
The First High-Quality Genome Assembly of Freshwater Pearl Mussel Sinohyriopsis cumingii: New Insights into Pearl Biomineralization
 , and
, and
Abstract
:1. Introduction
2. Results
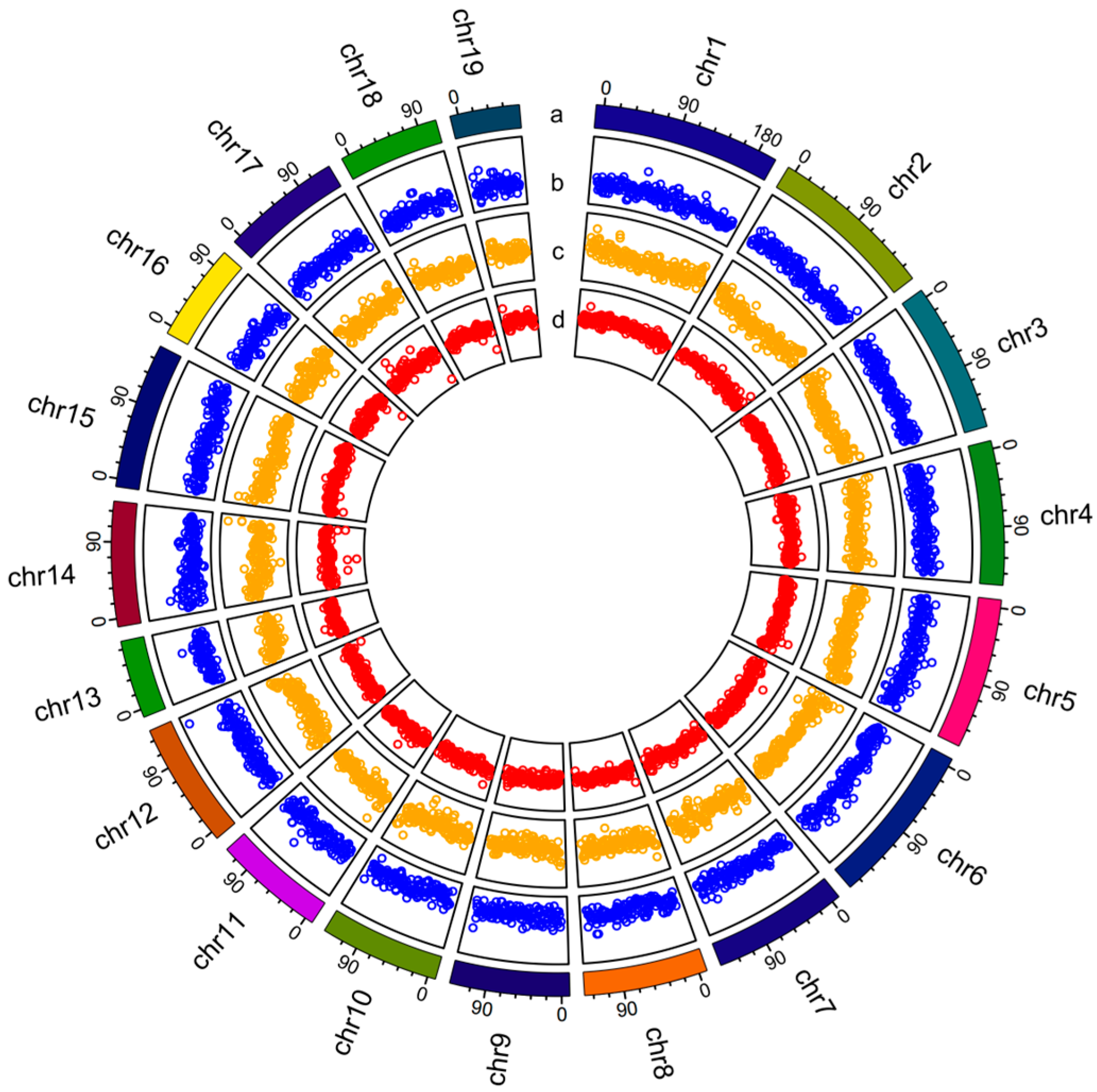
2.1. De Novo Sequencing and Genomic Characterization of S. cumingii
2.2. Repeat Element Identification and Genome Annotation
2.3. Gene Family Identification and Phylogenetic Analyses
2.4. Expansion and Contraction of Gene Families and Positively Selected Genes in the S. cumingii Genome
2.5. Fibrillin Family Genes in S. cumingii’s Genome
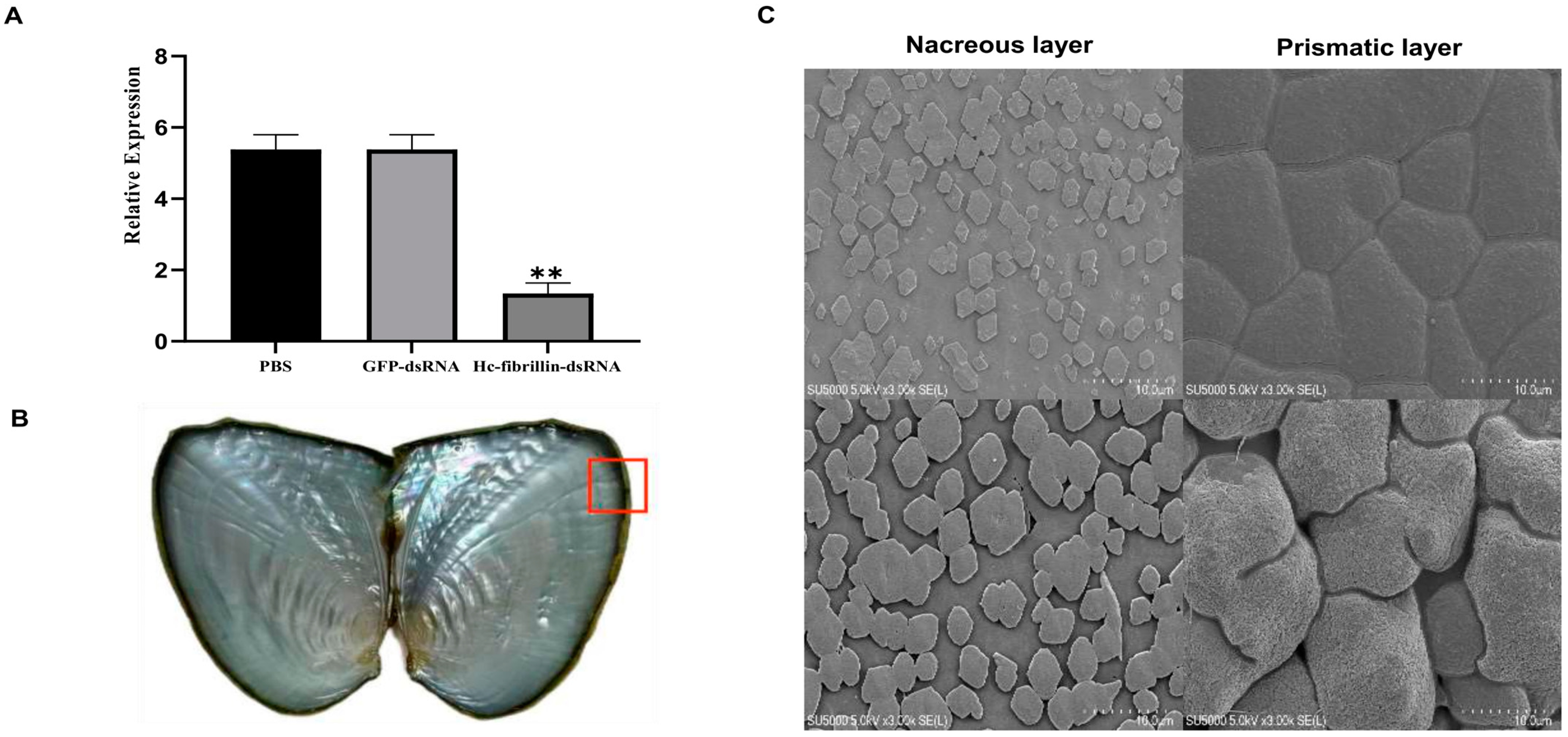
2.6. Function Analysis of One Key Fibrillin Gene Associated with Biomineralization in Sinohyriopsis cumingii
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and Sequencing
4.2. Repetitive Sequence Identification and Genome Annotation
4.3. Gene Family Identification and Phylogenetic Analysis
4.4. Gene Family Expansion and Contraction, and Positive Selection Analysis
4.5. Identification of Fibrillin Gene Family Members and Phylogenetic Analysis
4.6. Transcriptome Analysis
4.7. RNA Silencing Assay
4.8. In Vitro Calcium Carbonate Crystallization Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Taylor, J.; Strack, E. Pearl Production. In The Pearl Oyster; Southgate, P.C., Lucas, J.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 273–302. ISBN 9780444529763. [Google Scholar]
- Li, J.; Wang, D.; Bai, Z.; Guan, Y.; Wu, C.; Chen, L. Report on the Development of Freshwater Pearl Culture Industry in China. China Aquac. 2019, 23–29. [Google Scholar]
- Li, J.; Wang, G.; Bai, Z. Genetic Variability in Four Wild and Two Farmed Stocks of the Chinese Freshwater Pearl Mussel (Hyriopsis cumingii) Estimated by Microsatellite DNA Markers. Aquaculture 2009, 287, 286–291. [Google Scholar] [CrossRef]
- Griffith, A.W.; Gobler, C.J. Harmful Algal Blooms: A Climate Change Co-Stressor in Marine and Freshwater Ecosystems. Harmful Algae 2020, 91, 101590. [Google Scholar] [CrossRef]
- Addadi, L.; Joester, D.; Nudelman, F.; Weiner, S. Mollusk Shell Formation: A Source of New Concepts for Understanding Biomineralization Processes. Chem.-Eur. J. 2006, 12, 980–987. [Google Scholar] [CrossRef]
- Marin, F.; Luquet, G.; Marie, B.; Medakovic, D. Molluscan Shell Proteins: Primary Structure, Origin, and Evolution. Curr. Top. Dev. Biol. 2007, 80, 209–276. [Google Scholar] [CrossRef]
- Furuhashi, T.; Schwarzinger, C.; Miksik, I.; Smrz, M.; Beran, A. Molluscan Shell Evolution with Review of Shell Calcification Hypothesis. Comp. Biochem. Physiol.-B Biochem. Mol. Biol. 2009, 154, 351–371. [Google Scholar] [CrossRef]
- Kenneth, S.; Wilbur, K.M. Biomineralization: Cell Biology and Mineral Deposition. Q. Rev. Biol. 1989, 1, 257–264. [Google Scholar]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Bevelander, G.; Nakahara, H. An Electron Microscope Study of the Formation of the Nacreous Layer in the Shell of Certain Bivalve Molluscs. Calcif. Tissue Res. 1969, 3, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Takeuchi, T.; Luo, Y.J.; Ishikawa, A.; Kobayashi, T.; Koyanagi, R.; Villar-Briones, A.; Yamada, L.; Sawada, H.; Iwanaga, S.; et al. Dual Gene Repertoires for Larval and Adult Shells Reveal Molecules Essential for Molluscan Shell Formation. Mol. Biol. Evol. 2018, 35, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; McDougall, C.; Woodcroft, B.; Moase, P.; Rose, R.A.; Kube, M.; Reinhardt, R.; Rokhsar, D.S.; Montagnani, C.; Joubert, C.; et al. Parallel Evolution of Nacre Building Gene Sets in Molluscs. Mol. Biol. Evol. 2010, 27, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Marin, F. Mollusc Shellomes: Past, Present and Future. J. Struct. Biol. 2020, 212, 107583. [Google Scholar] [CrossRef]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The Oyster Genome Reveals Stress Adaptation and Complexity of Shell Formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.; Hu, X.; Xun, X.; Zhang, J.; Guo, X.; Jiao, W.; Zhang, L.; Liu, W.; Wang, J.; et al. Scallop Genome Reveals Molecular Adaptations to Semi-Sessile Life and Neurotoxins. Nat. Commun. 2017, 8, 1721. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Jiao, W.; Li, J.; Xun, X.; Sun, Y.; Guo, X.; Huan, P.; Dong, B.; Zhang, L.; et al. Scallop Genome Provides Insights into Evolution of Bilaterian Karyotype and Development. Nat. Ecol. Evol. 2017, 1, 120. [Google Scholar] [CrossRef]
- Du, X.; Fan, G.; Jiao, Y.; Zhang, H.; Guo, X.; Huang, R.; Zheng, Z.; Bian, C.; Deng, Y.; Wang, Q.; et al. The Pearl Oyster Pinctada Fucata martensii Genome and Multi-Omic Analyses Provide Insights into Biomineralization. GigaScience 2017, 6, gix059. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, F.; Mu, H.; Huang, M.; Bao, Y.; Wang, L.; Wong, N.K.; Xiao, S.; Dai, H.; Xiang, Z.; et al. The Genome of Nautilus pompilius Illuminates Eye Evolution and Biomineralization. Nat. Ecol. Evol. 2021, 5, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Calcino, A.D.; De Oliveira, A.L.; Simakov, O.; Schwaha, T.; Zieger, E.; Wollesen, T.; Wanninger, A. The Quagga Mussel Genome and the Evolution of Freshwater Tolerance. DNA Res. 2019, 26, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Guerra, D.; Hoeh, W.R.; Stewart, D.T.; Bogan, A.E.; Ghiselli, F.; Milani, L.; Passamonti, M.; Breton, S. Genome Survey of the Freshwater Mussel Venustaconcha ellipsiformis (Bivalvia: Unionida) Using a Hybrid de Novo Assembly Approach. Genome Biol. Evol. 2018, 10, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.L.; Grizzard, S.L.; Titus-McQuillan, J.E.; Bockrath, K.; Patel, S.; Wares, J.P.; Garner, J.T.; Moore, C.C. Gene Family Amplification Facilitates Adaptation in Freshwater Unionid Bivalve Megalonaias nervosa. Mol. Ecol. 2020, 30, 1155–1173. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.H. A High-Quality Reference Genome for a Parasitic Bivalve with Doubly Uniparental inheritance (Bivalvia: Unionida). Genome Biol. Evol. 2021, 13, evab029. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Dos-Santos, A.; Lopes-Lima, M.; Machado, A.M.; Marcos Ramos, A.; Usié, A.; Bolotov, I.N.; Vikhrev, I.V.; Breton, S.; Castro, L.F.C.; Da Fonseca, R.R.; et al. The Crown Pearl: A Draft Genome Assembly of the European Freshwater Pearl Mussel Margaritifera margaritifera (Linnaeus, 1758). DNA Res. 2021, 28, dsab002. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to Deep-Sea Chemosynthetic Environments as Revealed by Mussel Genomes. Nat. Ecol. Evol. 2017, 1, 121. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Dondero, F.; Dan Otto, T.; Costa, I.; Lima, N.C.B.; Americo, J.A.; Mazzoni, C.J.; Prosdocimi, F.; Rebelo, M.d.F. A Hybrid-Hierarchical Genome Assembly Strategy to Sequence the Invasive Golden Mussel, Limnoperna fortunei. GigaScience 2018, 7, gix128. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Liu, B.; Ma, B.; Liu, F.; Liu, G.; Shi, Q.; Wang, C. Draft Genome of the Peruvian Scallop Argopecten purpuratus. GigaScience 2018, 7, giy031. [Google Scholar] [CrossRef]
- Powell, D.; Subramanian, S.; Suwansa-Ard, S.; Zhao, M.; O’Connor, W.; Raftos, D.; Elizur, A.; Kohara, Y. The Genome of the Oyster Saccostrea Offers Insight into the Environmental Resilience of Bivalves. DNA Res. 2018, 25, 655–665. [Google Scholar] [CrossRef]
- Ran, Z.; Li, Z.; Yan, X.; Liao, K.; Kong, F.; Zhang, L.; Cao, J.; Zhou, C.; Zhu, P.; He, S.; et al. Chromosome-Level Genome Assembly of the Razor Clam Sinonovacula constricta (Lamarck, 1818). Mol. Ecol. Resour. 2019, 19, 1647–1658. [Google Scholar] [CrossRef]
- Yan, X.; Nie, H.; Huo, Z.; Ding, J.; Li, Z.; Yan, L.; Jiang, L.; Mu, Z.; Wang, H.; Meng, X.; et al. Clam Genome Sequence Clarifies the Molecular Basis of Its Benthic Adaptation and Extraordinary Shell Color Diversity. iScience 2019, 19, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.M.; Xin, L.S.; Rosani, U.; Wu, B.; Wang, Q.C.; Duan, X.K.; Liu, Z.H.; Wang, C.M. Chromosomal-Level Assembly of the Blood Clam, Scapharca (Anadara) broughtonii, Using Long Sequence Reads and Hi-C. GigaScience 2019, 8, giz067. [Google Scholar] [CrossRef] [PubMed]
- Thai, B.T.; Lee, Y.P.; Gan, H.M.; Austin, C.M.; Croft, L.J.; Trieu, T.A.; Tan, M.H. Whole Genome Assembly of the Snout Otter Clam, Lutraria rhynchaena, Using Nanopore and Illumina Data, Benchmarked Against Bivalve Genome Assemblies. Front. Genet. 2019, 10, 1158. [Google Scholar] [CrossRef]
- Wang, X.; Xu, W.; Wei, L.; Zhu, C.; He, C.; Song, H.; Cai, Z.; Yu, W.; Jiang, Q.; Li, L.; et al. Nanopore Sequencing and De Novo Assembly of a Black-Shelled Pacific Oyster (Crassostrea gigas) Genome. Front. Genet. 2019, 10, 1211. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, W.; Lu, J.; Zhang, Z.; Mu, C.; Song, W.; Migaud, H.; Wang, C.; Bekaert, M. The Whole-Genome Sequencing and Hybrid Assembly of Mytilus coruscus. Front. Genet. 2020, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.J.; McCarthy, S.A.; Dudchenko, O.; James, K.; Betteridge, E.; Corton, C.; Dolucan, J.; Mead, D.; Oliver, K.; Omer, A.D.; et al. The Gene-Rich Genome of the Scallop Pecten maximus. GigaScience 2020, 9, giaa037. [Google Scholar] [CrossRef]
- Wei, M.; Ge, H.; Shao, C.; Yan, X.; Nie, H.; Duan, H.; Liao, X.; Zhang, M.; Chen, Y.; Zhang, D.; et al. Chromosome-Level Clam Genome Helps Elucidate the Molecular Basis of Adaptation to a Buried Lifestyle. iScience 2020, 23, 101148. [Google Scholar] [CrossRef]
- Dong, Y.; Zeng, Q.; Ren, J.; Yao, H.; Lv, L.; He, L.; Ruan, W.; Xue, Q.; Bao, Z.; Wang, S.; et al. The Chromosome-Level Genome Assembly and Comprehensive Transcriptomes of the Razor Clam (Sinonovacula constricta). Front. Genet. 2020, 11, 664. [Google Scholar] [CrossRef]
- Yang, J.L.; Feng, D.D.; Liu, J.; Xu, J.K.; Chen, K.; Li, Y.F.; Zhu, Y.T.; Liang, X.; Lu, Y. Chromosome-Level Genome Assembly of the Hard-Shelled Mussel Mytilus coruscus, a Widely Distributed Species from the Temperate Areas of East Asia. GigaScience 2021, 10, giab024. [Google Scholar] [CrossRef]
- Teng, W.; Xie, X.; Nie, H.; Sun, Y.; Liu, X.; Yu, Z.; Zheng, J.; Liu, H.; Li, D.; Zhang, M.; et al. Chromosome-Level Genome Assembly of Scapharca kagoshimensis Reveals the Expanded Molecular Basis of Heme Biosynthesis in Ark Shells. Mol. Ecol. Resour. 2022, 22, 295–306. [Google Scholar] [CrossRef]
- Peñaloza, C.; Gutierrez, A.P.; Eöry, L.; Wang, S.; Guo, X.; Archibald, A.L.; Bean, T.P.; Houston, R.D. A Chromosome-Level Genome Assembly for the Pacific Oyster Crassostrea gigas. GigaScience 2021, 10, giab020. [Google Scholar] [CrossRef]
- Bao, Y.; Zeng, Q.; Wang, J.; Zhang, Z.; Zhang, Y.; Wang, S.; Wong, N.K.; Yuan, W.; Huang, Y.; Zhang, W.; et al. Genomic Insights into the Origin and Evolution of Molluscan Red-Bloodedness in the Blood Clam Tegillarca granosa. Mol. Biol. Evol. 2021, 38, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Liu, J.; Wang, C.; Wang, H.; Zhang, L.; Hu, J.; Bao, L.; Wang, S. High-Quality Reannotation of the King Scallop Genome Reveals No ‘Gene-Rich’ Feature and Evolution of Toxin Resistance. Comput. Struct. Biotechnol. J. 2021, 19, 4954–4960. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Suzuki, Y.; Watabe, S.; Nagai, K.; Masaoka, T.; Fujie, M.; Kawamitsu, M.; Satoh, N.; Myers, E.W. A High-Quality, Haplotype-Phased Genome Reconstruction Reveals Unexpected Haplotype Diversity in a Pearl Oyster. DNA Res. 2022, 29, dsac035. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Gyoja, F.; Tanaka, M.; Ikuta, T.; Shoguchi, E.; Fujiwara, M.; Shinzato, C.; Hisata, K.; et al. Draft Genome of the Pearl Oyster Pinctada fucata: A Platform for Understanding Bivalve Biology. DNA Res. 2012, 19, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Koyanagi, R.; Gyoja, F.; Kanda, M.; Hisata, K.; Fujie, M.; Goto, H.; Yamasaki, S.; Nagai, K.; Morino, Y.; et al. Bivalve-Specific Gene Expansion in the Pearl Oyster Genome: Implications of Adaptation to a Sessile Lifestyle. Zool. Lett. 2016, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Murgarella, M.; Puiu, D.; Novoa, B.; Figueras, A.; Posada, D.; Canchaya, C. A First Insight into the Genome of the Filter-Feeder Mussel Mytilus galloprovincialis. PLoS ONE 2016, 11, e0151561. [Google Scholar] [CrossRef]
- Mun, S.; Kim, Y.J.; Markkandan, K.; Shin, W.; Oh, S.; Woo, J.; Yoo, J.; An, H.; Han, K. The Whole-Genome and Transcriptome of the Manila Clam (Ruditapes philippinarum). Genome Biol. Evol. 2017, 9, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, L.; Hu, J.; Wang, J.; Bao, Z.; Wang, S. The Evo-Devo of Molluscs: Insights from a Genomic Perspective. Evol. Dev. 2020, 22, 409–424. [Google Scholar] [CrossRef]
- Brosius, J. Retroposons-Seeds of Evolution. Science 1991, 251, 753. [Google Scholar] [CrossRef]
- Juretic, N.; Hoen, D.R.; Huynh, M.L.; Harrison, P.M.; Bureau, T.E. The Evolutionary Fate of MULE-Mediated Duplications of Host Gene Fragments in Rice. Genome Res. 2005, 15, 1292–1297. [Google Scholar] [CrossRef]
- Harris, R.M.; Hofmann, H.A. Seeing Is Believing: Dynamic Evolution of Gene Families. Proc. Natl. Acad. Sci. USA 2015, 112, 1252–1253. [Google Scholar] [CrossRef]
- Fang, Z.; Yan, Z.; Li, S.; Wang, Q.; Cao, W.; Xu, G.; Xiong, X.; Xie, L.; Zhang, R. Localization of Calmodulin and Calmodulin-like Protein and Their Functions in Biomineralization in P. fucata. Prog. Nat. Sci. 2008, 18, 405–412. [Google Scholar] [CrossRef]
- Fernandez, M.; Arriagada, K.; Arias, J. SEM Localization of Proteoglycans in Abalone Shell (Haliotis rufescens). Microsc. Microanal. 2007, 13, 1462–1463. [Google Scholar] [CrossRef]
- Schwaner, C.; Farhat, S.; Haley, J.; Pales Espinosa, E.; Allam, B. Transcriptomic, Proteomic, and Functional Assays Underline the Dual Role of Extrapallial Hemocytes in Immunity and Biomineralization in the Hard Clam Mercenaria mercenaria. Front. Immunol. 2022, 13, 838530. [Google Scholar] [CrossRef]
- Mount, A.S.; Wheeler, A.P.; Paradkar, R.P.; Snider, D. Hemocyte-Mediated Shell Mineralization in the Eastern Oyster. Science 2004, 304, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, S.; Ramirez, F. Fibrillin Microfibrils in Bone Physiology. Matrix Biol. 2016, 52–54, 191–197. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R. Fibrillin Protein Pleiotropy: Acromelic Dysplasias. Matrix Biol. 2019, 80, 6–13. [Google Scholar] [CrossRef]
- Handford, P.A.; Baron, M.; Mayhew, M.; Willis, A.; Beesley, T.; Brownlee, G.G.; Campbell, I.D. The First EGF-like Domain from Human Factor IX Contains a High-Affinity Calcium Binding Site. EMBO J. 1990, 9, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.H.; Cheng, H.; Sweeney, W.V.; Pardi, A.; Tam, J.P. Sequence-Specific 1H NMR Assignments, Secondary Structure, and Location of the Calcium Binding Site in the First Epidermal Growth Factor Like Domain of Blood Coagulation Factor IX. Biochemistry 1991, 30, 7402–7409. [Google Scholar] [CrossRef]
- Liu, Y.; Annis, D.S.; Mosher, D.F. Interactions among the Epidermal Growth Factor-like Modules of Thrombospondin-1. J. Biol. Chem. 2009, 284, 22206–22212. [Google Scholar] [CrossRef] [PubMed]
- Valcarce, C.; Selander-Sunnerhagen, M.; Tamlitz, A.M.; Drakenberg, T.; Bjork, I.; Stenflo, J. Calcium Affinity of the NH2-Terminal Epidermal Growth Factor-like Module of Factor, X. Effect of the γ-Carboxyglutamic Acid-Containing Module. J. Biol. Chem. 1993, 268, 26673–26678. [Google Scholar] [CrossRef]
- Jin, C.; Zhao, J.; Pu, J.; Liu, X.; Li, J. Hichin, a Chitin Binding Protein Is Essential for the Self-Assembly of Organic Frameworks and Calcium Carbonate during Shell Formation. Int. J. Biol. Macromol. 2019, 135, 745–751. [Google Scholar] [CrossRef]
- Zhang, X.; Xia, Z.; Liu, X.; Li, J. The Novel Matrix Protein Hic7 of Hyriopsis cumingii Participates in the Formation of the Shell and Pearl. Comp. Biochem. Physiol. Part-B Biochem. Mol. Biol. 2021, 256, 110640. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, Z.; Ma, Z.; Liang, J.; Zhang, Z.; Yao, L.; Chen, X.; Liu, X.; Zhang, R. Shell Matrix Protein N38 of Pinctada fucata, Inducing Vaterite Formation, Extends the DING Protein to the Mollusca World. Mar. Biotechnol. 2022, 24, 531–541. [Google Scholar] [CrossRef]
- Lakshminarayanan, R.; Chi-Jin, E.O.; Loh, X.J.; Kini, R.M.; Valiyaveettil, S. Purification and Characterization of a Vaterite-Inducing Peptide, Pelovaterin, from the Eggshells of Pelodiscus sinensis (Chinese Soft-Shelled Turtle). Biomacromolecules 2005, 6, 1429–1437. [Google Scholar] [CrossRef]
- Natoli, A.; Wiens, M.; Schröder, H.C.; Stifanic, M.; Batel, R.; Soldati, A.L.; Jacob, D.E.; Müller, W.E.G. Bio-Vaterite Formation by Glycoproteins from Freshwater Pearls. Micron 2010, 41, 359–366. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, D.; Yang, X.; Liu, C.; Xie, J.; Zheng, G.; Xie, L.; Zhang, R. A Novel Matrix Protein, PfY2, Functions as a Crucial Macromolecule during Shell Formation. Sci. Rep. 2017, 7, 6021. [Google Scholar] [CrossRef]
- Guillaume, M.; Carl, K. A Fast, Lock-free Approach for Efficient Parallel Counting of Occurrences of K-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Coombe, L.; Warren, R.L.; Jackman, S.D.; Yang, C.; Vandervalk, B.P.; Moore, R.A.; Pleasance, S.; Coope, R.J.; Bohlmann, J.; Holt, R.A.; et al. Assembly of the Complete Sitka spruce Chloroplast Genome Using 10X Genomics’ GemCode Sequencing Data. PLoS ONE 2016, 11, 0163059. [Google Scholar] [CrossRef]
- Bai, Z.Y.; Han, X.K.; Liu, X.J.; Li, Q.Q.; Li, J. Le Construction of a High-Density Genetic Map and QTL Mapping for Pearl Quality-Related Traits in Hyriopsis cumingii. Sci. Rep. 2016, 6, 32608. [Google Scholar] [CrossRef]
- Bai, Z.; Han, X.; Luo, M.; Lin, J.; Wang, G.; Li, J. Constructing a Microsatellite-Based Linkage Map and Identifying QTL for Pearl Quality Traits in Triangle Pearl Mussel (Hyriopsis cumingii). Aquaculture 2015, 437, 102–110. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Shi, L.; Gong, D.; Zhang, S.; Zhao, Q.; Zhan, D.; Vasseur, L.; Wang, Y.; Yu, J.; et al. Haplotype-Resolved Genome Assembly Provides Insights into Evolutionary History of the Tea Plant Camellia Sinensis. Nat. Genet. 2021, 53, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 4–9. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 1–14. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Robin, C.R.; Wortman, J.R. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A Web Server for Annotation and Identification of Enriched Pathways and Diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Bielawski, J.P.; Baker, J.L.; Mingrone, J. Inference of Episodic Changes in Natural Selection Acting on Protein Coding Sequences via CODEML. Curr. Protoc. Bioinform. 2016, 2016, 1–32. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Liu, F.; Li, Y.; Yu, H.; Zhang, L.; Hu, J.; Bao, Z.; Wang, S. MolluscDB: An Integrated Functional and Evolutionary Genomics Database for the Hyper-Diverse Animal Phylum Mollusca. Nucleic Acids Res. 2021, 49, D988–D997. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile Hidden Markov Models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB Bioinformatics Resource Portal. Nucleic Acids Res. 2012, 40, 597–603. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Value |
|---|---|
| Number of contigs | 1808 |
| Contig N50 (bp) | 5,295,426 |
| Contig N90 (bp) | 1,144,919 |
| Total length of contigs (bp) | 2,904,942,185 |
| Element | Number of Elements | Length Occupied (bp) | Percentage |
|---|---|---|---|
| Retroelements | 781,650 | 359,759,912 | 12.38% |
| SINEs | 0 | 0 | 0.00% |
| Penelope | 18,874 | 7,315,525 | 0.25% |
| LINEs | 258,394 | 160,402,819 | 5.52% |
| CRE/SLACS | 0 | 0 | 0.00% |
| L2/CR1/Rex | 83,861 | 48,684,191 | 1.68% |
| R1/LOA/Jockey | 38,560 | 33,760,425 | 1.16% |
| R2/R4/NeSL | 3247 | 596,989 | 0.02% |
| RTE/Bov-B | 69,197 | 46,843,959 | 1.61% |
| L1/CIN4 | 1401 | 864,374 | 0.03% |
| LTR elements | 523,256 | 199,357,093 | 6.86% |
| BEL/Pao | 3768 | 1,264,899 | 0.04% |
| Ty1/Copia | 22 | 6808 | 0.00% |
| Gypsy/DIRS1 | 152,335 | 87,172,551 | 3.00% |
| Retroviral | 0 | 0 | 0.00% |
| DNA transposons | 400,216 | 383,834,032 | 13.21% |
| hobo-Activator | 64,261 | 23,061,802 | 0.79% |
| Tc1-IS630-Pogo | 34,110 | 12,365,403 | 0.43% |
| En-Spm | 0 | 0 | 0.00% |
| MuDR-IS905 | 0 | 0 | 0.00% |
| PiggyBac | 3422 | 1,122,485 | 0.04% |
| Tourist/Harbinger | 0 | 0 | 0.00% |
| Other (Mirage, P-element, Transib) | 6018 | 2,447,569 | 0.08% |
| Rolling-circles | 0 | 0 | 0.00% |
| Unclassified | 3,228,829 | 670,454,699 | 23.08% |
| Total interspersed repeats | 1,414,048,643 | 48.68% | |
| Small RNA | 0 | 0 | 0.00% |
| Satellites | 1 | 413 | 0.00% |
| Simple repeats | 922,774 | 60,734,831 | 2.09% |
| Low complexity | 54,102 | 2,669,708 | 0.09% |
| Total number of elements | 5,387,572 | 1,477,453,595 | 100.00% |
| Section | Results |
|---|---|
| Genome size (bp) | 2,904,942,185 |
| Repeat sequence (bp) | 1,477,453,595 |
| Number of genes | 37,696 |
| Gene average length (CDS) | 1820.50 |
| Gene average length (DNA) | 37,988.60 |
| Exon number per gene | 6.77 |
| Exon average length (bp) | 218.78 |
| Genome GC content (%) | 36.07% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Z.; Lu, Y.; Hu, H.; Yuan, Y.; Li, Y.; Liu, X.; Wang, G.; Huang, D.; Wang, Z.; Mao, Y.; et al. The First High-Quality Genome Assembly of Freshwater Pearl Mussel Sinohyriopsis cumingii: New Insights into Pearl Biomineralization. Int. J. Mol. Sci. 2024, 25, 3146. https://doi.org/10.3390/ijms25063146
Bai Z, Lu Y, Hu H, Yuan Y, Li Y, Liu X, Wang G, Huang D, Wang Z, Mao Y, et al. The First High-Quality Genome Assembly of Freshwater Pearl Mussel Sinohyriopsis cumingii: New Insights into Pearl Biomineralization. International Journal of Molecular Sciences. 2024; 25(6):3146. https://doi.org/10.3390/ijms25063146
Chicago/Turabian StyleBai, Zhiyi, Ying Lu, Honghui Hu, Yongbin Yuan, Yalin Li, Xiaojun Liu, Guiling Wang, Dandan Huang, Zhiyan Wang, Yingrui Mao, and et al. 2024. "The First High-Quality Genome Assembly of Freshwater Pearl Mussel Sinohyriopsis cumingii: New Insights into Pearl Biomineralization" International Journal of Molecular Sciences 25, no. 6: 3146. https://doi.org/10.3390/ijms25063146