

Electrophoretic Deposition of Calcium Phosphates on Carbon–Carbon Composite Implants: Morphology, Phase/Chemical Composition and Biological Reactions
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
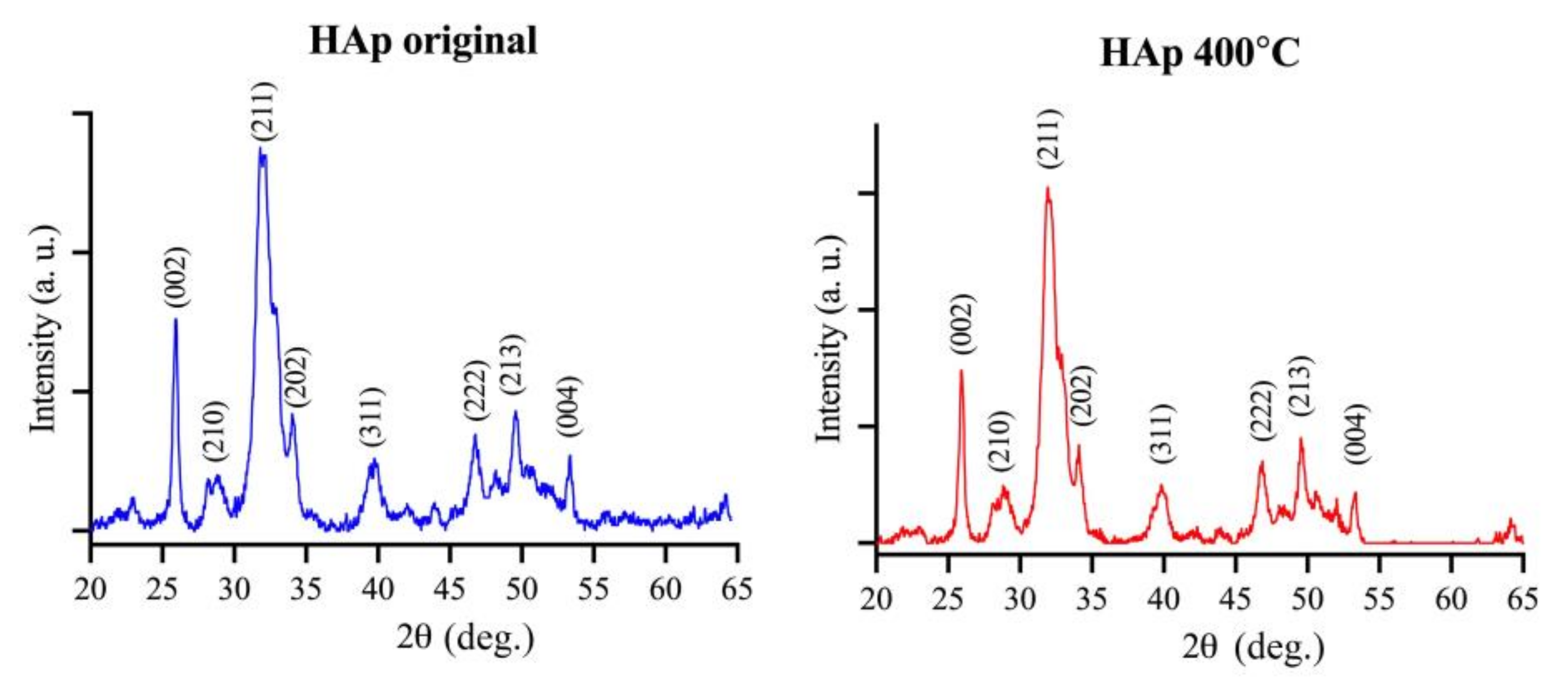
2.1. Morphology and Properties of as-Prepared C-C Composites and HAp Powder
2.2. Morphology and Properties of Ca-P-Coated C-C Composites
2.3. Results of In Vitro Tests
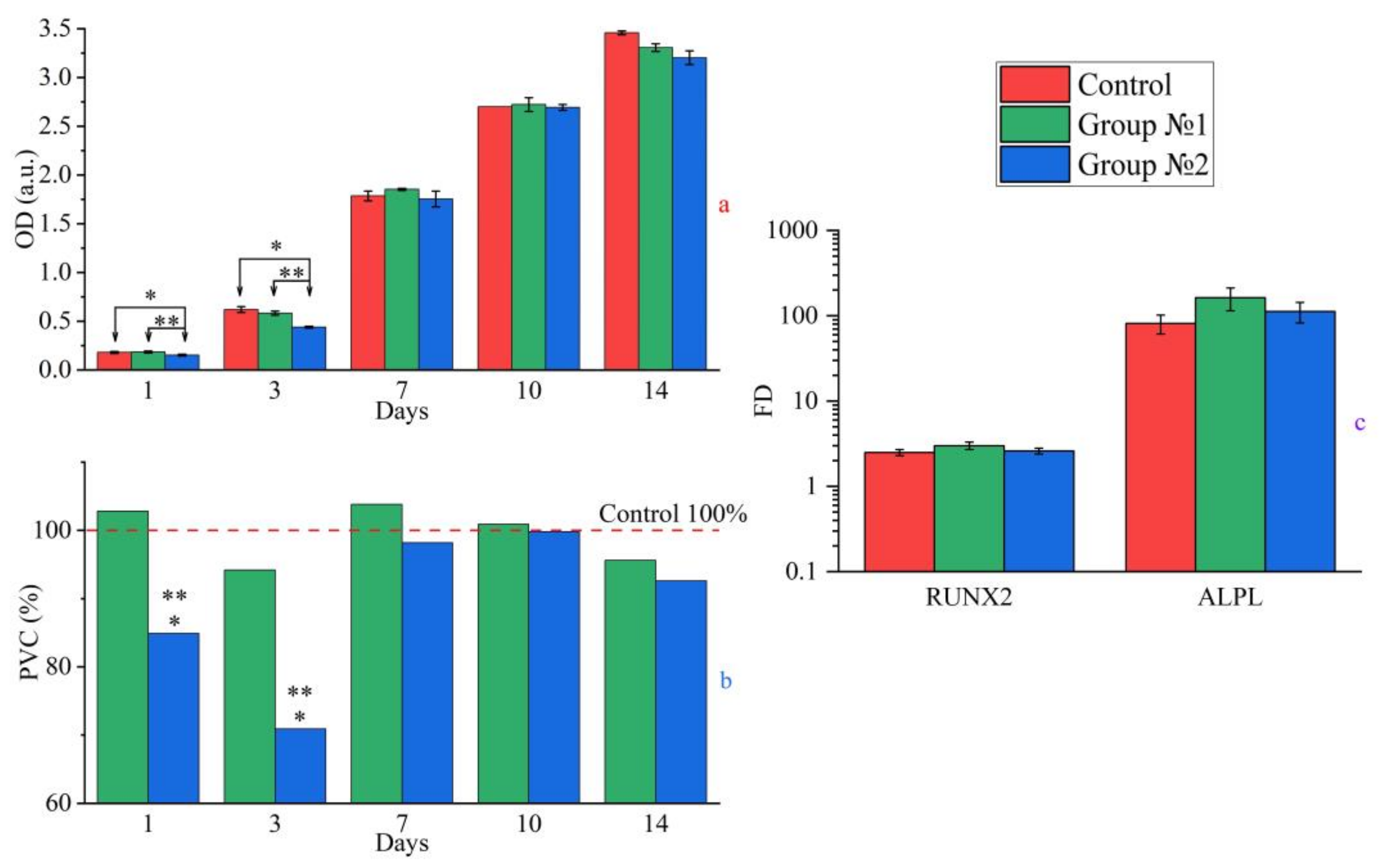
2.3.1. Cytotoxicity
2.3.2. Cytocompatibility
2.3.3. Osteogenic Differentiation
2.4. In Vivo Tests
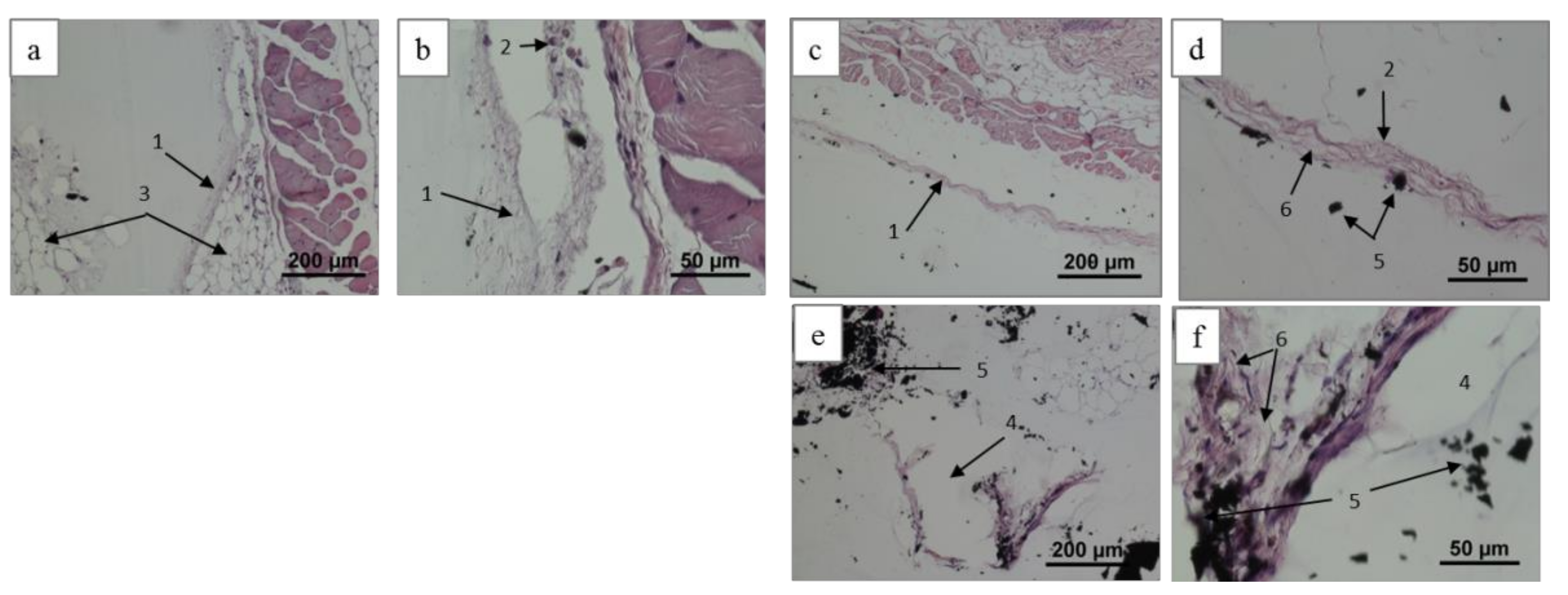
2.4.1. Histological Patterns
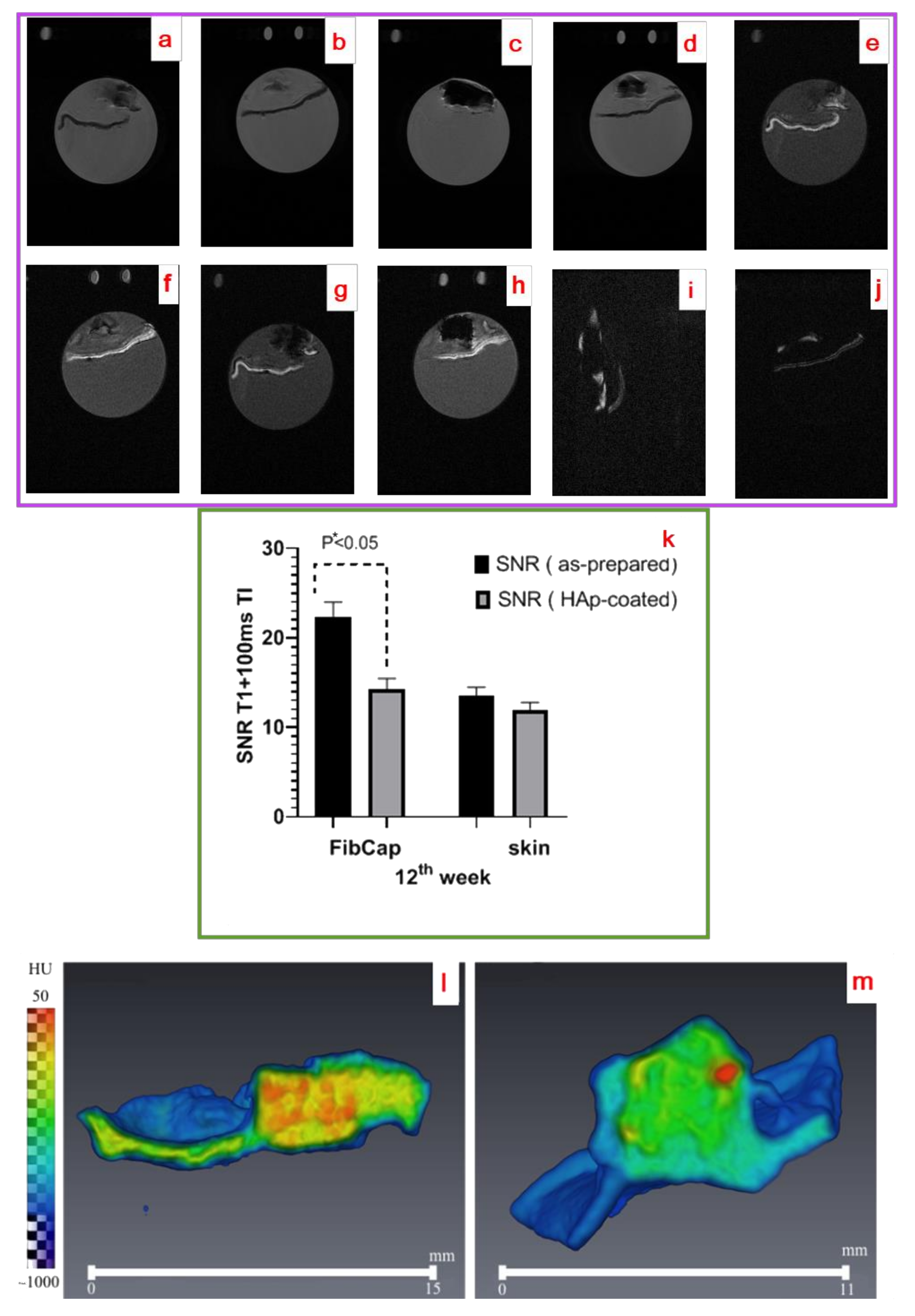
2.4.2. Radiological Studies
3. Discussion
4. Materials and Methods
4.1. C-C Composites and HAp Powder
4.2. Deposition Procedure
4.3. Coating Characterization
4.4. In Vitro Tests
4.4.1. Sample Sterilization
4.4.2. Cell Cultures
4.4.3. Cytotoxicity Tests
Indirect Contact Test
Direct Contact Test
4.4.4. Cytocompatibility Test
4.4.5. Assessment of Cell Culture Viability
Assessment of Cell Culture Viability Using the Quantitative MTT Assay
Assessment of Cell Culture Viability Using a Live/Dead Kit
4.4.6. Osteogenic Differentiation of MSCs on Composites Using PCR
PCR Test
4.4.7. Statistical Analysis
4.5. In Vivo Tests
4.5.1. Implantation and Histology Procedures
4.5.2. Magnetic Resonance Imaging and X-ray Computer Tomography
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Amirtharaj Mosas, K.K.; Chandrasekar, A.R.; Dasan, A.; Pakseresht, A.; Galusek, D. Recent Advancements in Materials and Coatings for Biomedical Implants. Gels 2022, 8, 323. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R. (Ed.) Biomedical Materials; Springer Science + Business Media: New York, NY, USA, 2009. [Google Scholar]
- Mano, J.F.; Sousa, R.A.; Boesel, L.F.; Neves, N.M.; Reis, R.L. Bioinert, biodegradable and injectable polymeric matrix composites for hard tissue replacement: State of the art and recent developments. Compos. Sci. Technol. 2004, 64, 789–817. [Google Scholar] [CrossRef]
- Yeung, C.M.; Bhashyam, A.R.; Patel, S.S.; Ortiz-Cruz, E.; Lozano-Calderón, S.A. Carbon Fiber Implants in Orthopaedic Oncology. J. Clin. Med. 2022, 11, 4959. [Google Scholar] [CrossRef] [PubMed]
- Babaie, E.; Bhaduri, S.B. Fabrication Aspects of Porous Biomaterials in Orthopedic Applications: A Review. ACS Biomater. Sci. Eng. 2018, 4, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Heimann, R.B. Structure, properties, and biomedical performance of osteoconductive bioceramic coatings. Surf. Coat. Technol. 2013, 233, 27–38. [Google Scholar] [CrossRef]
- Siddiqui, H.A.; Pickering, K.L.; Mucalo, M.R. A Review on the Use of Hydroxyapatite-Carbonaceous Structure Composites in Bone Replacement Materials for Strengthening Purposes. Materials 2018, 11, 1813. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.W.; Anestopoulos, I.; Panayiotidis, M.I.; Birkett, M. Soft diamond-like carbon coatings with superior biocompatibility for medical applications. Ceram. Int. 2023, 49, 17203–17211. [Google Scholar] [CrossRef]
- Alves-Sampaio, A.; Del-Cerro, P.; Collazos-Castro, J.E. Composite Fibrin/Carbon Microfiber Implants for Bridging Spinal Cord Injury: A Translational Approach in Pigs. Int. J. Mol. Sci. 2023, 24, 11102. [Google Scholar] [CrossRef]
- Olivier, F.; Bonnamy, S.; Rochet, N.; Drouet, C. Activated Carbon Fiber Cloth/Biomimetic Apatite: A Dual Drug Delivery System. Int. J. Mol. Sci. 2021, 22, 12247. [Google Scholar] [CrossRef]
- Alavi, S.E.; Gholami, M.; Shahmabadi, H.E.; Reher, P. Resorbable GBR Scaffolds in Oral and Maxillofacial Tissue Engineering: Design, Fabrication, and Applications. J. Clin. Med. 2023, 12, 6962. [Google Scholar] [CrossRef]
- Steinberg, E.L.; Rath, E.; Shlaifer, A.; Chechik, O.; Maman, E.; Salai, M. Carbon fiber reinforced PEEK Optima—A composite material biomechanical properties and wear/debris characteristics of CF-PEEK composites for orthopedic trauma implants. J. Mech. Behav. Biomed. Mater. 2013, 17, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.C.M.; Pinho, S.S.; Braz, M.P.; Silva, F.S.; Henriques, B. Carbon fiber-reinforced PEEK in implant dentistry: A scoping review on the finite element method. Comput. Methods Biomech. Biomed. 2021, 24, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, H.; A, L.; Yang, S.; Zhang, J.; Wang, H.; Zhou, Z.; Zhou, Y.; Zhao, J.; Jiang, Z. Enhanced bioactivity and osteogenic property of carbon fiber reinforced polyetheretherketone composites modified with amino groups. Colloids Surf. B 2020, 193, 111098. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, W.; Shi, R.; Yang, S.; Zhang, J.; Han, X.; Zhou, Z.; Gao, W.; Li, Y.; Zhao, J. Osseointegration behavior of carbon fiber reinforced polyetheretherketone composites modified with amino groups: An in vivo study. J. Biomed. Mater. Res. 2023, 111, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. (Ed.) Biological and Biomedical Coating Handbook: Processing and characterization; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Anderson, J.M. Inflammation, Wound Healing and Foreign-Body Response. In Biomaterials Science, 2nd ed.; Rater, B., Hoffman, A., Schoen, F.J., Lemons, J.E., Eds.; Elsevier: San Diego, CA, USA, 2004; pp. 296–303. [Google Scholar]
- Zhao, X.; Liu, M.; Wang, P.; Guan, J.; Wang, X. Highly efficient preparation of multi-angle continuous carbon fiber reinforced hydroxyapatite composites by electrostatic splitting method. J. Eur. Ceram. 2022, 42, 7631–7647. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. There Are over 60 Ways to Produce Biocompatible Calcium Orthophosphate (CaPO4) Deposits on Various Substrates. J. Compos. Sci. 2023, 7, 273. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium Orthophosphate (CaPO4)-Based Bioceramics: Preparation, Properties, and Applications. Coatings 2022, 12, 1380. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Wang, W.; Zhang, J.; Zhang, L.; He, F.; Yang, J. Controllable preparation of a nano-hydroxyapatite coating on carbon fibers by electrochemical deposition and chemical treatment. Mater. Sci. Eng. C 2016, 63, 96–105. [Google Scholar] [CrossRef]
- Ang, A.; Ahmed, R.; Berndt, C.C. Biomaterials: Thermal Spray Processes and Applications. J. Therm. Spray Technol. 2018, 27, 1205–1211. [Google Scholar] [CrossRef]
- Sui, J.-L.; Li, M.-S.; Lü, Y.-P.; Yin, L.-W.; Song, Y.-J. Plasma-sprayed hydroxyapatite coatings on carbon/carbon composites. Surf. Coat. Technol. 2004, 176, 188–192. [Google Scholar] [CrossRef]
- Su, Y.; Li, K.; Zhang, L.; Wang, C.; Zhang, Y. Effect of the hydroxyapatite particle size on the properties of sprayed coating. J. Therm. Spray Technol. 2018, 352, 619–626. [Google Scholar] [CrossRef]
- Heimann, R.B. Plasma-sprayed hydroxylapatite coatings as biocompatible intermediaries between inorganic implant surfaces and living tissue. J. Therm. Spray Technol. 2018, 27, 1212–1237. [Google Scholar] [CrossRef]
- Tsygankov, P.A.; Skriabin, A.S.; Telekh, V.D.; Loktionov, E.Y.; Chelmodeev, R.I. Interaction between Dusty Shock Waves and Three-Dimensional Scaffolds of Carbon Nanocomposites upon the Deposition of Biocompatible Coatings. Bull. Russ. Acad. Sci. Phys. 2018, 82, 380–385. [Google Scholar] [CrossRef]
- Tsygankov, P.A.; Parada, F.B.; Skriabin, A.S. Experimental study of heat conductivity of carbon composite implants with calcium phosphate based coatings. AIP Conf. Proc. 2022, 2383, 040007. [Google Scholar] [CrossRef]
- Skryabin, A.S.; Tsygankov, P.A.; Vesnin, V.R.; Parshin, B.A.; Zaitsev, V.V.; Lukina, Y.S. Physicochemical properties and osseointegration of titanium implants with bioactive calcium phosphate coatings produced by detonation spraying. Inorg. Mater. 2022, 58, 71–77. [Google Scholar] [CrossRef]
- Khvostov, M.V.; Bulina, N.V.; Zhukova, N.A.; Morenkova, E.G.; Rybin, D.K.; Makarova, S.V.; Leonov, S.V.; Gorodov, V.G.; Ulianitsky, V.Y.; Tolstikova, T.G. A study on biological properties of titanium implants coated with multisubstituted hydroxyapatite. Ceram. Int. 2022, 48, 34780–34792. [Google Scholar] [CrossRef]
- Skriabin, A.S.; Shakurov, A.V.; Vesnin, V.R.; Lukina, Y.S.; Tsygankov, P.A.; Bionyshev-Abramov, L.L.; Serejnikova, N.B.; Vorob’ev, E.V. Titanium membranes with hydroxyapatite/titania bioactive ceramic coatings: Characterization and in vivo biocompatibility testing. ACS Omega 2022, 7, 47880–47891. [Google Scholar] [CrossRef]
- Mohan, L.; Durgalakshmi, D.; Geetha, M.; Sankara Narayanan, T.S.N.; Asokamani, R. Electrophoretic deposition of nanocomposite (HAp + TiO2) on titanium alloy for biomedical applications. Ceram. Int. 2012, 38, 3435–3443. [Google Scholar] [CrossRef]
- Hamai, R.; Tsuchiya, K.; Suzuki, O. Adsorption of serum albumin onto octacalcium phosphate in supersaturated solutions regarding calcium phosphate phases. Materials 2019, 12, 2333. [Google Scholar] [CrossRef]
- Nosenko, V.V.; Yaremko, A.M.; Dzhagan, V.M.; Vorona, I.P.; Romanyuk, Y.A.; Zatovsky, I.V. Nature of some features in Raman spectra of hydroxyapatite-containing materials. J. Raman Spectrosc. 2016, 47, 726–730. [Google Scholar] [CrossRef]
- Venugopal, B.; Fernandez, F.B.; Babu, S.S.; Harikrishnan, V.S.; Varma, H.; John, A. Adipogenesis on biphasic calcium phosphate using rat adipose-derived mesenchymal stem cells: In vitro and in vivo. J. Biomed. Mater. Res. Part A 2012, 100, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Major, R.; Grajoszek, A.; Byrski, A.; Szawiraacz, K.; Jerzy Barski, J.; Major, Ł.; Gawlikowski, M.; Kopernik, M.; Kot, M.; Dyner, A.; et al. Evaluation of In Vivo Biocompatibility in Preclinical Studies of a Finger Implant Medical Device Correlated with Mechanical Properties and Microstructure. ACS Appl. Mater. Interfaces 2024, 16, 376–388. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Guan, K.; Wang, Z.; Wang, Y.; Li, H.; Lei, X.; Zhang, W.; Wang, K.; Hu, Y. Boosting bonding strength of hydroxyapatite coating for carbon/carbon composites via applying tree-planting interface structure. Ceram. Int. 2021, 47, 11922–11928. [Google Scholar] [CrossRef]
- Xiong, X.; Liu, L.; Ma, J.; Ni, X.; Li, Y.; Zeng, X. A simplified process for preparing adhesive hydroxyapatite coatings on carbon/carbon composites. Surf. Coat. Technol. 2019, 377, 124925. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Li, K.; Zhang, S.; Lu, J.; Li, W.; Cao, S.; Wang, B. Carbon foam/hydroxyapatite coating for carbon/carbon composites: Microstructure and biocompatibility. Appl. Surf. Sci. 2013, 286, 421–427. [Google Scholar] [CrossRef]
- Vimalraj, S.; Arumugam, B.; Miranda, P.J.; Selvamurugan, N. Runx2: Structure, function, and phosphorylation in osteoblast differentiation. Int. J. Biol. Macromol. 2015, 78, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Khor, K.A.; Cheang, P. Characterization of thermal sprayed hydroxyapatite powders and coatings. J. Therm. Spray Technol. 1994, 3, 45–50. [Google Scholar] [CrossRef]
- Demnati, I.; Grossin, D.; Combes, C.; Rey, C. Plasma-Sprayed Apatite Coatings: Review of Physical-Chemical Characteristics and Their Biological Consequences. J. Med. Biol. Eng. 2014, 34, 1–7. [Google Scholar] [CrossRef]
- Sneha, M.; Sundaram, N.M. Preparation and characterization of an iron oxide-hydroxyapatite nanocomposite for potential bone cancer therapy. Int. J. Nanomed. 2015, 10, 99–106. [Google Scholar] [CrossRef]
- Hench, L.; Best, S. Ceramics, Glasses and Glass-Ceramics. In Biomaterials Science, 2nd ed.; Rater, B., Hoffman, A., Schoen, F.J., Lemons, J.E., Eds.; Elsevier: San Diego, CA, USA, 2004; pp. 73–83. [Google Scholar]
- Nuss, K.M.; von Rechenberg, B. Biocompatibility issues with modern implants in bone—A review for clinical orthopedics. Open J. Orthop. 2008, 2, 66–78. [Google Scholar] [CrossRef]
- Kader, A.; Kaufmann, J.O.; Mangarova, D.B.; Moeckel, J.; Adams, L.C.; Brangsch, J.; Heyl, J.L.; Zhao, J.; Verlemann, C.; Karst, U.; et al. Collagen-Specific Molecular Magnetic Resonance Imaging of Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 711. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, M.; Thelwall, P.E.; Stanisz, G.J.; Blackband, S.J. Chemical Fixation Alters the Water Microenvironment in Rat Cortical Brain Slices—Implications for MRI Contrast. Proc. Int. Soc. Magn. Reson. Med. 2005, 13, 619. [Google Scholar]
- Junker, R.; Dimakis, A.; Thoneick, M.; Jansen, J.A. Effects of implant surface coatings and composition on bone integration: A systematic review. Clin. Oral Implant. Res. 2009, 20, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, E.; Iaquinta, M.R.; Lanzillotti, C.; Mazziotta, C.; Maritati, M.; Montesi, M.; Sprio, S.; Tampieri, A.; Tognon, M.; Martini, F. Bioactive Materials for Soft Tissue Repair. Front. Bioeng. Biotechnol. 2021, 9, 613787. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Andersson, M.; Wigren, S.; Pivodic, A.; Flynn, M.; Nannmark, U. Soft Tissue Integration of Hydroxyapatite-Coated Abutments for Bone Conduction Implants. Clin. Implant Dent. Relat. Res. 2015, 17, e730–e735. [Google Scholar] [CrossRef] [PubMed]
- Drevet, R.; Fauré, J.; Benhayoune, H. Bioactive Calcium Phosphate Coatings for Bone Implant Applications: A Review. Coatings 2023, 13, 1091. [Google Scholar] [CrossRef]
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone tissue engineering: Recent advances and challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef]
- Pang, X.; Lin, L.; Tang, B. Unraveling the role of Calcium ions in the mechanical properties of individual collagen fibrils. Sci. Rep. 2017, 7, 46042. [Google Scholar] [CrossRef]
- Gordeev, S.K. Nanoporous and nanofragmental carbon composite materials. In Nanostructured Carbon for Advanced Applications; Benedek, G., Milani, P., Ralchenko, V.G., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 71–88. [Google Scholar]
- PDF-2 2024. Phase Identification + Value. Available online: https://www.icdd.com/pdf-2/ (accessed on 21 February 2024).
- ISO 10993.5-99; State Standard: Medical Ware. Evaluation of the Biological Effect of Medical ware. Part 5: In Vitro Toxicity Tests. Standartinform: Moscow, Russia, 2010.
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (Group) | 24 h | 72 h | ||||
|---|---|---|---|---|---|---|
| OD, a.u. | PVC, % | TI, % | OD, a.u. | PVC, % | TI, % | |
| CGM (negative control group) | 0.252 ± 0.01 | 100.0 | 0.0 | 0.458 ± 0.004 | 100.0 | 0.0 |
| As-prepared C-C composite (group 1) | 0.245 ± 0.00 | 97.2 | 2.8 | 0.476 ± 0.013 | 103.9 | 0.0 |
| HAp-coated C-C composite (group 2) | 0.210 ± 0.00 *, ** | 83.3 | 16.7 | 0.389 ± 0.01 *, ** | 84.9 | 15.1 |
| 50% dimethyl sulfoxide (positive control group) | 0.041 ± 0.00 * | 16.3 | 83.7 | 0.040 ± 0.00 * | 8.7 | 91.3 |
| Symbol | Encoded Protein | Sequences F and R |
|---|---|---|
| RUNX2 | Runt- related transcription factor 2 (human) | F: tca-acg-atc-tga-gat-ttg-tgg-g R: ggg-gag-gat-ttg-tga-aga-cgg |
| ALPL | Alkaline phosphate (human) | F: acc-acc-acg-aga-gtg-aac-ca R: cgt-tgt-ctg-agt-acc-agt-ccc |
| GAPDH-1 | Glyceraldehyde-3-phosphate dehydrogenase, housekeeping gene (human) | F: gaa-ggt-gaa-ggt-cgg-agt-c R: gaa-gat-ggt-gat-ggg-att-tc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skriabin, A.S.; Tsygankov, P.A.; Vesnin, V.R.; Shakurov, A.V.; Skriabina, E.S.; Sviridova, I.K.; Sergeeva, N.S.; Kirsanova, V.A.; Akhmedova, S.A.; Zherdeva, V.V.; et al. Electrophoretic Deposition of Calcium Phosphates on Carbon–Carbon Composite Implants: Morphology, Phase/Chemical Composition and Biological Reactions. Int. J. Mol. Sci. 2024, 25, 3375. https://doi.org/10.3390/ijms25063375
Skriabin AS, Tsygankov PA, Vesnin VR, Shakurov AV, Skriabina ES, Sviridova IK, Sergeeva NS, Kirsanova VA, Akhmedova SA, Zherdeva VV, et al. Electrophoretic Deposition of Calcium Phosphates on Carbon–Carbon Composite Implants: Morphology, Phase/Chemical Composition and Biological Reactions. International Journal of Molecular Sciences. 2024; 25(6):3375. https://doi.org/10.3390/ijms25063375
Chicago/Turabian StyleSkriabin, Andrei S., Petr A. Tsygankov, Vladimir R. Vesnin, Alexey V. Shakurov, Elizaveta S. Skriabina, Irina K. Sviridova, Natalia S. Sergeeva, Valentina A. Kirsanova, Suraya A. Akhmedova, Victoria V. Zherdeva, and et al. 2024. "Electrophoretic Deposition of Calcium Phosphates on Carbon–Carbon Composite Implants: Morphology, Phase/Chemical Composition and Biological Reactions" International Journal of Molecular Sciences 25, no. 6: 3375. https://doi.org/10.3390/ijms25063375
APA StyleSkriabin, A. S., Tsygankov, P. A., Vesnin, V. R., Shakurov, A. V., Skriabina, E. S., Sviridova, I. K., Sergeeva, N. S., Kirsanova, V. A., Akhmedova, S. A., Zherdeva, V. V., Lukina, Y. S., & Bionyshev-Abramov, L. L. (2024). Electrophoretic Deposition of Calcium Phosphates on Carbon–Carbon Composite Implants: Morphology, Phase/Chemical Composition and Biological Reactions. International Journal of Molecular Sciences, 25(6), 3375. https://doi.org/10.3390/ijms25063375