

Alpha-Synuclein and GM1 Ganglioside Co-Localize in Neuronal Cytosol Leading to Inverse Interaction—Relevance to Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
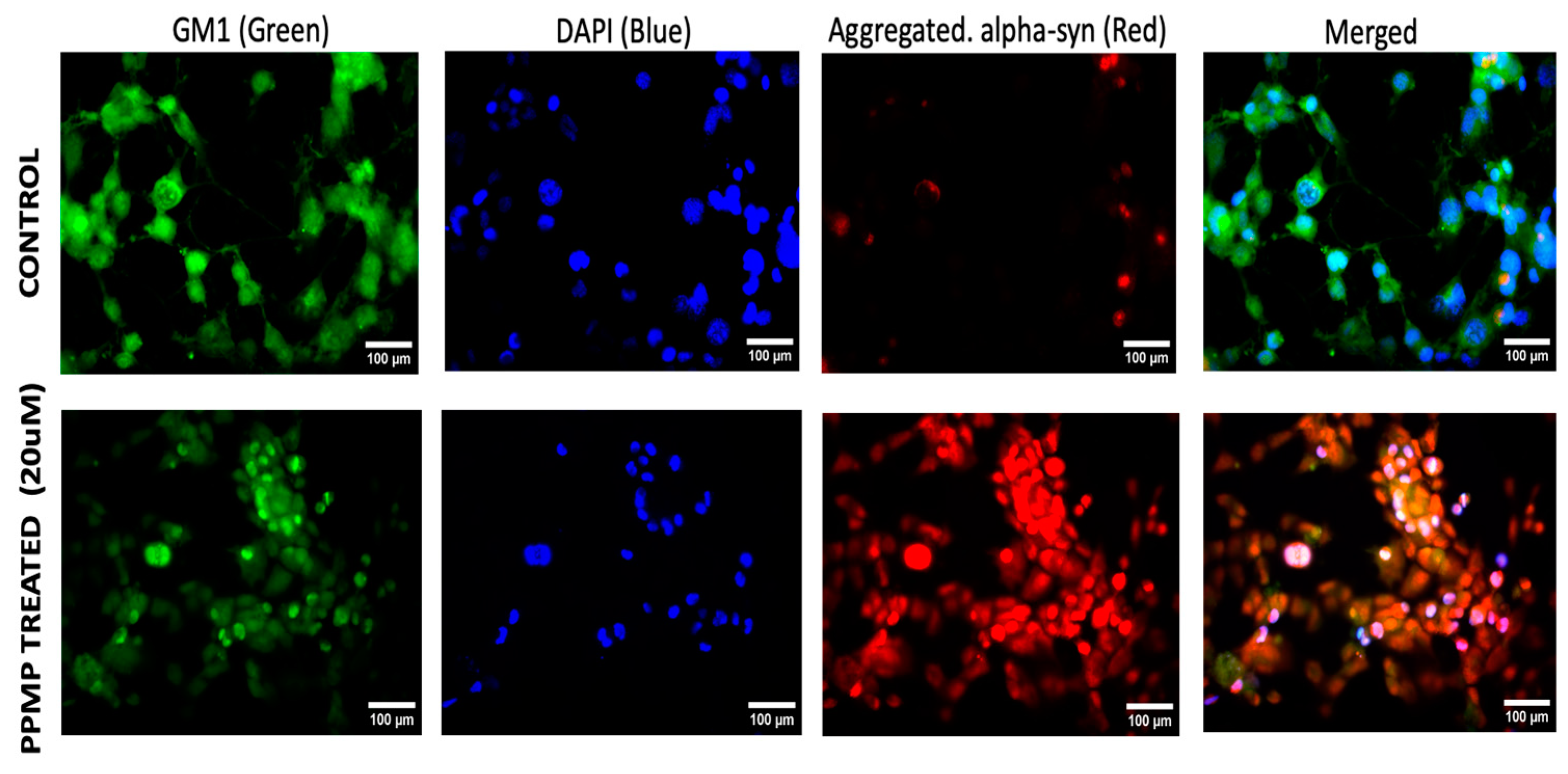
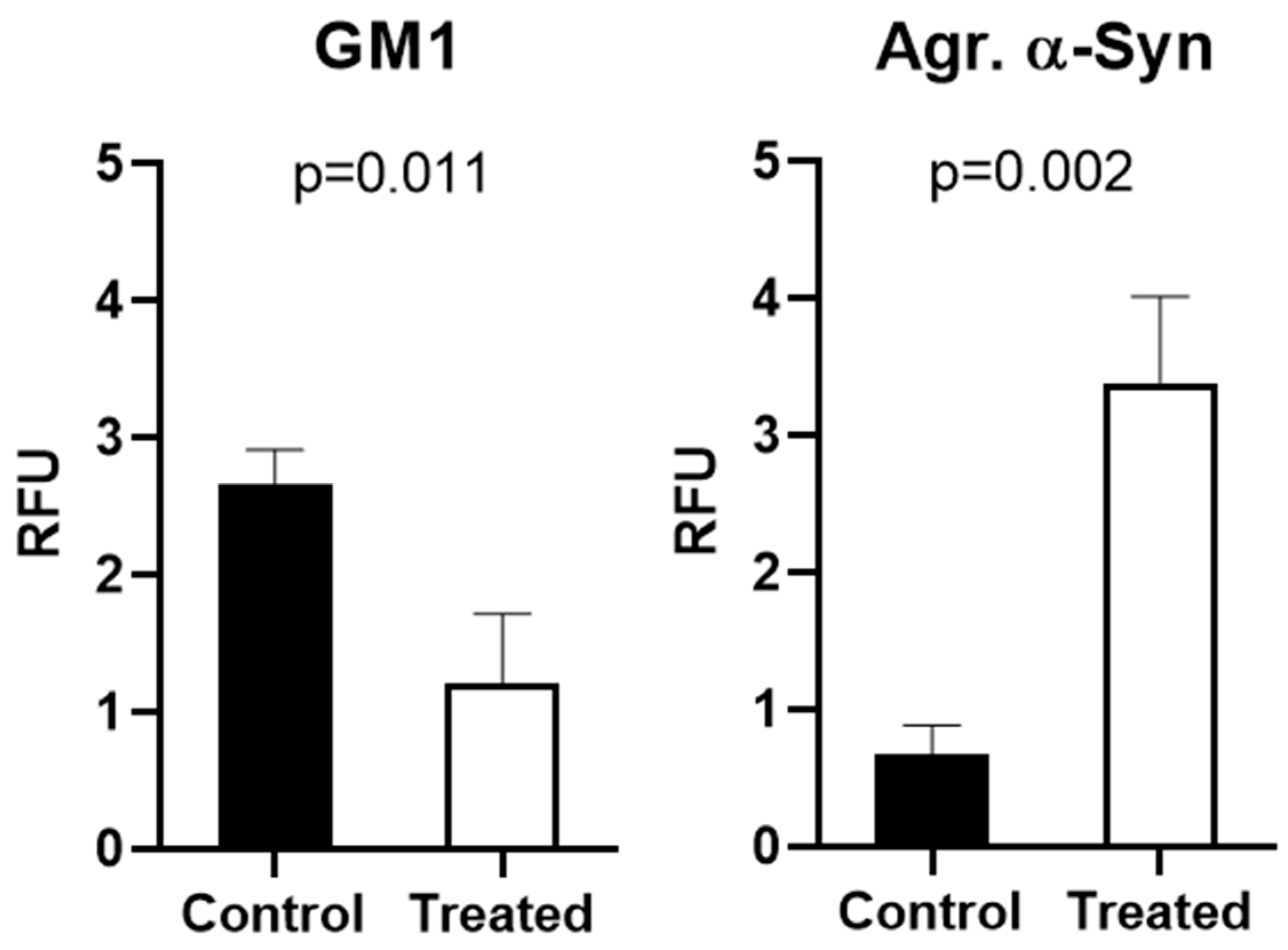
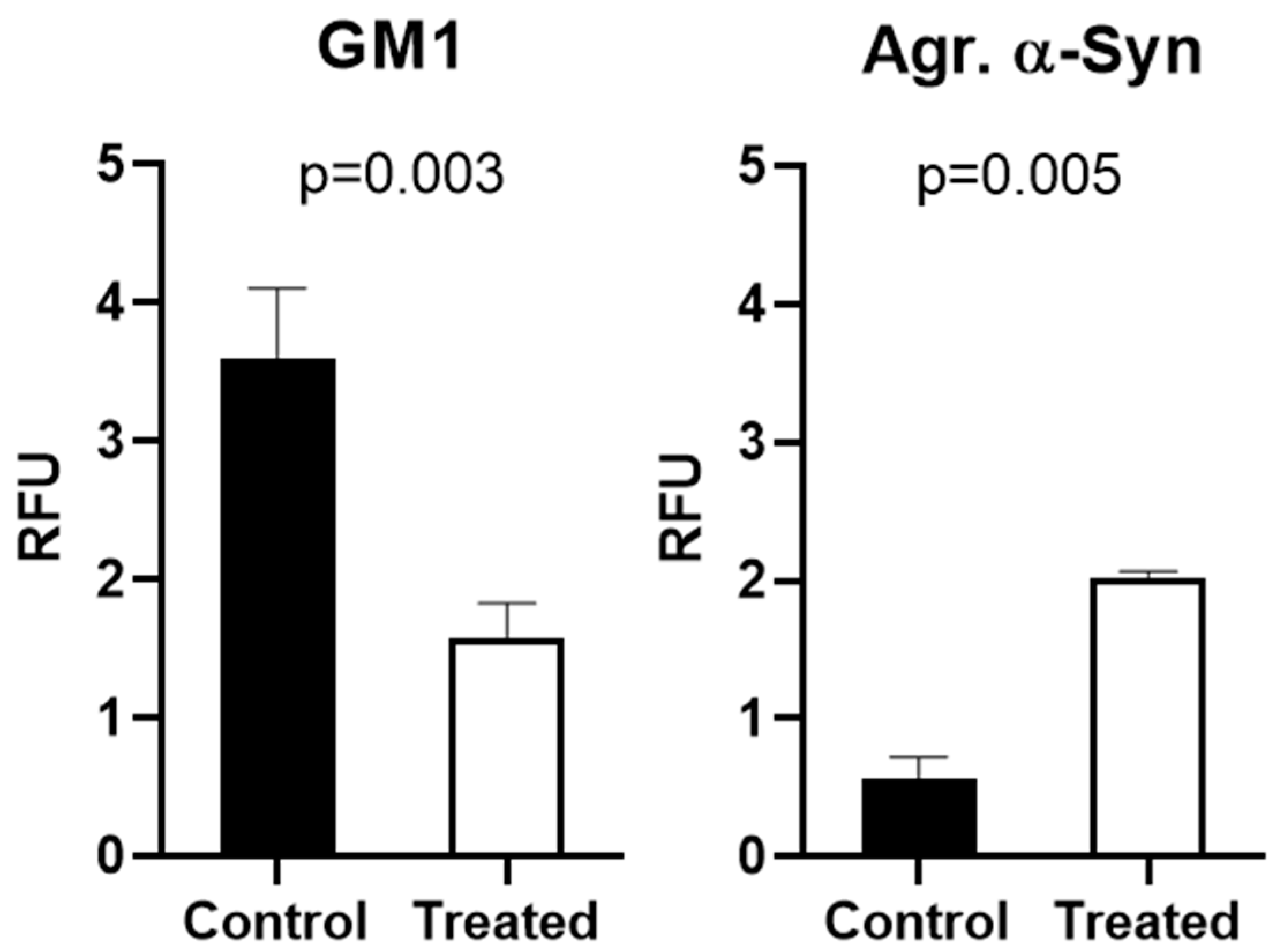
2.1. Alpha-Synuclein Aggregation and GM1 in Cultured Cells
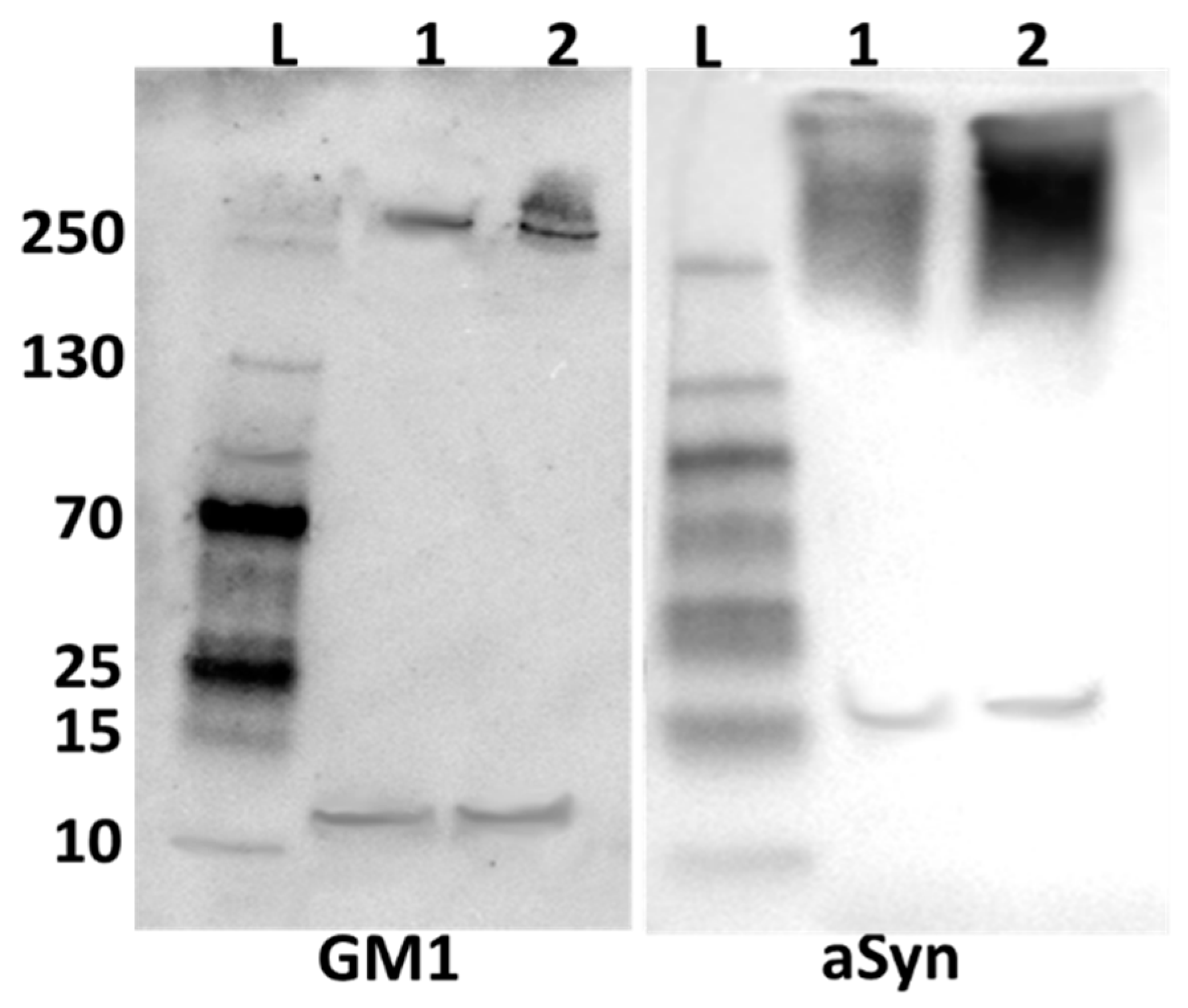
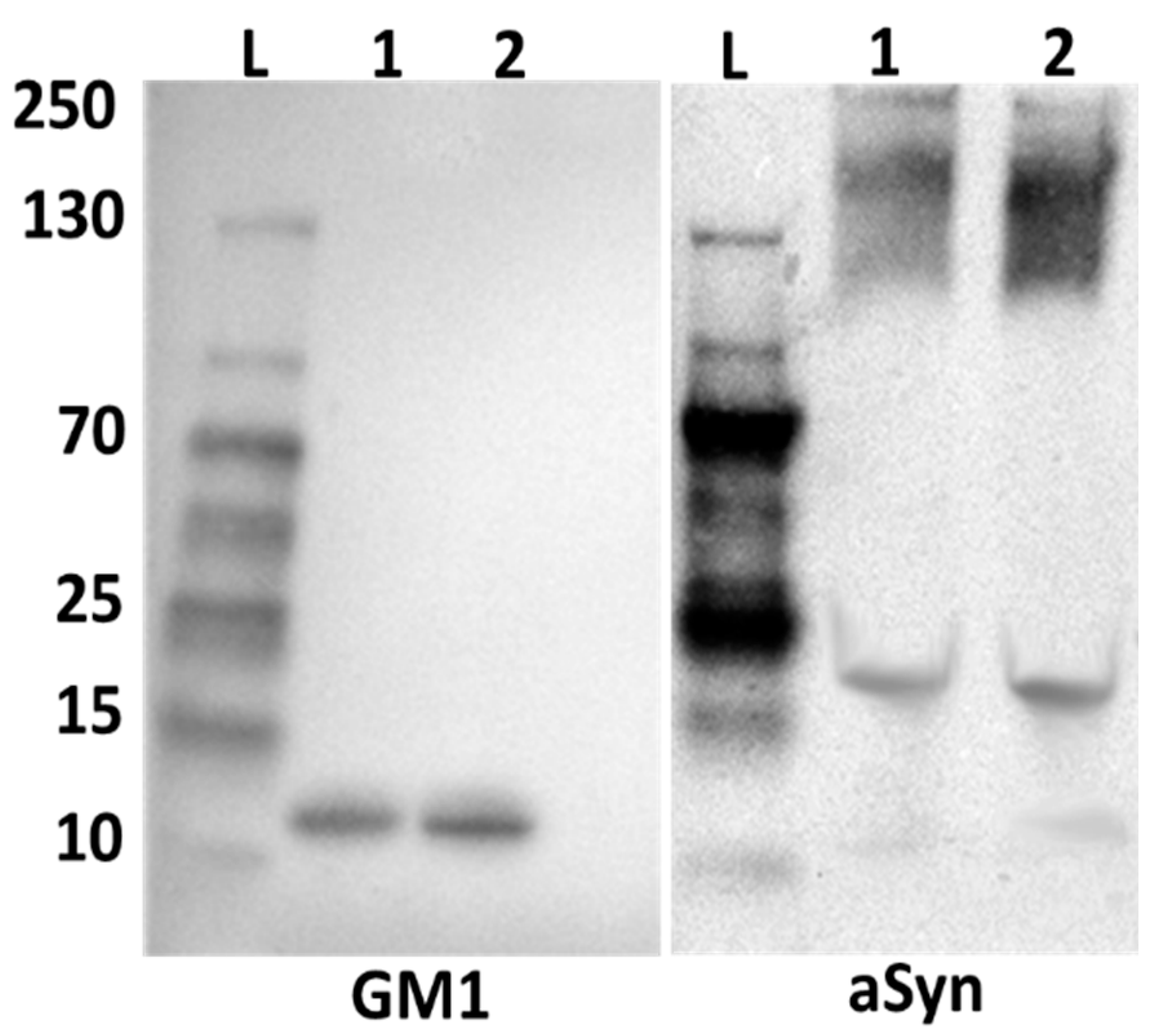
2.2. Alpha-Synuclein and GM1 in the Neuronal Cytosol
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. NG108-15 and SH-SY5Y Cellular Studies
4.3. Western Blot Analysis
4.4. Image Quantification and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galpern, W.R.; Lang, A.E. Interface between tauopathies and synucleinopathies: A tale of two proteins. Ann. Neurol. 2006, 59, 449–458. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Lecture, B.H.S.F. the staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord 2006, 21, 2042–2051. [Google Scholar]
- Pellicano, C.; Benincasa, D.; Pisani, V.; Buttarelli, F.R.; Giovannelli, M.; Pontieri, F.E. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 145–152. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454. [Google Scholar] [PubMed]
- Fazzari, M.; Di Biase, E.; Lunghi, G.; Mauri, L.; Chiricozzi, E.; Sonnino, S. Novel insights on GM1 and Parkinson’s disease: A critical review. Glycoconj. J. 2022, 39, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S. The relationship between depletion of brain GM1 ganglioside and Parkinson’s disease. FEBS Open Bio 2023, 13, 1548–1557. [Google Scholar] [CrossRef]
- Ledeen, R.; Chowdhury, S.; Lu, Z.-H.; Chakraborty, M.; Wu, G. Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 2022, 39, 75–82. [Google Scholar] [CrossRef]
- Alselehdar, S.K.; Chakraborty, M.; Chowdhury, S.; Alcalay, R.N.; Surface, M.; Ledeen, R. Subnormal GM1 in PBMCs: Promise for early diagnosis of Parkinson’s disease? Int. J. Mol. Sci. 2021, 22, 11522. [Google Scholar] [CrossRef]
- Schneider, J.S. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS ONE 2018, 13, e0199189. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 ganglioside modifies α-synuclein toxicity and is neuroprotective in a rat α-synuclein model of Parkinson’s disease. Sci. Rep. 2019, 9, 8362. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Mauri, L.; Ciampa, M.G.; Prinetti, A.; Toffano, G.; Secchieri, C.; Sonnino, S. GM1 ganglioside: Past studies and future potential. Mol. Neurobiol. 2016, 53, 1824–1842. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.; Lunghi, G.; Chiricozzi, E.; Mauri, L.; Sonnino, S. Gangliosides and the treatment of neurodegenerative diseases: A long italian tradition. Biomedicines 2022, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the brain: Physiology, pathophysiology and therapeutic applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in membrane organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar]
- Ravichandra, B.; Joshi, P.G. Regulation of transmembrane signaling by ganglioside GM1: Interaction of anti-GM1 with Neuro2a cells. J. Neurochem. 1999, 73, 557–567. [Google Scholar] [CrossRef]
- Fong, T.G.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside improves spatial learning and memory of aged rats. Behav. Brain Res. 1997, 85, 203–211. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G.; Lu, Z.H.; Kozireski-Chuback, D.; Fang, Y. The Role of GM1 and Other Gangliosides in Neuronal Differentiation Overview and New Findingsa. Ann. N. Y. Acad. Sci. 1998, 845, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015, 40, 407–418. [Google Scholar] [CrossRef]
- Ferrari, G.; Anderson, B.L.; Stephens, R.M.; Kaplan, D.R.; Greene, L.A. Prevention of Apoptotic Neuronal Death by GM1 Ganglioside: INVOLVEMENT OF Trk NEUROTROPHIN RECEPTORS (∗). J. Biol. Chem. 1995, 270, 3074–3080. [Google Scholar] [CrossRef]
- Todeschini, A.R.; Hakomori, S.-I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Svennerholm, L.; Boström, K.; Fredman, P.; Månsson, J.-E.; Rosengren, B.; Rynmark, B.-M. Human brain gangliosides: Developmental changes from early fetal stage to advanced age. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1989, 1005, 109–117. [Google Scholar] [CrossRef]
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef] [PubMed]
- Toffano, G.; Savoini, G.; Moroni, F.; Lombardi, G.; Calza, L.; Agnati, L. GM1 ganglioside stimulates the regeneration of dopaminergic neurons in the central nervous system. Brain Res. 1983, 261, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, T.; Tokuda, A.; Miyadai, T.; Hamaguchi, M.; Fujiki, N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. USA 1995, 92, 5087–5091. [Google Scholar] [CrossRef]
- Pitto, M.; Mutoh, T.; Kuriyama, M.; Ferraretto, A.; Palestini, P.; Masserini, M. Influence of endogenous GM1 ganglioside on TrkB activity, in cultured neurons. FEBS Lett. 1998, 439, 93–96. [Google Scholar] [CrossRef]
- Wales, P.; Pinho, R.; Lázaro, D.F.; Outeiro, T.F. Limelight on alpha-synuclein: Pathological and mechanistic implications in neurodegeneration. J. Park. Dis. 2013, 3, 415–459. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The synaptic function of α-synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef]
- Melki, R. Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Park. Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Kragh, C.L.; Ubhi, K.; Wyss-Corey, T.; Masliah, E. Autophagy in dementias. Brain Pathol. 2012, 22, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-L.; Duan, W.-J.; Lu, D.-H.; Ma, X.-H.; Li, X.-X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.-Z.; Li, Y.-F. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. Intrinsically disordered proteins in synaptic vesicle trafficking and release. J. Biol. Chem. 2019, 294, 3325–3342. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell Jr, R.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.M.; Finke, R.G. α-Synuclein aggregation variable temperature and variable pH kinetic data: A re-analysis using the Finke–Watzky 2-step model of nucleation and autocatalytic growth. Biophys. Chem. 2009, 140, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β-and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.-Y. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar]
- Presgraves, S.P.; Ahmed, T.; Borwege, S.; Joyce, J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2003, 5, 579–598. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Kovács, P.; Pintér, M.; Csaba, G. Effect of glucosphingolipid synthesis inhibitor (PPMP and PDMP) treatment on Tetrahymena pyriformis: Data on the evolution of the signaling system. Cell Biochem. Funct. Cell. Biochem. Modul. Act. Agents Dis. 2000, 18, 269–280. [Google Scholar]
- Wang, X.; Zhou, R.; Sun, X.; Li, J.; Wang, J.; Yue, W.; Wang, L.; Liu, H.; Shi, Y.; Zhang, D. Preferential Regulation of Γ-Secretase-Mediated Cleavage of APP by Ganglioside GM1 Reveals a Potential Therapeutic Target for Alzheimer’s Disease. Adv. Sci. 2023, 10, 2303411. [Google Scholar] [CrossRef]
- Byrne, M.C.; Farooq, M.; Sbaschnig-Agler, M.; Norton, W.T.; Ledeen, R.W. Ganglioside content of astroglia and neurons isolated from maturing rat brain: Consideration of the source of astroglial gangliosides. Brain Res. 1988, 461, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sbaschnig-Agler, M.; Dreyfus, H.; Norton, W.T.; Sensenbrenner, M.; Farooq, M.; Byrne, M.C.; Ledeen, R.W. Gangliosides of cultured astroglia. Brain Res. 1988, 461, 98–106. [Google Scholar] [CrossRef]
- Asou, H.; Brunngraber, E.G. Absence of ganglioside GM1 in astroglial cells from 21-day old rat brain: Immunohistochemical, histochemical, and biochemical studies. Neurochem. Res. 1983, 8, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Ghidoni, R.; Fiorilli, A.; Venerando, B.; Tettamanti, G. Cytosolic gangliosides of rat brain: Their fractionation into protein-bound complexes of different ganglioside compositions. J. Neurosci. Res. 1984, 12, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.; Skrivanek, J.; Tirri, L.; Margolis, R.; Margolis, R. Gangliosides of the neuron: Localization and origin. Ganglioside Funct. Biochem. Pharmacol. Implic. 1976, 71, 83–103. [Google Scholar]
- Maroteaux, L.; Scheller, R. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Mol. Brain Res. 1991, 11, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Ledeen, R. The key role of GM1 ganglioside in Parkinson’s disease. Biomolecules 2022, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Hadjiconstantinou, N.; Rossetti, Z.; Paxton, R.; Neff, N. Administration of GMI. ganglioside restores the dopamine content in striatum after chronic treatment with MPTP. Neuropharmacology 1986, 25, 1075–1077. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment avenues in neurodegenerative disease. Front. Neurol. 2019, 10, 859. [Google Scholar] [CrossRef]
- Schneider, J.; Di Stefano, L. Response of the damaged dopamine system to GM1 and semisynthetic gangliosides: Effects of dose and extent of lesion. Neuropharmacology 1995, 34, 489–493. [Google Scholar] [CrossRef]
- Schneider, J. Gangliosides and glycolipids in neurodegenerative disorders. Glycobiol. Nerv. Syst. 2014, 9, 449–461. [Google Scholar]
- Robert, K.Y.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides-an overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar]
- Chiricozzi, E.; Mauri, L.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Maggioni, M.; Valsecchi, M.; Prioni, S.; Loberto, N.; Pomè, D.Y. Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1+/− mouse model. Sci. Rep. 2019, 9, 19330. [Google Scholar] [CrossRef]
- Chowdhury, S.; Wu, G.; Lu, Z.-H.; Kumar, R.; Ledeen, R. Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease. Biomedicines 2023, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.-H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.; Di Biase, E.; Zaccagnini, L.; Henriques, A.; Callizot, N.; Ciampa, M.G.; Mauri, L.; Carsana, E.V.; Loberto, N.; Aureli, M. GM1 oligosaccharide efficacy against α-synuclein aggregation and toxicity in vitro. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2023, 1868, 159350. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Chowdhury, S.; Ledeen, R. Alpha-Synuclein and GM1 Ganglioside Co-Localize in Neuronal Cytosol Leading to Inverse Interaction—Relevance to Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 3323. https://doi.org/10.3390/ijms25063323
Kumar R, Chowdhury S, Ledeen R. Alpha-Synuclein and GM1 Ganglioside Co-Localize in Neuronal Cytosol Leading to Inverse Interaction—Relevance to Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(6):3323. https://doi.org/10.3390/ijms25063323
Chicago/Turabian StyleKumar, Ranjeet, Suman Chowdhury, and Robert Ledeen. 2024. "Alpha-Synuclein and GM1 Ganglioside Co-Localize in Neuronal Cytosol Leading to Inverse Interaction—Relevance to Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 6: 3323. https://doi.org/10.3390/ijms25063323