
Unveiling Prognostic RNA Biomarkers through a Multi-Cohort Study in Colorectal Cancer


Abstract
:1. Introduction
2. Results
2.1. Characteristics of Each Cohort
2.2. Cox Regression Analysis Test Result
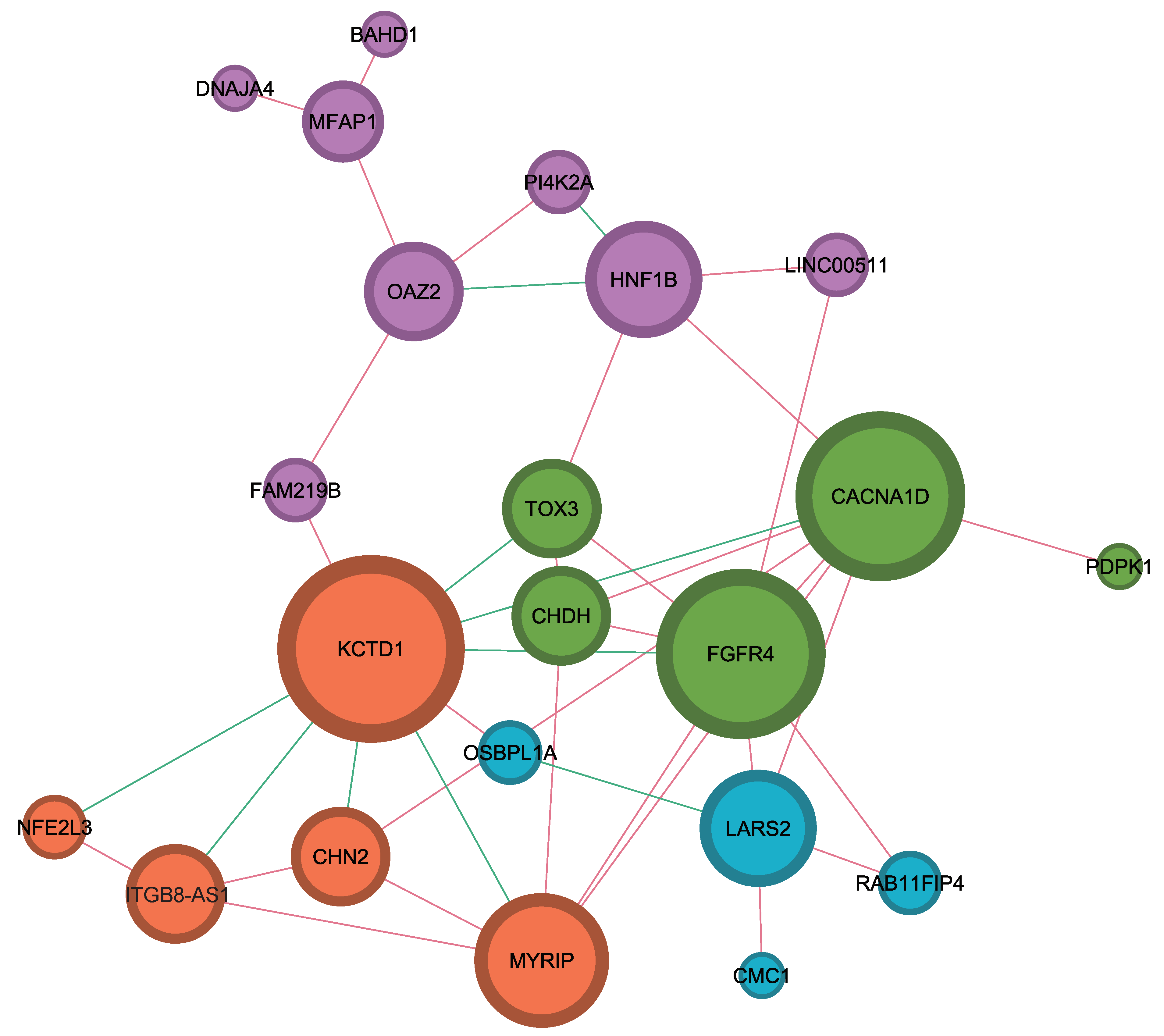
2.3. Identification of Interaction among Hub Genes Using Network Analysis
2.4. Development of Prognostic Prediction Model and Performance Evaluation Using Pivotal RNA Biomarkers
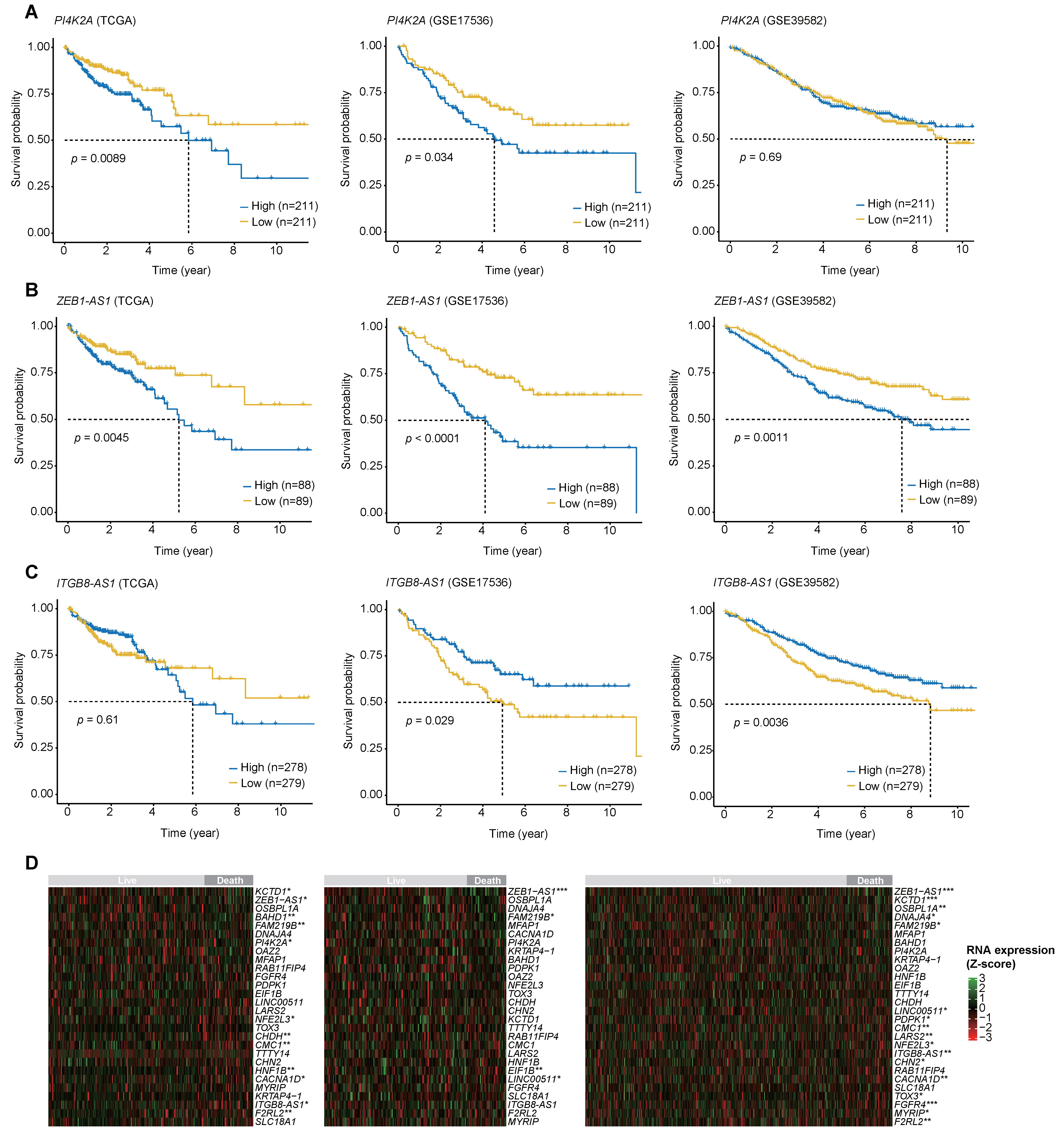
2.5. The Log-Rank Test Results for the Three Biomarkers with the Highest MDG Values and the RNA Expression Heatmaps
3. Discussion
4. Materials and Methods
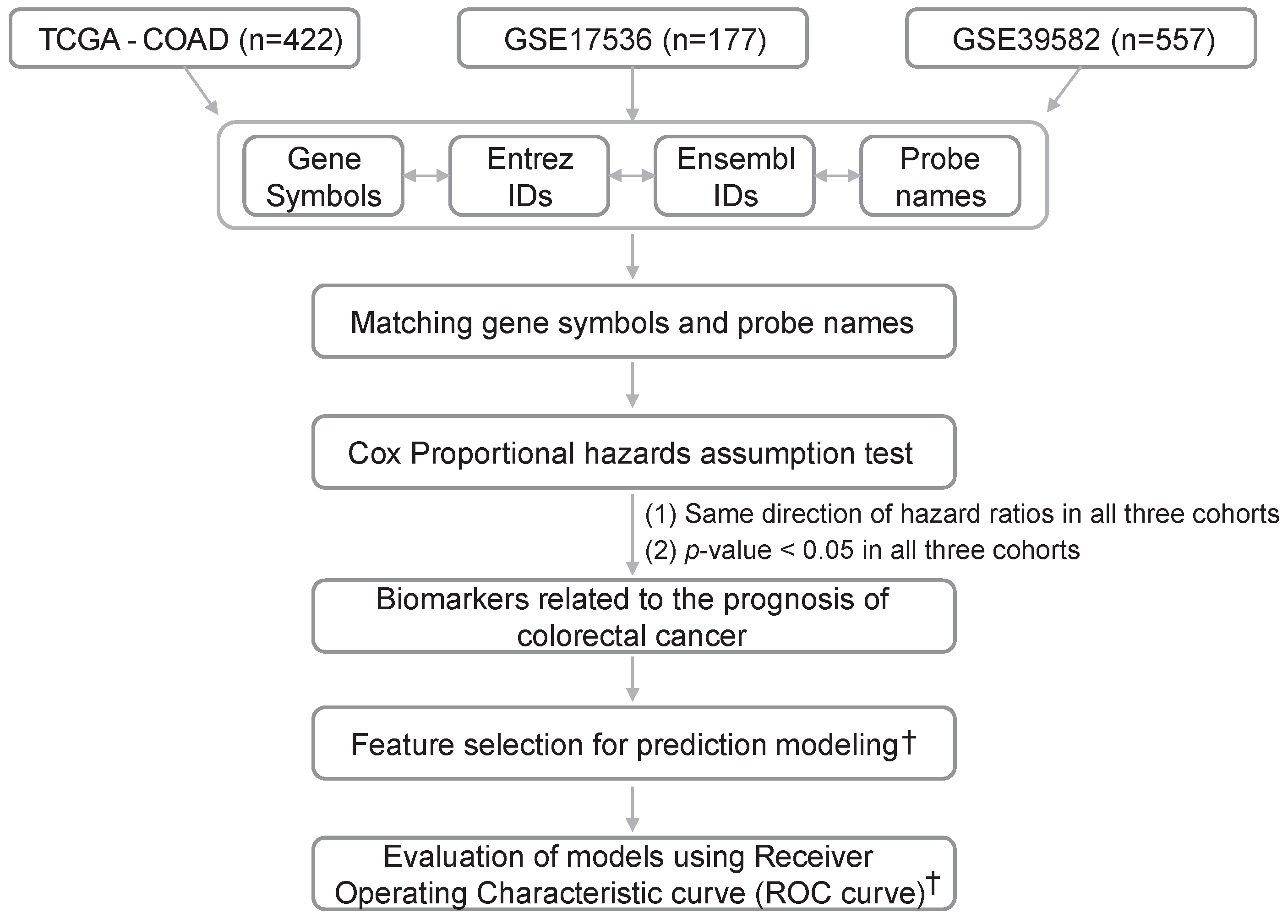
4.1. Data Acquisition and Preprocessing of Gene Expression Data
4.2. Selecting a List of Genes Satisfying the Cox Proportional Hazard Assumption
4.3. Network Analysis of Genes with Statistical Significance for Survival
4.4. Feature Selection for Survival Prediction
4.5. Evaluation of Predicting Accuracy
4.6. Pathway Analysis
4.7. Statistical Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Marley, A.R.; Nan, H. Epidemiology of colorectal cancer. Int. J. Mol. Epidemiol. Genet. 2016, 7, 105–114. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Binefa, G.; Rodriguez-Moranta, F.; Teule, A.; Medina-Hayas, M. Colorectal cancer: From prevention to personalized medicine. World J. Gastroenterol. 2014, 20, 6786–6808. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Nakamura, Y. Cancer precision medicine: From cancer screening to drug selection and personalized immunotherapy. Trends Pharmacol. Sci. 2017, 38, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; on behalf of the IBCD-Faculty; Moore, H.; Martens, J.W.M.; Lively, T.; Malik, S.; McDermott, U.; Michiels, S.; Moscow, J.A.; Tejpar, S.; et al. Steps forward for cancer precision medicine. Nat. Rev. Drug Discov. 2018, 17, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Parmigiani, G.; Garrett, E.S.; Irizarry, R.A.; Zeger, S.L. The Analysis of Gene Expression Data: An Overview of Methods and Software. In The Analysis of Gene Expression Data: Methods and Software; Parmigiani, G., Garrett, E.S., Irizarry, R.A., Zeger, S.L., Eds.; Springer New York: New York, NY, USA, 2003; pp. 1–45. [Google Scholar]
- Kathleen Kerr, M.; AChurchill, G. Statistical design and the analysis of gene expression microarray data. Genet. Res. 2001, 77, 123–128. [Google Scholar] [CrossRef]
- Smyth, G.K. limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer New York: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Zahurak, M.; Parmigiani, G.; Yu, W.; Scharpf, R.B.; Berman, D.; Schaeffer, E.; Shabbeer, S.; Cope, L. Pre-processing Agilent microarray data. BMC Bioinform. 2007, 8, 142. [Google Scholar] [CrossRef]
- Archer, K.J.; Reese, S.E. Detection call algorithms for high-throughput gene expression microarray data. Brief. Bioinform. 2009, 11, 244–252. [Google Scholar] [CrossRef]
- Schurmann, C.; Heim, K.; Schillert, A.; Blankenberg, S.; Carstensen, M.; Dörr, M.; Endlich, K.; Felix, S.B.; Gieger, C.; Grallert, H.; et al. Analyzing Illumina Gene Expression Microarray Data from Different Tissues: Methodological Aspects of Data Analysis in the MetaXpress Consortium. PLoS ONE 2012, 7, e50938. [Google Scholar] [CrossRef] [PubMed]
- Gohlmann, H.; Talloen, W. Gene Expression Studies Using Affymetrix Microarrays; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Jiang, N.; Leach, L.J.; Hu, X.; Potokina, E.; Jia, T.; Druka, A.; Waugh, R.; Kearsey, M.J.; Luo, Z.W. Methods for evaluating gene expression from Affymetrix microarray datasets. BMC Bioinform. 2008, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wilhelm, B.T.; Landry, J.R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef]
- Barnes, M.; Freudenberg, J.; Thompson, S.; Aronow, B.; Pavlidis, P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res. 2005, 33, 5914–5923. [Google Scholar] [CrossRef]
- Narrandes, S.; Xu, W. Gene Expression Detection Assay for Cancer Clinical Use. J. Cancer 2018, 9, 2249–2265. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Kapushesky, M.; Emam, I.; Holloway, E.; Kurnosov, P.; Zorin, A.; Malone, J.; Rustici, G.; Williams, E.; Parkinson, H.; Brazma, A. Gene expression atlas at the European bioinformatics institute. Nucleic Acids Res. 2010, 38, D690–D698. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; A Fonseca, N.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Morera, D.S.; Hasanali, S.L.; Belew, D.; Ghosh, S.; Klaassen, Z.; Jordan, A.R.; Wang, J.; Terris, M.K.; Bollag, R.J.; Merseburger, A.S.; et al. Clinical Parameters Outperform Molecular Subtypes for Predicting Outcome in Bladder Cancer: Results from Multiple Cohorts, Including TCGA. J. Urol. 2020, 203, 62–72. [Google Scholar] [CrossRef]
- Ren, Q.; Zhang, P.; Zhang, X.; Feng, Y.; Li, L.; Lin, H.; Yu, Y. A fibroblast-associated signature predicts prognosis and immunotherapy in esophageal squamous cell cancer. Front. Immunol. 2023, 14, 1199040. [Google Scholar] [CrossRef]
- Du, J.; Yang, M.; Chen, S.; Li, D.; Chang, Z.; Dong, Z. PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene 2016, 35, 3314–3323. [Google Scholar] [CrossRef]
- Li, D.; Mullinax, J.E.; Aiken, T.; Xin, H.; Wiegand, G.; Anderson, A.; Thorgeirsson, S.; Avital, I.; Rudloff, U. Loss of PDPK1 abrogates resistance to gemcitabine in label-retaining pancreatic cancer cells. BMC Cancer 2018, 18, 772. [Google Scholar] [CrossRef]
- Levine, K.M.; Ding, K.; Chen, L.; Oesterreich, S. FGFR4: A promising therapeutic target for breast cancer and other solid tumors. Pharmacol. Ther. 2020, 214, 107590. [Google Scholar] [CrossRef]
- Park, S.W.; Kang, J.; Kim, H.S.; Yoon, S.; Kim, B.S.; Lim, C.; Lee, D.; Kim, Y.H. Predicting prognosis through the discovery of specific biomarkers according to colorectal cancer lymph node metastasis. Am. J. Cancer Res. 2023, 13, 3221–3233. [Google Scholar]
- García-Alfonso, P.; García-González, G.; Gallego, I.; Peligros, M.I.; Ortega, L.; Pérez-Solero, G.T.; Sandoval, C.; Martin, A.M.; Codesido, M.B.; Ferrándiz, A.C.; et al. Prognostic value of molecular biomarkers in patients with metastatic colorectal cancer: A real-world study. Clin. Transl. Oncol. 2021, 23, 122–129. [Google Scholar] [CrossRef]
- Shahjaman, M.; Rahman, M.R.; Islam, S.M.S.; Mollah, M.N.H. A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs. Medicina 2019, 55, 269. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, X.; Gao, S. Identification of potential biomarkers for pathogenesis of Alzheimer’s disease. Hereditas 2021, 158, 23. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hung, M.C. Nuclear EGFR signalling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; Wang, B.; Wu, Y.; Chen, Z.; Lv, M.; Lin, Y.; Yang, J. Potential biomarkers for anti-EGFR therapy in metastatic colorectal cancer. Tumour Biol. 2016, 37, 11645–11655. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.E.; Zorbas, M.A.; Danels, Y.J.; Garcia, R.; Gallick, G.E.; Olive, M.; Brattain, M.G.; Boman, B.M.; Yeoman, L.C. Cellular growth response to epidermal growth factor in colon carcinoma cells with an amplified epidermal growth factor receptor derived from a familial adenomatous polyposis patient. Cancer Res. 1991, 51, 1452–1459. [Google Scholar] [PubMed]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Radinsky, R.; Risin, S.; Fan, D.; Dong, Z.; Bielenberg, D.; Bucana, C.D.; Fidler, I.J. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin. Cancer Res. 1995, 1, 19–31. [Google Scholar]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Gibson, T.B.; Ranganathan, A.; Grothey, A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin. Color. Cancer 2006, 6, 29–31. [Google Scholar] [CrossRef]
- Kasprzak, A.; Adamek, A. Insulin-Like Growth Factor 2 (IGF2) Signaling in Colorectal Cancer-From Basic Research to Potential Clinical Applications. Int. J. Mol. Sci. 2019, 20, 4915. [Google Scholar] [CrossRef]
- Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276. [Google Scholar] [CrossRef]
- Lamonerie, T.; Lavialle, C.; de Galle, B.; Binoux, M.; Brison, O. Constitutive or Inducible Overexpression of the IGF-2 Gene in Cells of a Human Colon Carcinoma Cell Line. Exp. Cell Res. 1995, 216, 342–351. [Google Scholar] [CrossRef]
- Zarrilli, R.; Pignata, S.; Romano, M.; Gravina, A.; Casola, S.; Bruni, C.B.; Acquaviva, A.M. Expression of insulin-like growth factor (IGF)-II and IGF-I receptor during proliferation and differentiation of CaCo-2 human colon carcinoma cells. Cell Growth Differ. 1994, 5, 1085–1091. [Google Scholar]
- Lee, H.; Kim, N.; Yoo, Y.J.; Kim, H.; Jeong, E.; Choi, S.; Moon, S.U.; Oh, S.H.; Mills, G.B.; Yoon, S.; et al. β-catenin/TCF activity regulates IGF-1R tyrosine kinase inhibitor sensitivity in colon cancer. Oncogene 2018, 37, 5466–5475. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Harper, J.; Burns, J.L.; Foulstone, E.J.; Pignatelli, M.; Zaina, S.; Hassan, A.B. Soluble IGF2 Receptor Rescues Apc (Min/+) Intestinal Adenoma Progression Induced by Igf2 Loss of Imprinting. Cancer Res. 2006, 66, 1940–1948. [Google Scholar] [CrossRef]
- Peters, G.; Gongoll, S.; Langner, C.; Mengel, M.; Piso, P.; Klempnauer, J.; Rüschoff, J.; Kreipe, H.; von Wasielewski, R. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Arch. 2003, 443, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.G.; Tirro, E.; Pennisi, M.S.; Massimino, M.; Stella, S.; Romano, C.; Manzella, L. The Insulin/IGF System in Colorectal Cancer Development and Resistance to Therapy. Front. Oncol. 2015, 5, 230. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. (Eds.) Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | TCGA (n = 422) | GSE17536 (n = 177) | GSE39582 (n = 557) | |
|---|---|---|---|---|
| Case Number (Proportion) | ||||
| Age (Mean and SD) | 66.4 ± 12.9 | 65.5 ± 13.1 | 66.8 ± 13.3 | |
| Gender | Male | 256 (53.6%) | 96 (54.2%) | 305 (54.8%) |
| Female | 196 (46.4%) | 81 (45.8%) | 252 (45.2%) | |
| Stage | I | 73 (17.3%) | 24 (13.6%) | 31 (5.6%) |
| II | 165 (39.1%) | 57 (32.2%) | 262 (47.0%) | |
| III | 123 (29.1%) | 57 (32.2%) | 204 (36.6%) | |
| IV | 61 (14.5%) | 39 (22.0%) | 60 (10.8%) | |
| MSI | Yes (or dMMR) | 11 (2.6%) | N/A | 71 (12.7%) |
| No (or pMMR) | 79 (18.7%) | N/A | 440 (79.0%) | |
| Histologic type | Adenocarcinoma | 356 (84.4%) | N/A | N/A |
| Mucinous adenocarcinoma | 61 (14.5%) | N/A | N/A | |
| Other types | 5 (1.1%) | N/A | N/A | |
| Gene Symbol (Probe Name) | TCGA-COAD (n = 422) | GSE17536 (n = 177) | GSE39582 (n = 557) | |||
|---|---|---|---|---|---|---|
| HR | p-Value | HR | p-Value | HR | p-Value | |
| PI4K2A (209345_s_at) | 1.766 | 0.03 | 9.269 | 0 | 1.571 | 0.022 |
| BAHD1 (203051_at) | 1.936 | 0.011 | 4.052 | 0.005 | 1.67 | 0.031 |
| MFAP1 (203406_at) | 1.84 | 0.022 | 2.561 | 0.006 | 1.407 | 0.049 |
| OAZ2 (201365_at) | 1.829 | 0.041 | 2.392 | 0.026 | 1.51 | 0.043 |
| FAM219B (224804_s_at) | 1.815 | 0.011 | 2.348 | 0.016 | 1.51 | 0.019 |
| KCTD1 (226246_at) | 1.3 | 0.004 | 2.583 | 0.009 | 1.303 | 0.014 |
| DNAJA4 (1554334_a_at) | 1.6 | 0.049 | 2.069 | 0.002 | 1.297 | 0.024 |
| ZEB1-AS1 (229090_at) | 1.383 | 0.018 | 1.839 | 0.015 | 1.463 | 0.001 |
| OSBPL1A (209485_s_at) | 1.262 | 0.036 | 1.723 | 0.006 | 1.238 | 0.007 |
| MYRIP (214156_at) | 0.901 | 0.016 | 0.69 | 0.002 | 0.866 | 0.017 |
| TOX3 (215108_x_at) | 0.854 | 0.033 | 0.726 | 0.009 | 0.844 | 0.017 |
| F2RL2 (230147_at) | 0.869 | 0.026 | 0.687 | 0.016 | 0.815 | 0.001 |
| FGFR4 (204579_at) | 0.831 | 0.046 | 0.673 | 0.049 | 0.852 | 0.047 |
| KRTAP4-1 (234635_at) | 0.937 | 0.043 | 0.547 | 0.009 | 0.762 | 0.025 |
| CHN2 (207486_x_at) | 0.774 | 0.013 | 0.527 | 0.013 | 0.78 | 0.023 |
| NFE2L3 (236471_at) | 0.7 | 0.041 | 0.625 | 0.004 | 0.754 | 0.001 |
| TTTY14 (207063_at) | 0.899 | 0.013 | 0.384 | 0.046 | 0.774 | 0.05 |
| CACNA1D (1555993_at) | 0.738 | 0 | 0.518 | 0.037 | 0.776 | 0.021 |
| SLC18A1 (207074_s_at) | 0.939 | 0.045 | 0.251 | 0.007 | 0.775 | 0.025 |
| CMC1 (228283_at) | 0.644 | 0.028 | 0.519 | 0.035 | 0.706 | 0.012 |
| ITGB8-AS1 (230446_at) | 0.8 | 0.021 | 0.416 | 0.002 | 0.638 | 0.001 |
| LARS2 (204016_at) | 0.621 | 0.019 | 0.459 | 0.027 | 0.661 | 0.017 |
| RAB11FIP4 (225739_at) | 0.681 | 0.042 | 0.466 | 0.003 | 0.576 | 0 |
| LINC00511 (230812_at) | 0.798 | 0.043 | 0.302 | 0.036 | 0.611 | 0.015 |
| CHDH (1559591_s_at) | 0.739 | 0.01 | 0.314 | 0.004 | 0.648 | 0.008 |
| HNF1B (208135_at) | 0.842 | 0.037 | 0.053 | 0 | 0.598 | 0.008 |
| PDPK1 (221244_s_at) | 0.469 | 0.001 | 0.181 | 0.012 | 0.582 | 0.027 |
| EIF1B (237988_at) | 0.602 | 0.017 | 0.025 | 0.006 | 0.333 | 0.015 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Z.; Lee, J.; Yoon, Y.E.; Yun, J.W. Unveiling Prognostic RNA Biomarkers through a Multi-Cohort Study in Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 3317. https://doi.org/10.3390/ijms25063317
Kim Z, Lee J, Yoon YE, Yun JW. Unveiling Prognostic RNA Biomarkers through a Multi-Cohort Study in Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(6):3317. https://doi.org/10.3390/ijms25063317
Chicago/Turabian StyleKim, Zehwan, Jaebon Lee, Ye Eun Yoon, and Jae Won Yun. 2024. "Unveiling Prognostic RNA Biomarkers through a Multi-Cohort Study in Colorectal Cancer" International Journal of Molecular Sciences 25, no. 6: 3317. https://doi.org/10.3390/ijms25063317