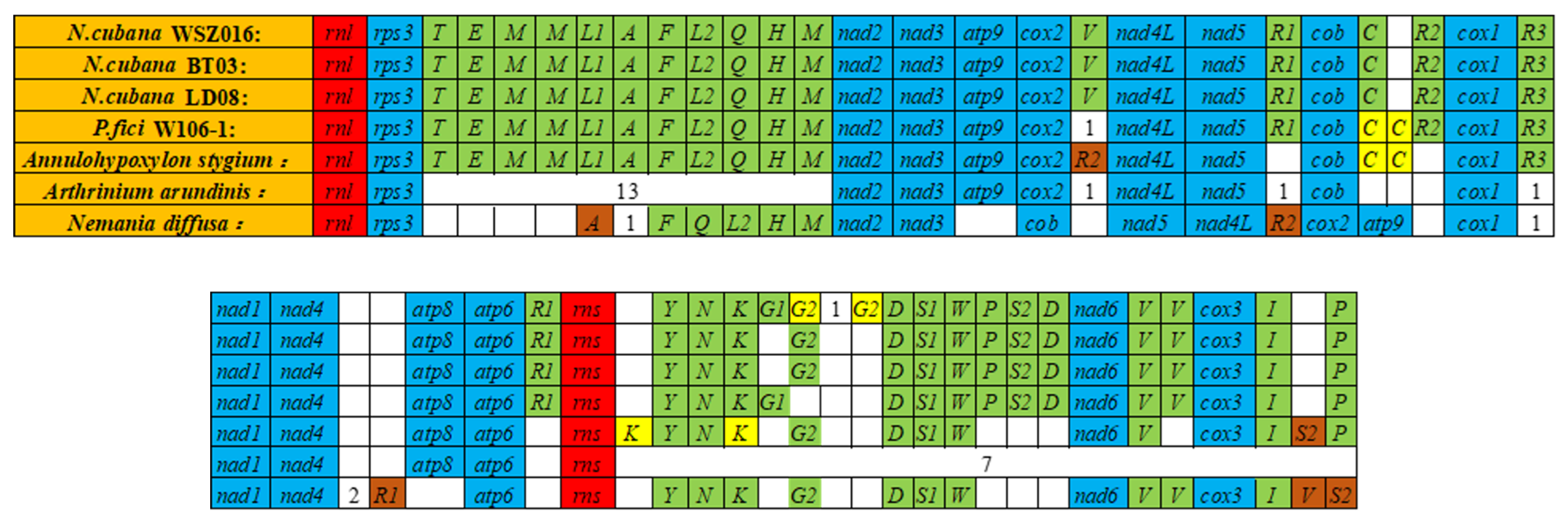
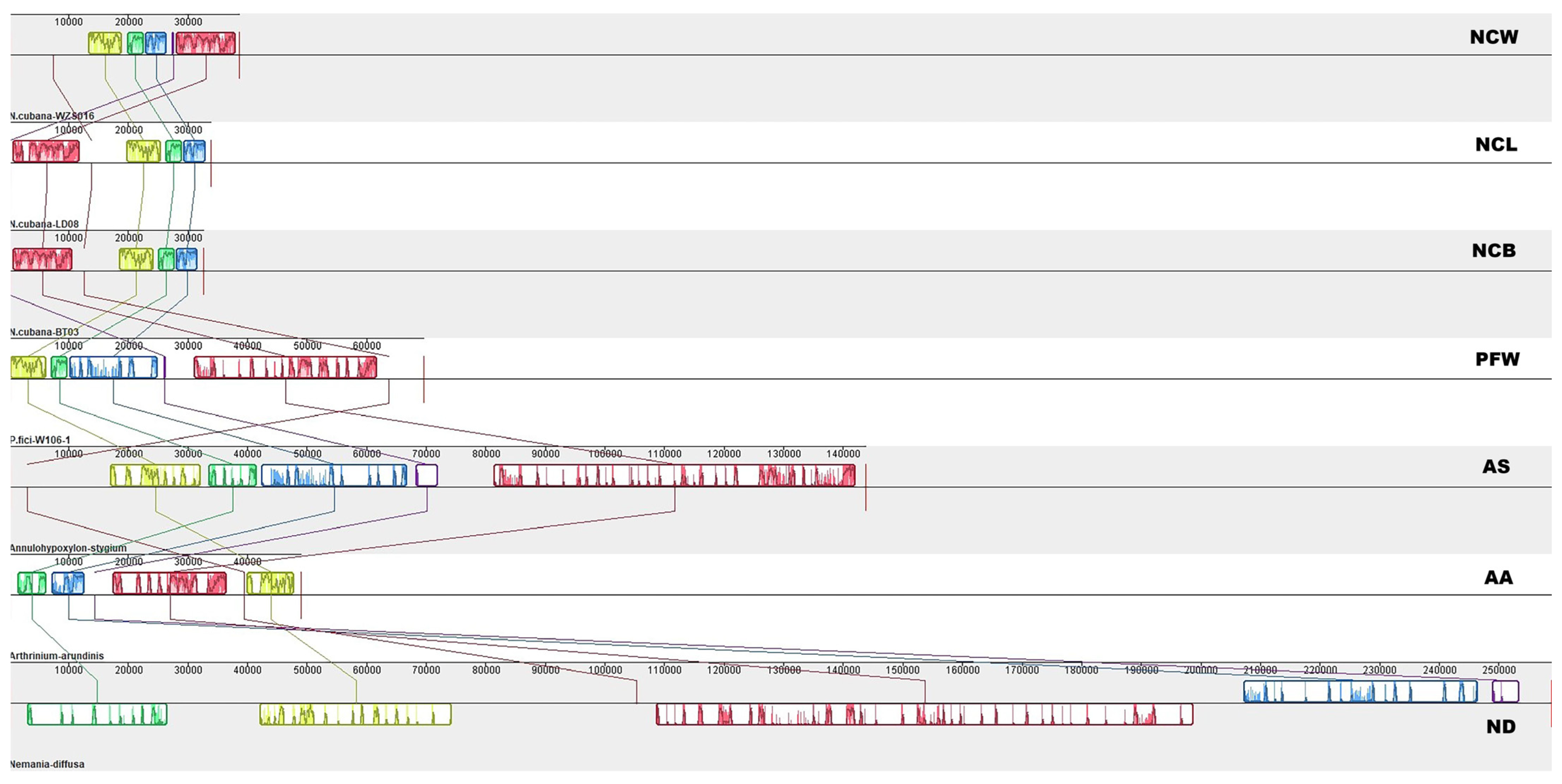
2.5. Gene Rearrangement in the Mitochondrial Genome
2.6. Variations in Core PCGs
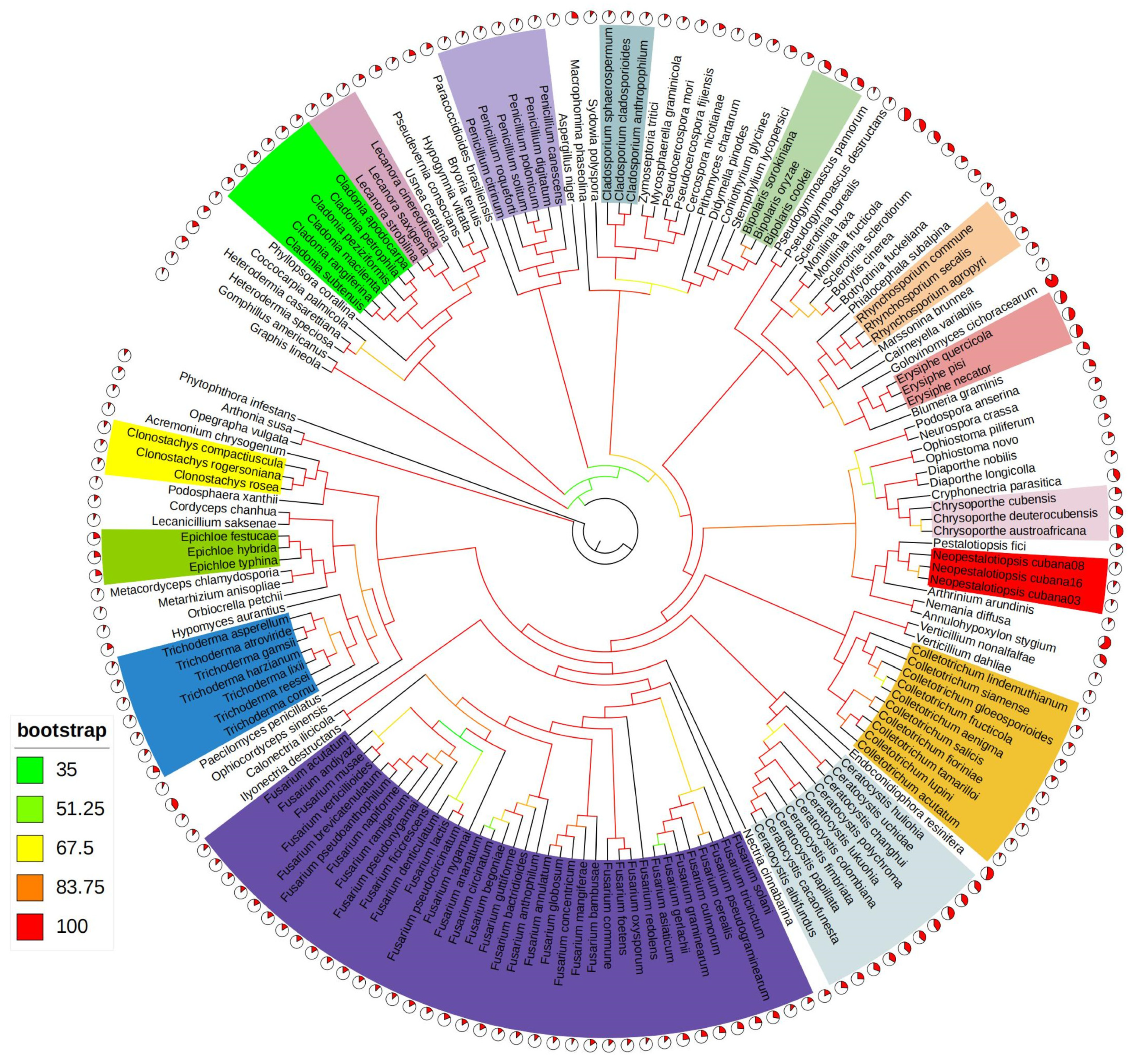
2.7. Phylogenetic Analysis
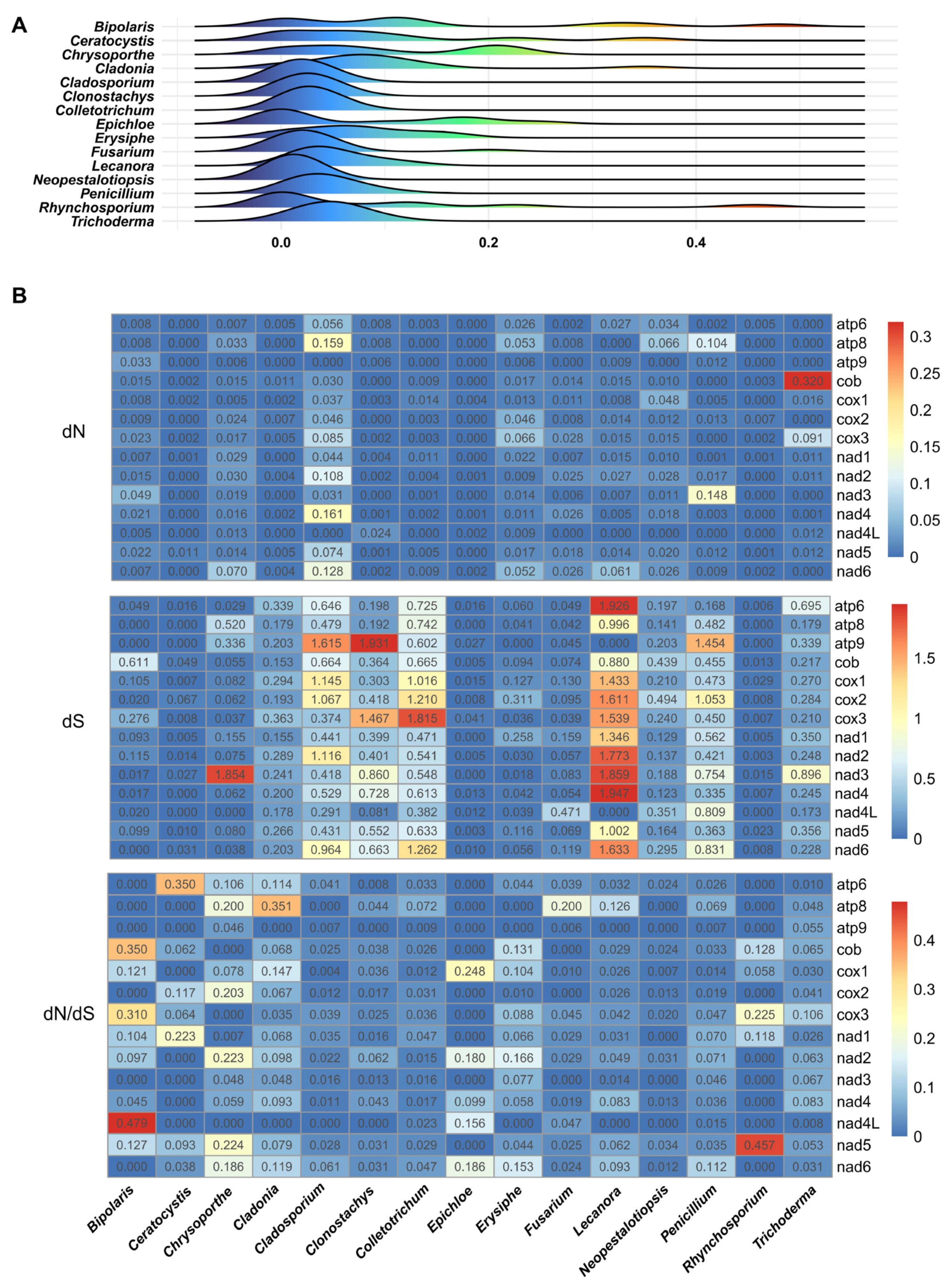
2.8. Evolutionary Adaptation of Core PCGs
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. Sequencing, Assembly, and Annotation
4.3. Comparative Analysis of the Mitogenomes of a Variety of Fungi
4.4. Phylogenetic Analysis
4.5. Availability of Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sandor, S.; Zhang, Y.; Xu, J. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 2018, 102, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, D.; Goremykin, V.; Xiao, J.; Xu, Y.; Garg, S.; Zhang, C.; Martin, W.F.; Zhu, R. Phylogenetic analyses with systematic taxon sampling show that mitochondria branch within Alphaproteobacteria. Nat. Ecol. Evol. 2020, 4, 1213–1219. [Google Scholar]
- Aguileta, G.; de Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldon, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Tian, Y.; Liu, W.; Lin, C.; He, F.; Yang, J.; Miao, W.; Li, Z. Mitochondrial characteristics of the powdery mildew genus Erysiphe revealed an extraordinary evolution in protein-coding genes. Int. J. Biol. Macromol. 2023, 230, 123153. [Google Scholar] [CrossRef] [PubMed]
- Abuduaini, A.; Wang, Y.-B.; Zhou, H.-Y.; Kang, R.-P.; Ding, M.-L.; Jiang, Y.; Suo, F.-Y.; Huang, L.-D. The complete mitochondrial genome of Ophiocordyceps gracilis and its comparison with related species. IMA Fungus 2021, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, G.; Ozkilinc, H. Pan-mitogenomics approach discovers diversity and dynamism in the prominent brown rot fungal pathogens. Front. Microbiol. 2021, 12, 647989. [Google Scholar] [CrossRef]
- Li, Q.; Li, L.; Zhang, T.; Xiang, P.; Wu, Q.; Tu, W.; Bao, Z.; Zou, L.; Chen, C. The first two mitochondrial genomes for the genus Ramaria reveal mitochondrial genome evolution of Ramaria and phylogeny of Basidiomycota. IMA Fungus 2022, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Wang, D.; Wang, Y.; Mustafa, G.; Yu, H. Mitogenomic and Phylogenetic Analysis of the Entomopathogenic Fungus Ophiocordyceps lanpingensis and Comparative Analysis with Other Ophiocordyceps Species. Genes 2023, 14, 710. [Google Scholar] [CrossRef]
- Fang, J.; Mamut, R.; Wang, L.; Anwar, G. De novo mitochondrial genome sequencing of Cladonia subulata and phylogenetic analysis with other dissimilar species. PLoS ONE 2023, 18, e0285818. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.-D.; Yang, R.-H.; Hsiang, T.; Wang, K.; Liang, D.-Q.; Liang, F.; Cao, D.-M.; Zhou, F.; Wen, G. Complete mitochondrial genome of the medicinal fungus Ophiocordyceps sinensis. Sci. Rep. 2015, 5, 13892. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.-J.; Li, Z.-L. Complete mitogenome of the entomopathogenic fungus Sporothrix insectorum RCEF 264 and comparative mitogenomics in Ophiostomatales. Appl. Microbiol. Biotechnol. 2019, 103, 5797–5809. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, J.; Wang, Y.-H.; Wang, K.; Wang, R.-L.; Zhao, X.-C.; Yao, Y.-J. Complete mitochondrial genome of Pleurocordyceps sinensis (Hypocreales, Ascomycota), a species with uncertain family-level taxonomic assignment. Qual. Assur. Saf. Crops Foods 2022, 14, 212–226. [Google Scholar] [CrossRef]
- Zhong, C.; Jin, J.; Zhou, R.; Liu, H.; Xie, J.; Wan, D.; Xiao, S.; Zhang, S. Comparative analysis of the complete mitochondrial genomes of four cordyceps fungi. Ecol. Evol. 2022, 12, e8818. [Google Scholar] [CrossRef] [PubMed]
- Brankovics, B.; van Dam, P.; Rep, M.; de Hoog, G.S.; van der Lee, T.A.J.; Waalwijk, C.; van Diepeningen, A.D. Mitochondrial genomes reveal recombination in the presumed asexual Fusarium oxysporum species complex. BMC Genom. 2017, 18, 735. [Google Scholar] [CrossRef] [PubMed]
- Férandon, C.; Moukha, S.; Callac, P.; Benedetto, J.-P.; Castroviejo, M.; Barroso, G. The Agaricus bisporus cox1 gene: The longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS ONE 2010, 5, e14048. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, L.; Cai, Y.; Tao, W.; Zhang, Y.-J.; Huang, B. Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl. Microbiol. Biotechnol. 2019, 103, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Al-Reedy, R.M.; Malireddy, R.; Dillman, C.B.; Kennell, J.C. Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet. Biol. 2012, 49, 2–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhang, G.; Liu, X.; Wang, C.; Xu, J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 2015, 77, 95–107. [Google Scholar] [CrossRef]
- Zhang, S.; Hao, A.-J.; Zhao, Y.-X.; Zhang, X.-Y.; Zhang, Y.-J. Comparative mitochondrial genomics toward exploring molecular markers in the medicinal fungus Cordyceps militaris. Sci. Rep. 2017, 7, 40219. [Google Scholar] [CrossRef]
- Maharachchikumbura, S.S.; Hyde, K.D.; Jones, E.G.; McKenzie, E.H.; Huang, S.-K.; Abdel-Wahab, M.A.; Daranagama, D.A.; Dayarathne, M.; D’souza, M.J.; Goonasekara, I.D. Towards a natural classification and backbone tree for Sordariomycetes. Fungal Divers. 2015, 72, 199–301. [Google Scholar] [CrossRef]
- Li, G.J.; Hyde, K.D.; Zhao, R.L.; Hongsanan, S.; Abdel-Aziz, F.A.; Abdel-Wahab, M.A.; Alvarado, P.; Alves-Silva, G.; Ammirati, J.F.; Ariyawansa, H.A. Fungal diversity notes 253–366: Taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers. 2016, 78, 1–237. [Google Scholar] [CrossRef]
- Maharachchikumbura, S.S.; Larignon, P.; Hyde, K.D.; Al-Sadi, A.M.; Liu, Z.-Y. Characterization of Neopestalotiopsis, Pestalotiopsis and Truncatella species associated with grapevine trunk diseases in France. Phytopathol. Mediterr. 2016, 55, 380–390. [Google Scholar]
- Gerardo-Lugo, S.S.; Tovar-Pedraza, J.M.; Maharachchikumbura, S.S.; Apodaca-Sánchez, M.A.; Correia, K.C.; Sauceda-Acosta, C.P.; Camacho-Tapia, M.; Hyde, K.D.; Marraiki, N.; Elgorban, A.M. Characterization of Neopestalotiopsis species associated with mango grey leaf spot disease in Sinaloa, Mexico. Pathogens 2020, 9, 788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, X.N.; Zhang, X.L.; Liu, X.Z.; Zhang, Y.J. Complete mitochondrial genome of the endophytic fungus Pestalotiopsis fici: Features and evolution. Appl. Microbiol. Biotechnol. 2017, 101, 1593–1604. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Lang, B.F.; Beck, N.; Prince, S.; Sarrasin, M.; Rioux, P.; Burger, G. Mitochondrial genome annotation with MFannot: A critical analysis of gene identification and gene model prediction. Front. Plant Sci. 2023, 14, 1222186. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. [Google Scholar]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Xia, R. A painless way to customize Circos plot: From data preparation to visualization using TBtools. iMeta 2022, 1, e35. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, K.; Zhao, F.; Liu, W.; Li, L.; Hua, Z.; Zhou, X. itol.toolkit accelerates working with iTOL (Interactive Tree of Life) by an automated generation of annotation files. BMC Bioinform. 2023, 39, btad339. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Wang, H.; Huo, S.; Lu, J.; Norvienyeku, J.; Miao, W.; Qin, C.; Liu, W. Comparative Mitogenomics Analysis Revealed Evolutionary Divergence among Neopestalotiopsis Species Complex (Fungi: Xylariales). Int. J. Mol. Sci. 2024, 25, 3093. https://doi.org/10.3390/ijms25063093
Huang Y, Wang H, Huo S, Lu J, Norvienyeku J, Miao W, Qin C, Liu W. Comparative Mitogenomics Analysis Revealed Evolutionary Divergence among Neopestalotiopsis Species Complex (Fungi: Xylariales). International Journal of Molecular Sciences. 2024; 25(6):3093. https://doi.org/10.3390/ijms25063093
Chicago/Turabian StyleHuang, Yutao, Huanwei Wang, Siyan Huo, Jinpeng Lu, Justice Norvienyeku, Weiguo Miao, Chunxiu Qin, and Wenbo Liu. 2024. "Comparative Mitogenomics Analysis Revealed Evolutionary Divergence among Neopestalotiopsis Species Complex (Fungi: Xylariales)" International Journal of Molecular Sciences 25, no. 6: 3093. https://doi.org/10.3390/ijms25063093