

Functional Consequences of CFTR Interactions in Cystic Fibrosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. From Discovery and Diagnostics to Therapy in Cystic Fibrosis
1.2. History of Cystic Fibrosis
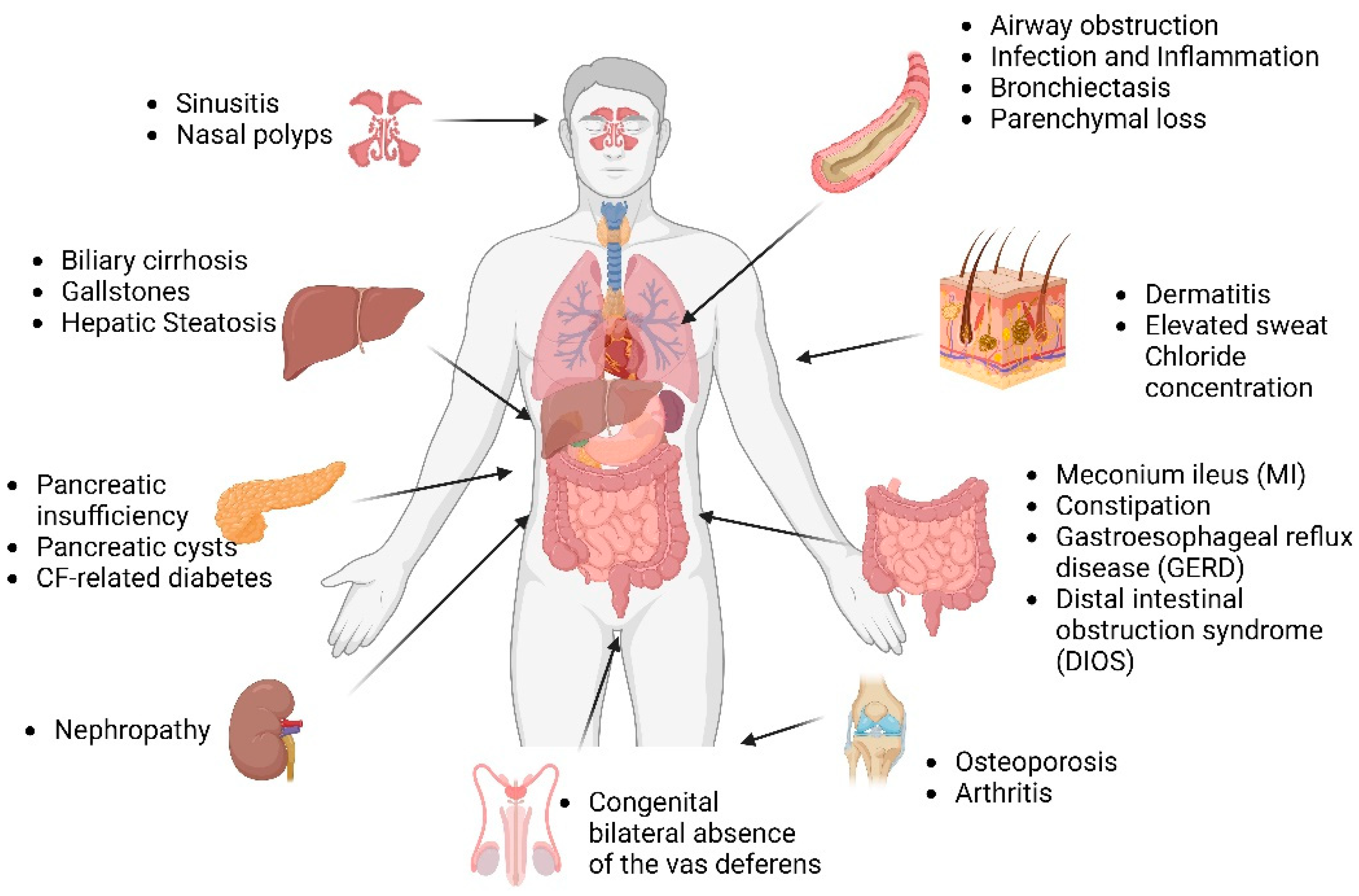
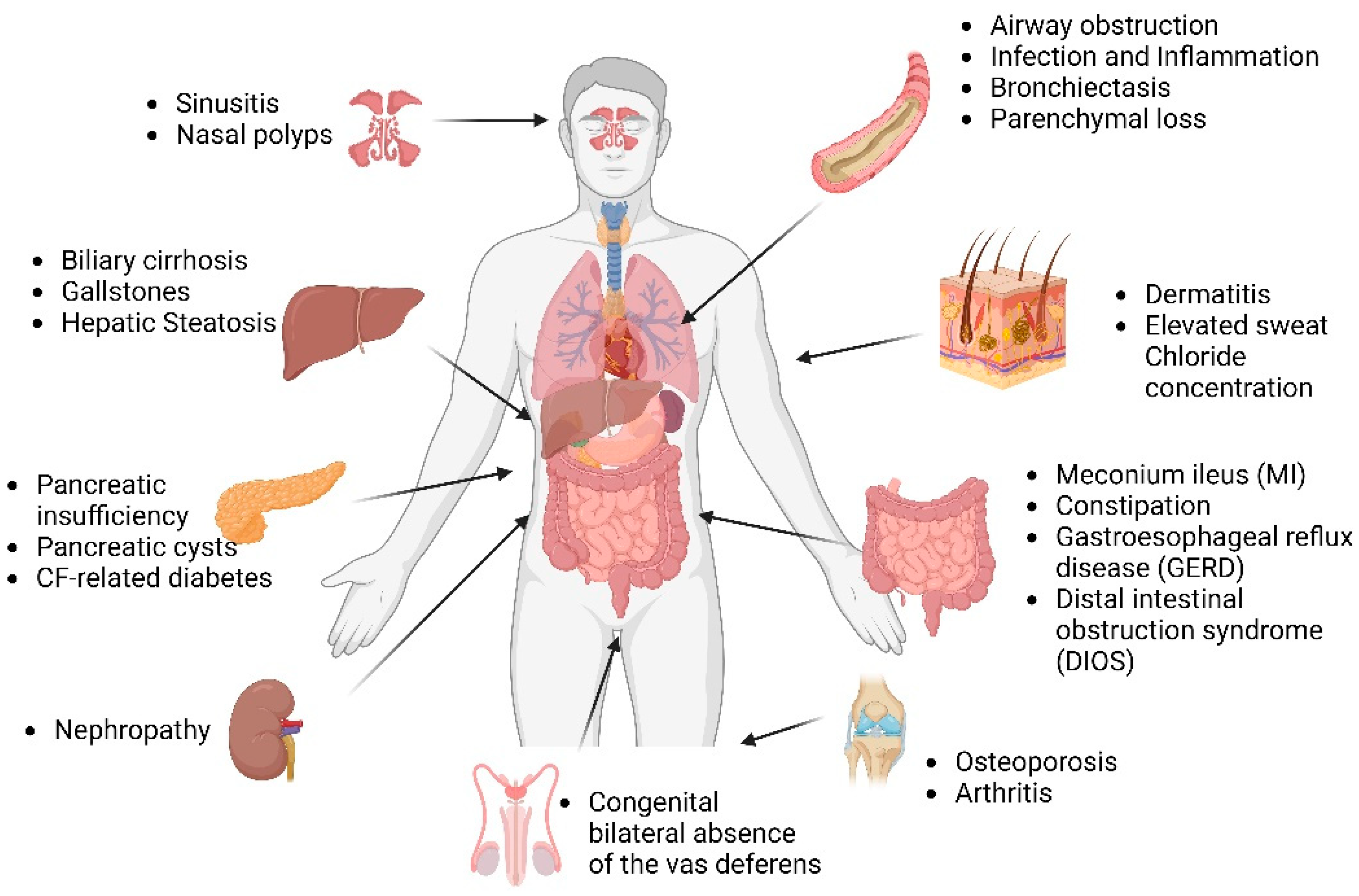
1.3. Multi-Organ Disease Manifestations in CF
1.3.1. CF Lung
1.3.2. CF Pancreas
1.3.3. CF Gut
1.4. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene
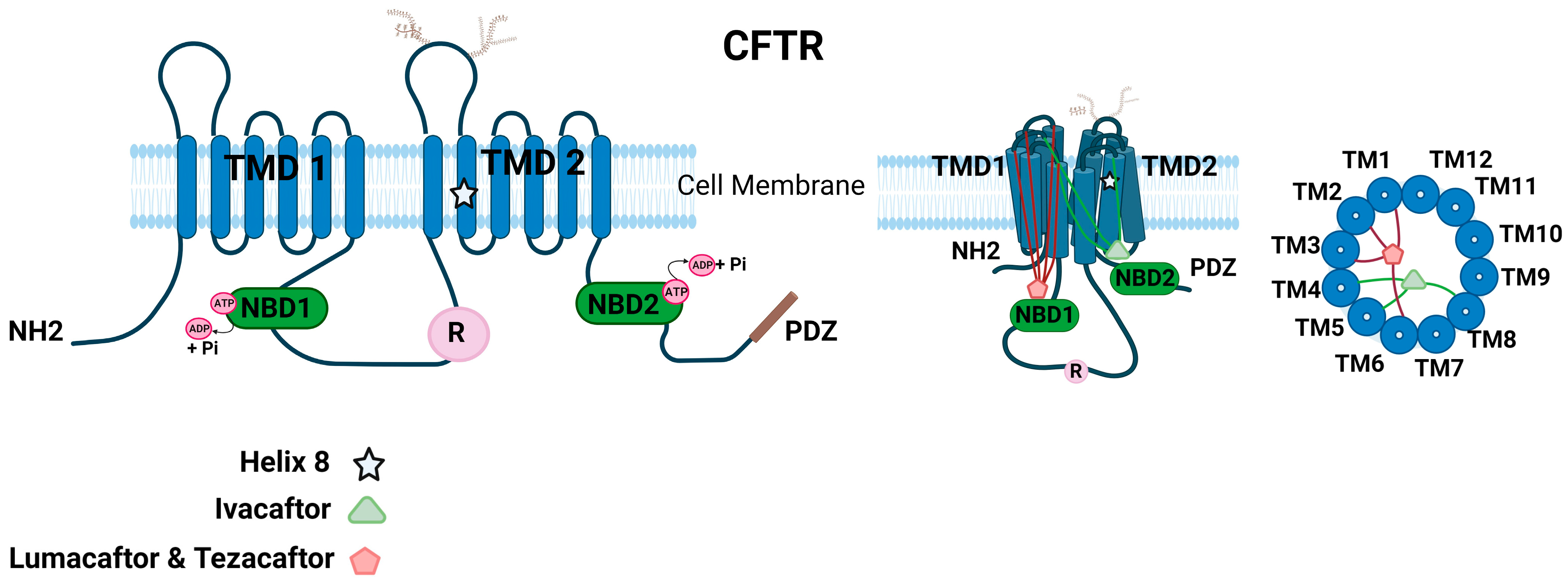
1.5. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Protein
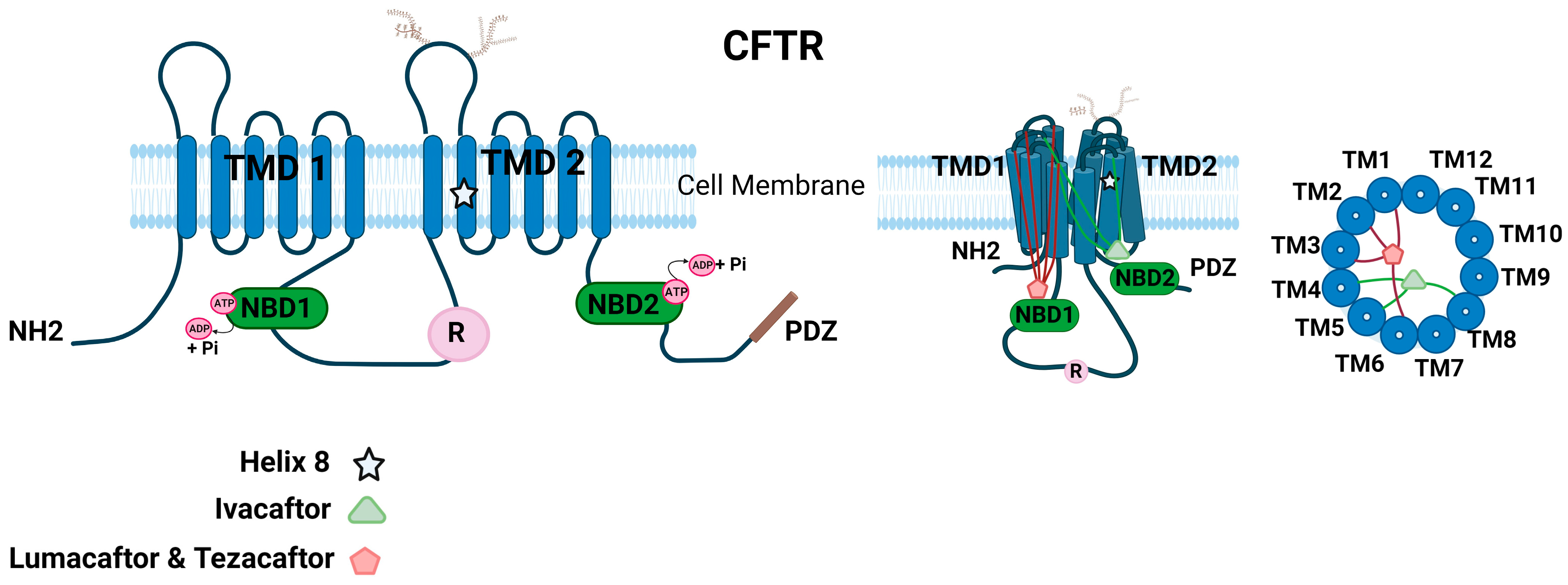
1.6. Molecular Structure of CFTR Ion Channel
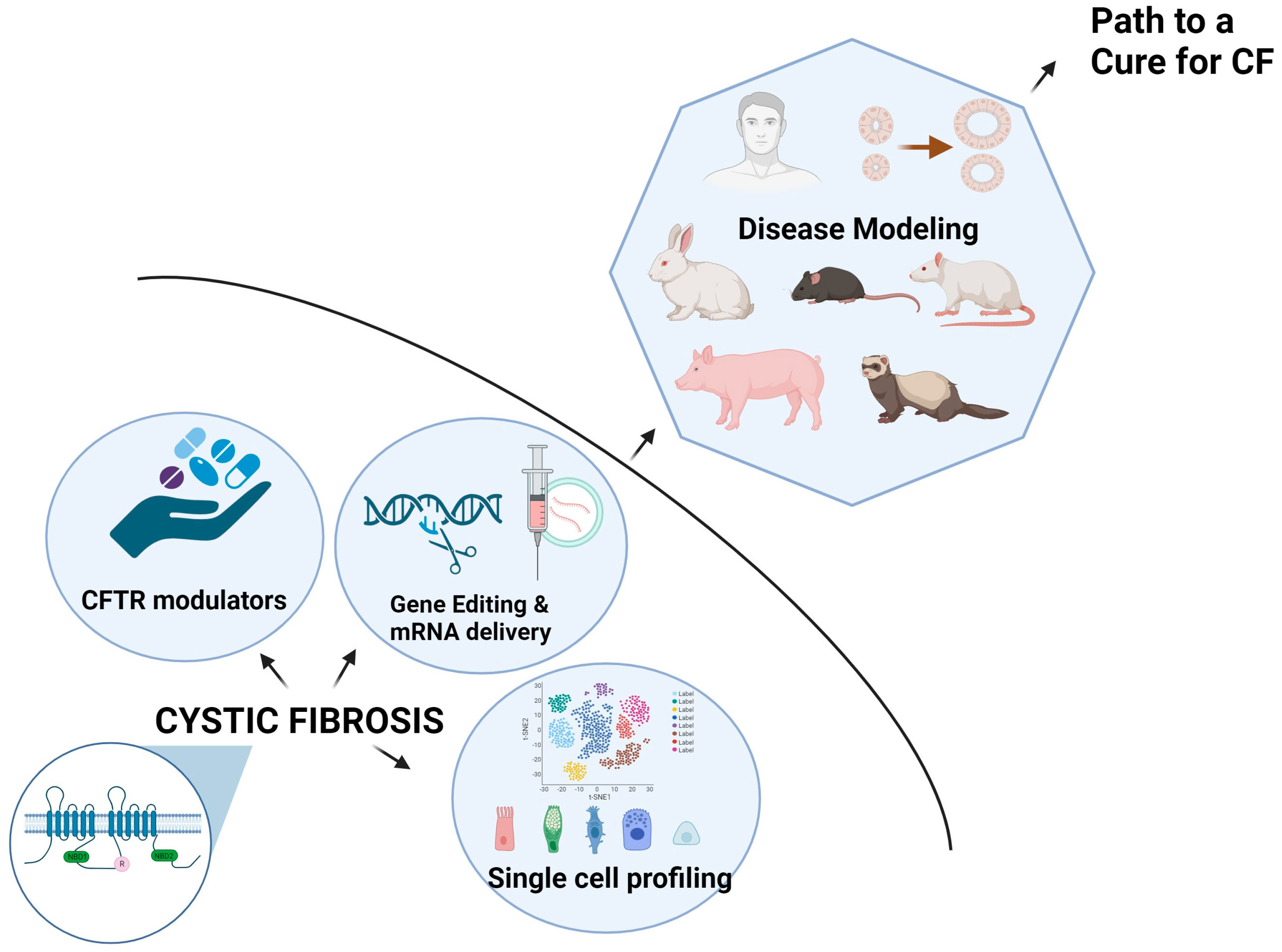
1.7. Addressing the CFTR Protein Defect Using Small Molecules-CFTR Modulators
1.7.1. CF Modulators in Clinical Application
1.7.2. Other CFTR Modulators for Potential Clinical Application
1.8. Non-CFTR Therapies in CF
1.9. Prospective Gene Modification Therapies in CF
1.10. CFTR Expression Defined Cellular Compositions in the Lung
1.10.1. Ionocytes
1.10.2. Secretory Cells
1.10.3. Basal Cells
1.10.4. Ciliated Cells
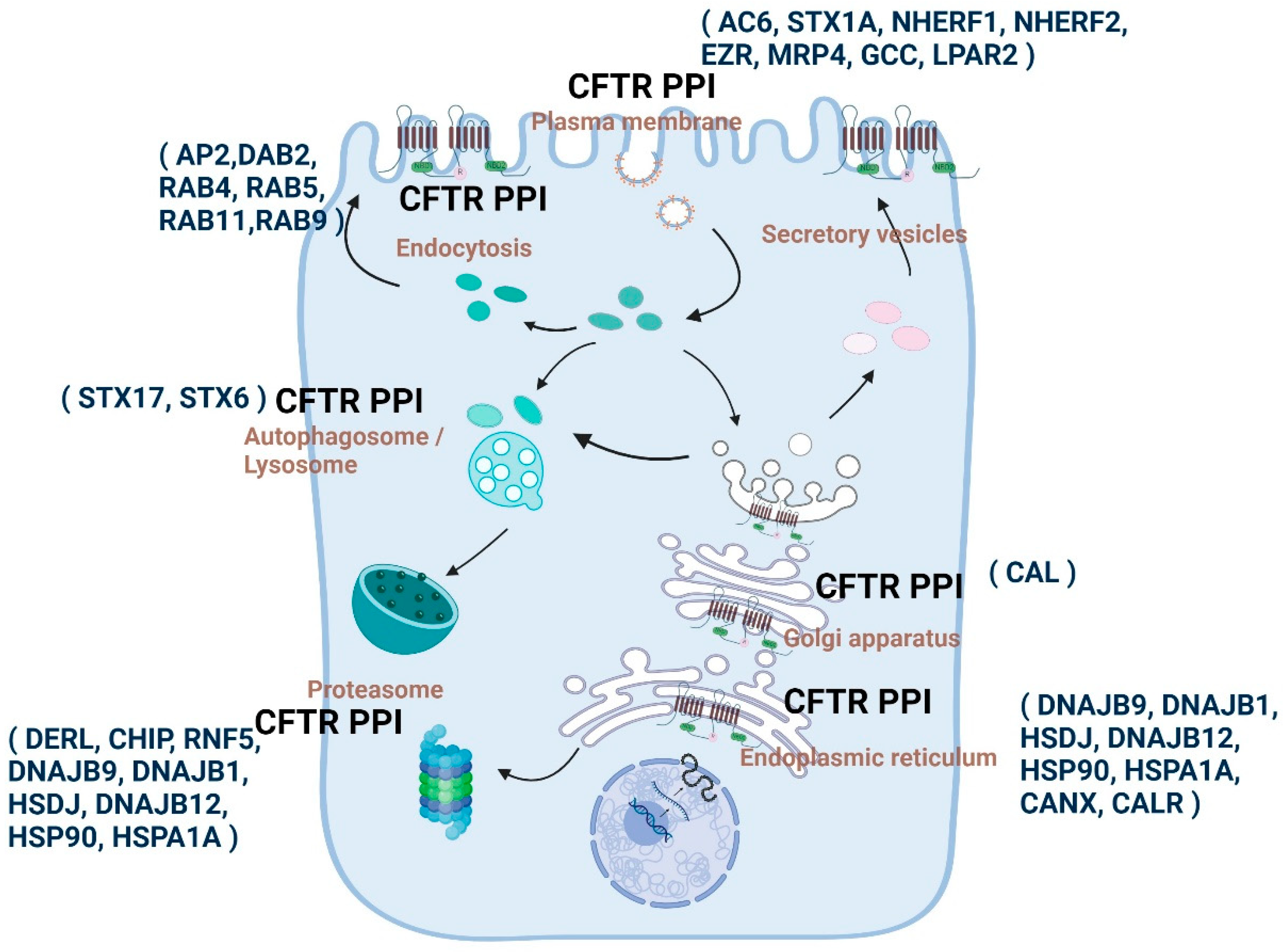
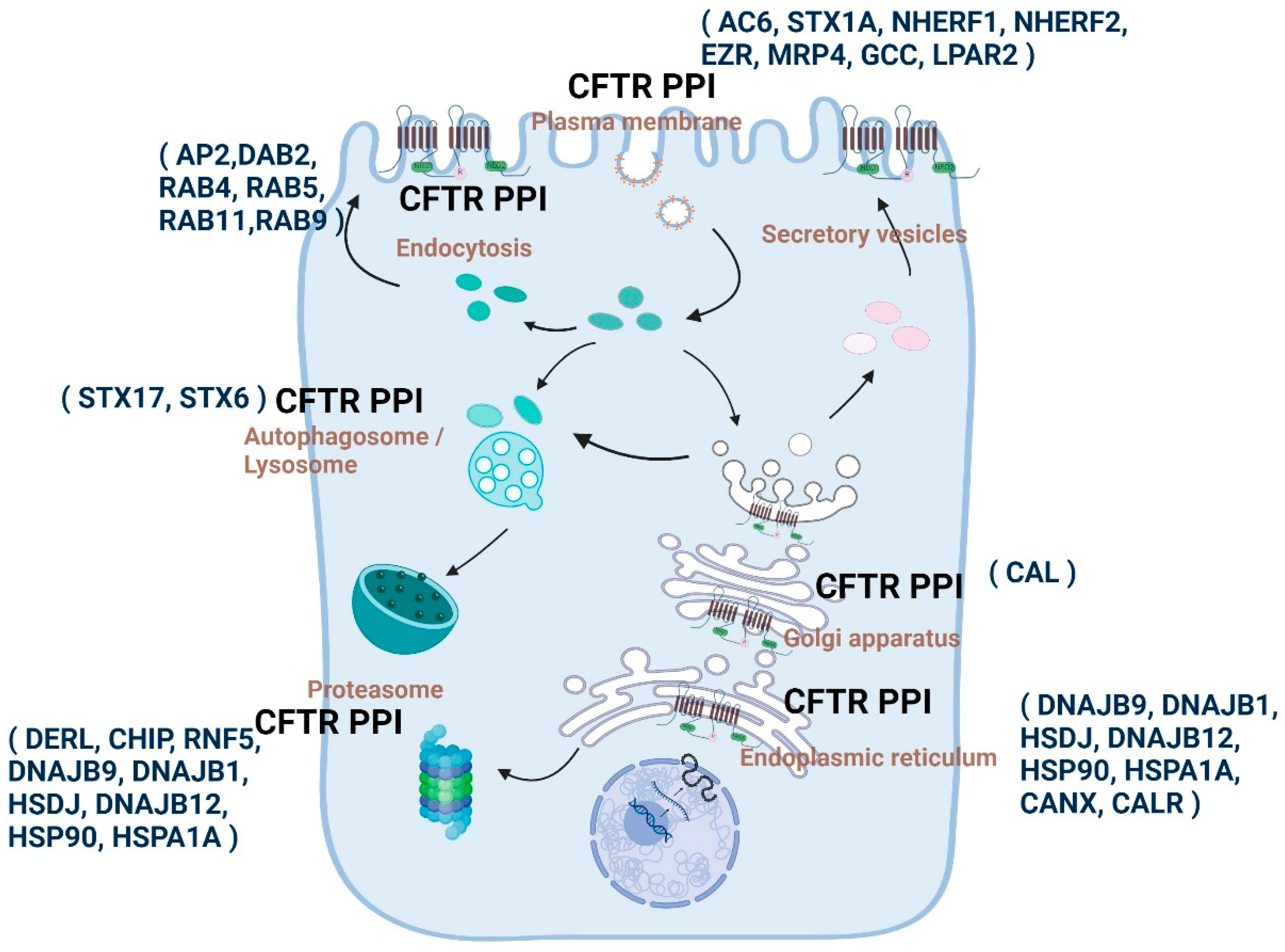
1.11. Altered Protein Interactions Determine the Etiological Course of CFTR-Related Disorders
1.11.1. Snapshot of the Molecular Communication between CFTR PPIs and CF by Various Proteomic Approaches
1.11.2. Cellular Sub-Compartmental State and Composition of CFTR PPIs and PPI Enabled Highly Compartmentalized CFTR Signalosomes
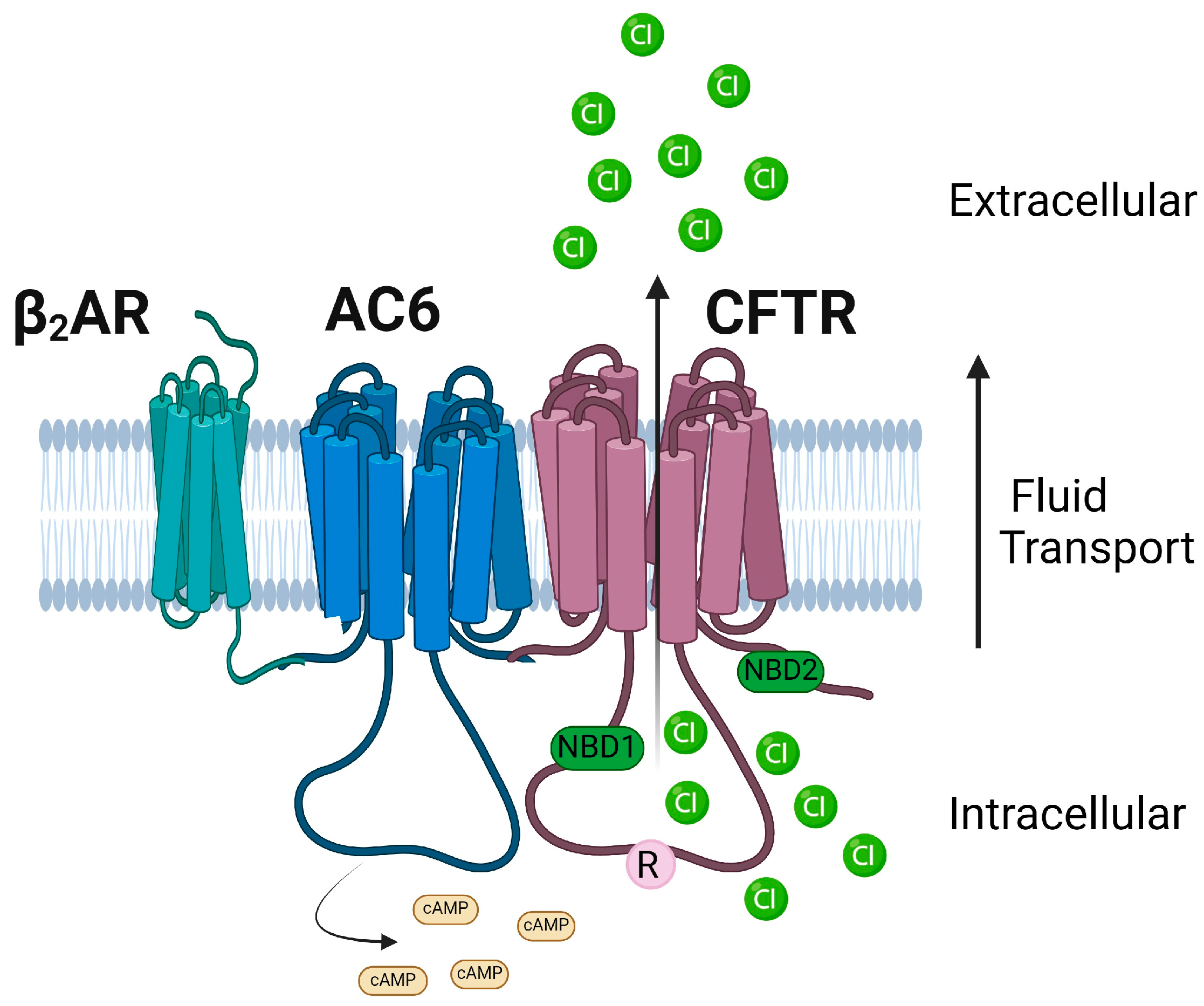
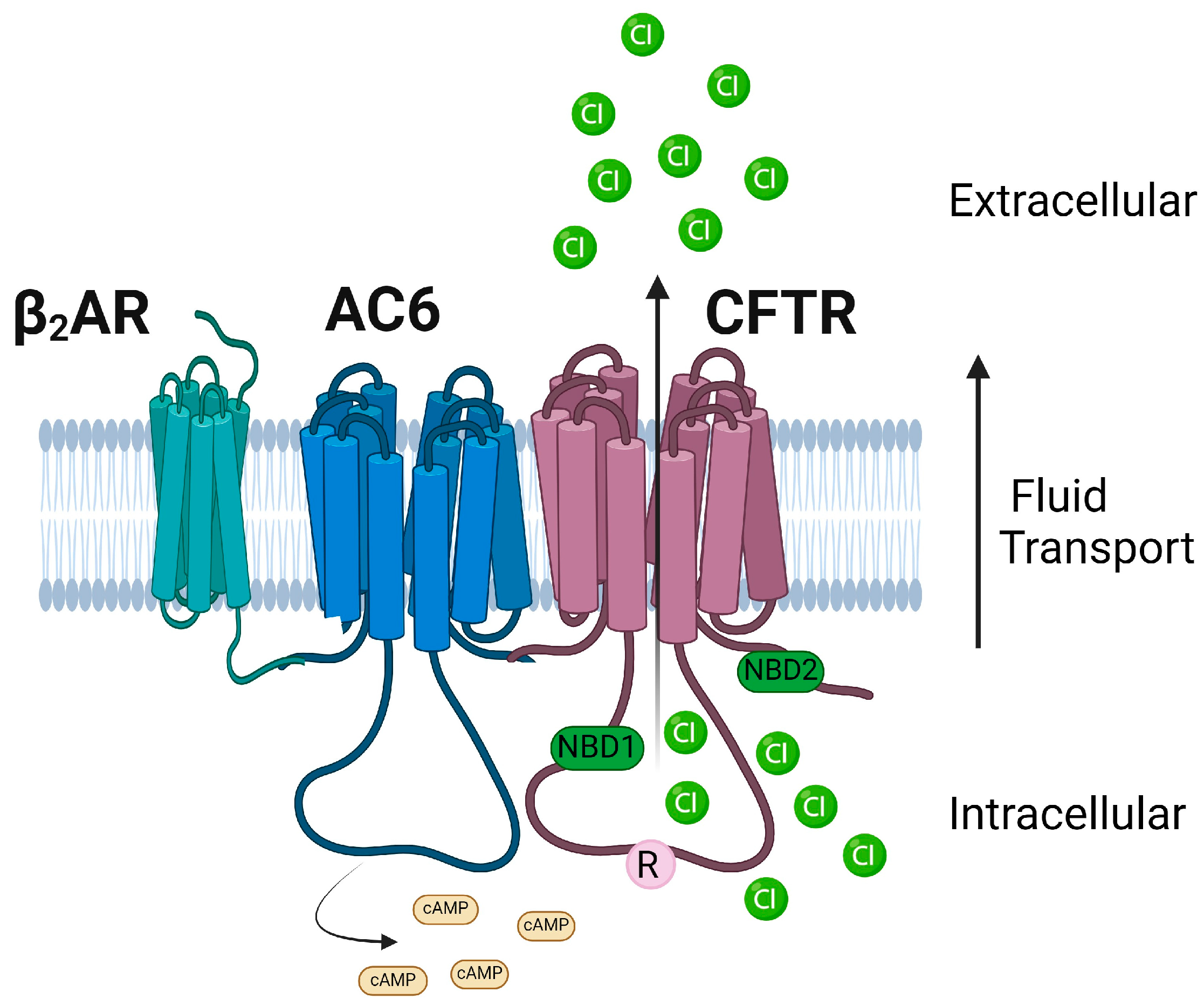
1.12. AC6
1.13. GCC
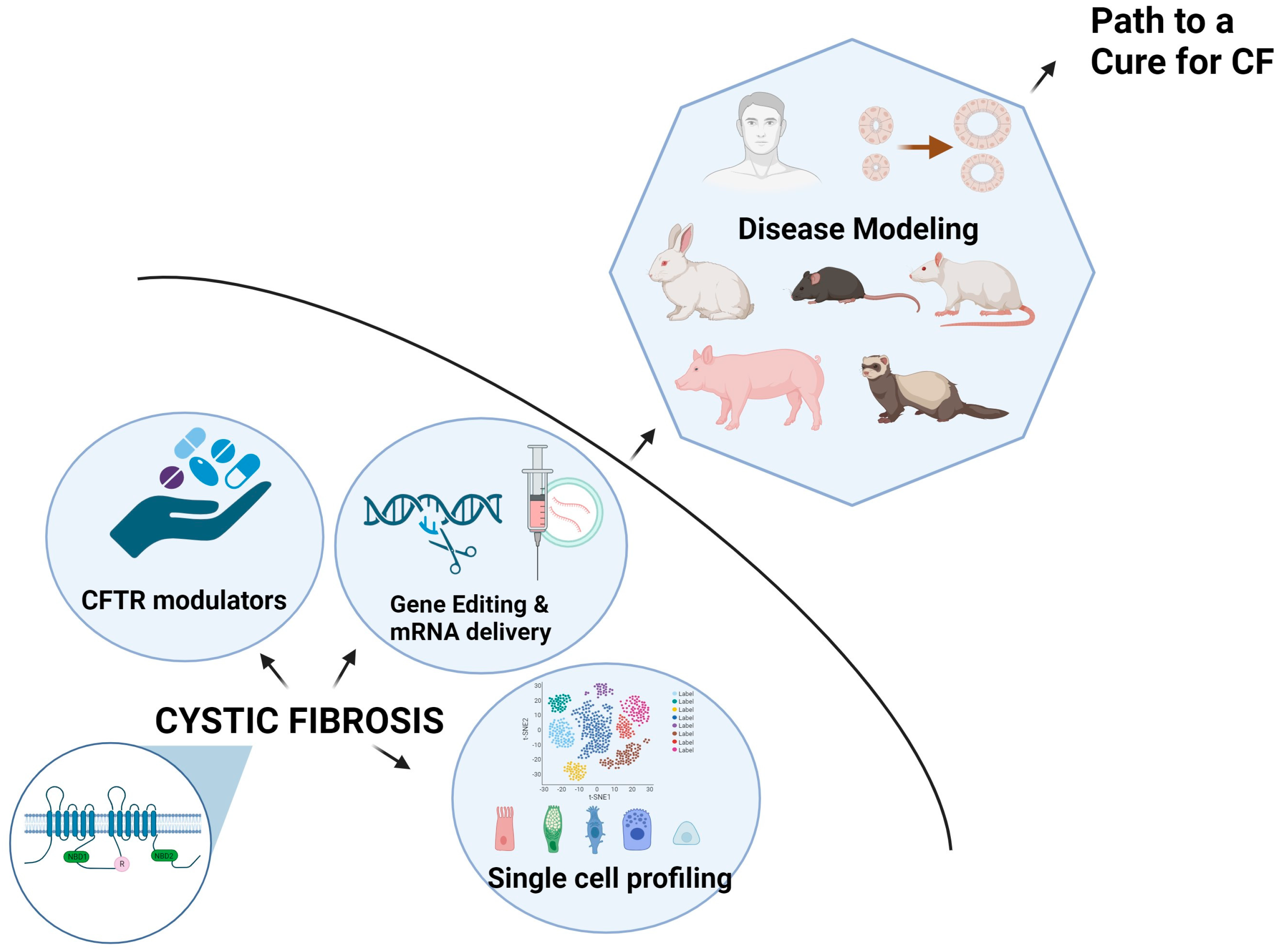
2. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.-S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.-C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-s.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Davis, S.D.; Rosenfeld, M.; Chmiel, J. Cystic Fibrosis: A Multi-Organ System Approach; Springer Nature: New York, NY, USA, 2020. [Google Scholar]
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Farber, S. Pancreatic function and disease in early life. V. Pathologic changes associated with pancreatic insufficiency in early life. Arch. Pathol. 1944, 37, 238–250. [Google Scholar]
- di Sant’Agnese, P.A.; Darling, R.C.; Perera, G.A.; Shea, E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: Clinical significance and relationship to the disease. Pediatrics 1953, 12, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.E.; Cooke, R.E. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959, 23, 545–549. [Google Scholar] [CrossRef]
- Matthews, L.W.; Doershuk, C.F.; Wise, M.; Eddy, G.; Nudelman, H.; Spector, S. A therapeutic regimen for patients with cystic fibrosis. J. Pediatr. 1964, 65, 558–575. [Google Scholar] [CrossRef]
- Anderson, M.P.; Rich, D.P.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Generation of cAMP-activated chloride currents by expression of CFTR. Science 1991, 251, 679–682. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmem-brane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular structure of the human CFTR ion channel. Cell 2017, 169, 85–95.e88. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Buchanan, S.G.; Burley, S.K.; Conners, K.; Dickey, M.; Dorwart, M.; Fowler, R.; Gao, X.; Guggino, W.B.; Hendrickson, W.A.; et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004, 23, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Naren, A.P. CFTR chloride channel in the apical compartments: Spatiotemporal coupling to its interacting partners. Integr. Biol. 2010, 2, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shen, Y.; Zheng, J. A review of cystic fibrosis: Basic and clinical aspects. Anim. Model. Exp. Med. 2021, 4, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, M. Molecular studies of CFTR interacting proteins. Pflug. Arch. 2001, 443 (Suppl. S1), S62–S64. [Google Scholar] [CrossRef] [PubMed]
- Heuser, T.; Raytchev, M.; Krell, J.; Porter, M.E.; Nicastro, D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 2009, 187, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.R.; Wallingford, J.B. Multiciliated cells. Curr. Biol. 2014, 24, R973–R982. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef]
- Whitsett, J.A. Airway epithelial differentiation and mucociliary clearance. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. S3), S143–S148. [Google Scholar] [CrossRef]
- Houtmeyers, E.; Gosselink, R.; Gayan-Ramirez, G.; Decramer, M. Regulation of mucociliary clearance in health and disease. Eur. Respir. J. 1999, 13, 1177–1188. [Google Scholar] [CrossRef]
- Zabner, J.; Smith, J.J.; Karp, P.H.; Widdicombe, J.H.; Welsh, M.J. Loss of CFTR Chloride Channels Alters Salt Absorption by Cystic Fibrosis Airway Epithelia In Vitro. Mol. Cell 1998, 2, 397–403. [Google Scholar] [CrossRef]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006, 290, C741–C749. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.; MacGregor, G.; Davis, M.; Innes, J.; Greening, A. Airways in cystic fibrosis are acidified: Detection by exhaled breath condensate. Thorax 2002, 57, 926–929. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; Van Eijk, M. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- McShane, D.; Davies, J.; Davies, M.; Bush, A.; Geddes, D.; Alton, E. Airway surface pH in subjects with cystic fibrosis. Eur. Respir. J. 2003, 21, 37–42. [Google Scholar] [CrossRef]
- Abou Alaiwa, M.H.; Beer, A.M.; Pezzulo, A.A.; Launspach, J.L.; Horan, R.A.; Stoltz, D.A.; Starner, T.D.; Welsh, M.J.; Zabner, J. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J. Cyst. Fibros. 2014, 13, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Muco-obstructive lung diseases. N. Engl. J. Med. 2019, 380, 1941–1953. [Google Scholar] [CrossRef]
- Tilley, A.E.; Walters, M.S.; Shaykhiev, R.; Crystal, R.G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015, 77, 379–406. [Google Scholar] [CrossRef]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef]
- Sun, X.; Olivier, A.K.; Liang, B.; Yi, Y.; Sui, H.; Evans, T.I.; Zhang, Y.; Zhou, W.; Tyler, S.R.; Fisher, J.T.; et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am. J. Respir. Cell Mol. Biol. 2014, 50, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Buxbaum, J. Gastrointestinal manifestations of cystic fibrosis. Dig. Dis. Sci. 2015, 60, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, H.; Durie, P.; Gaskin, K.; Weizman, Z.; Forstner, G. Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. N. Engl. J. Med. 1985, 312, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Shwachman, H.; Lebenthal, E.; Khaw, K.-T. Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics 1975, 55, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.L.; Dorney, S.F.; Gaskin, K.J.; Gruca, M.A.; O’Halloran, M.; Wilcken, B. Pancreatic function in infants identified as having cystic fibrosis in a neonatal screening program. N. Engl. J. Med. 1990, 322, 303–308. [Google Scholar] [CrossRef]
- McKay, I.R.; Ooi, C.Y. The exocrine pancreas in cystic fibrosis in the era of CFTR modulation: A mini review. Front. Pediatr. 2022, 10, 914790. [Google Scholar] [CrossRef]
- Flume, P.A.; Robinson, K.A.; O’Sullivan, B.P.; Finder, J.D.; Vender, R.L.; Willey-Courand, D.-B.; White, T.B.; Marshall, B.C.; Committee, C.P.G.f.P.T. Cystic fibrosis pulmonary guidelines: Airway clearance therapies. Respir. Care 2009, 54, 522–537. [Google Scholar]
- Ritivoiu, M.-E.; Drăgoi, C.M.; Matei, D.; Stan, I.V.; Nicolae, A.C.; Craiu, M.; Dumitrescu, I.-B.; Ciolpan, A.A. Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics 2023, 15, 162. [Google Scholar] [CrossRef]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic fibrosis-related diabetes. Front. Endocrinol. 2018, 9, 331856. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S. Gastrointestinal manifestations of cystic fibrosis. Gastroenterol. Hepatol. 2016, 12, 43. [Google Scholar]
- Van der Doef, H.P.; Kokke, F.; van der Ent, C.K.; Houwen, R.H. Intestinal obstruction syndromes in cystic fibrosis: Meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr. Gastroenterol. Rep. 2011, 13, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Corey, M.; Kerem, B.; Durie, P.; Tsui, L.C.; Levison, H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J. Pediatr. 1989, 114, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Chaudry, G.; Navarro, O.M.; Levine, D.S.; Oudjhane, K. Abdominal manifestations of cystic fibrosis in children. Pediatr. Radiol. 2006, 36, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.; Daneman, A.; Cutz, E.; Hagen, P.; Shandling, B. Abdominal calcification in cystic fibrosis with meconium ileus: Radiologic-pathologic correlation. Pediatr. Radiol. 1997, 27, 523–527. [Google Scholar] [CrossRef]
- Del Pin, C.A.; Czyrko, C.; Ziegler, M.M.; Scanlin, T.F.; Bishop, H.C. Management and survival of meconium ileus. A 30-year review. Ann. Surg. 1992, 215, 179. [Google Scholar] [CrossRef]
- Constantine, S.; Au, V.; Slavotinek, J. Abdominal manifestations of cystic fibrosis in adults: A review. Australas. Radiol. 2004, 48, 450–458. [Google Scholar] [CrossRef]
- McCarthy, V.A.; Harris, A. The CFTR gene and regulation of its expression. Pediatr. Pulmonol. 2005, 40, 1–8. [Google Scholar] [CrossRef]
- Zielenski, J.; Rozmahel, R.; Bozon, D.; Kerem, B.-s.; Grzelczak, Z.; Riordan, J.R.; Rommens, J.; Tsui, L.-C. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 1991, 10, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.-s.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- CFTR2. Cystic Fibrosis Foundation, John Hopkins Medicine; Sequenom Laboratories: San Diego, CA, SUA, 2023. [Google Scholar]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef] [PubMed]
- Parisi, G.F.; Mòllica, F.; Giallongo, A.; Papale, M.; Manti, S.; Leonardi, S. Cystic fibrosis transmembrane conductance regulator (CFTR): Beyond cystic fibrosis. Egypt. J. Med. Hum. Genet. 2022, 23, 94. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Amaral, M.D. CFTR and chaperones. J. Mol. Neurosci. 2004, 23, 41–48. [Google Scholar] [CrossRef]
- Tsui, L.-C. The cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Crit. Care Med. 1995, 151, S47. [Google Scholar] [CrossRef]
- Roxo-Rosa, M.; Xu, Z.; Schmidt, A.; Neto, M.; Cai, Z.; Soares, C.M.; Sheppard, D.N.; Amaral, M.D. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 17891–17896. [Google Scholar] [CrossRef]
- Gregory, R.J.; Rich, D.P.; Cheng, S.H.; Souza, D.; Paul, S.; Manavalan, P.; Anderson, M.P.; Welsh, M.J.; Smith, A.E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell. Biol. 1991, 11, 3886–3893. [Google Scholar] [PubMed]
- Seibert, F.S.; Linsdell, P.; Loo, T.W.; Hanrahan, J.W.; Clarke, D.M.; Riordan, J.R. Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 1996, 271, 15139–15145. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2014, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, N.A. CFTR and cystic fibrosis: A need for personalized medicine. In Studies of Epithelial Transporters and Ion Channels; Springer: New York, NY, USA, 2020; pp. 547–604. [Google Scholar]
- Lopes-Pacheco, M. CFTR modulators: Shedding light on precision medicine for cystic fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Beck, S.; Meyer, M.; Penque, D.; Cutting, G.R.; Amaral, M.D. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.R.; Picciano, J.A.; Bertrand, C.; Weixel, K.; Bridges, R.J.; Bradbury, N.A. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J. Biol. Chem. 2003, 278, 11554–11560. [Google Scholar] [CrossRef]
- Haardt, M.; Benharouga, M.; Lechardeur, D.; Kartner, N.; Lukacs, G.L. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: A novel class of mutation. J. Biol. Chem. 1999, 274, 21873–21877. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, G.; Chang, X.-B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 1993, 268, 21592–21598. [Google Scholar] [CrossRef]
- Gentzsch, M.; Riordan, J.R. Localization of sequences within the C-terminal domain of the cystic fibrosis transmembrane conductance regulator which impact maturation and stability. J. Biol. Chem. 2001, 276, 1291–1298. [Google Scholar] [CrossRef]
- Benharouga, M.; Haardt, M.; Kartner, N.; Lukacs, G.L. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell Biol. 2001, 153, 957–970. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Lewandowska, M.A.; Farinha, C.M.; Mendes, F.; Gonçalves, J.; Barreto, C.; Harris, A.; Amaral, M.D. Deletion of CFTR translation start site reveals functional isoforms of the protein in CF patients. Cell. Physiol. Biochem. 2009, 24, 335–346. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef]
- Pereira, S.V.-N.; Ribeiro, J.D.; Ribeiro, A.F.; Bertuzzo, C.S.; Marson, F.A.L. Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools. Sci. Rep. 2019, 9, 6234. [Google Scholar] [CrossRef] [PubMed]
- Deletang, K.; Taulan-Cadars, M. Splicing mutations in the CFTR gene as therapeutic targets. Gene Ther. 2022, 29, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Hwang, T.-C.; Nastiuk, K.L.; Nairn, A.C.; Gadsbyt, D.C. The protein kinase A-regulated cardiac CI− channel resembles the cystic fibrosis transmembrane conductance regulator. Nature 1992, 360, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.B.; Hou, Y.X.; Jensen, T.J.; Riordan, J.R. Mapping of cystic fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. J. Biol. Chem. 1994, 269, 18572–18575. [Google Scholar] [CrossRef]
- Li, C.; Naren, A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 2005, 108, 208–223. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Kota, P.; Aleksandrov, L.A.; He, L.; Jensen, T.; Cui, L.; Gentzsch, M.; Dokholyan, N.V.; Riordan, J.R. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J. Mol. Biol. 2010, 401, 194–210. [Google Scholar] [CrossRef]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991, 66, 1027–1036. [Google Scholar] [CrossRef]
- Anderson, M.P.; Berger, H.A.; Rich, D.P.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell 1991, 67, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Raghuram, V.; Kemp, B.E.; Witters, L.A.; Foskett, J.K. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J. Clin. Investig. 2000, 105, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Mehta, A. CFTR: A hub for kinases and crosstalk of c AMP and Ca2+. FEBS J. 2013, 280, 4417–4429. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Pato, M.D.; Riordan, J.R.; Hanrahan, J.W. Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am. J. Physiol. Cell Physiol. 1998, 274, C1397–C1410. [Google Scholar] [CrossRef] [PubMed]
- Vergani, P.; Lockless, S.W.; Nairn, A.C.; Gadsby, D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005, 433, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Naren, A.P. Methods for the study of intermolecular and intramolecular interactions regulating CFTR function. In Cystic Fibrosis Methods and Protocols; Springer: New York, NY, USA, 2002; pp. 175–186. [Google Scholar]
- Wilkinson, D.J.; Strong, T.V.; Mansoura, M.K.; Wood, D.L.; Smith, S.S.; Collins, F.S.; Dawson, D.C. CFTR activation: Additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 273, L127–L133. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Nitschke, R.; Greger, R.; Kunzelmann, K. The cystic fibrosis transmembrane conductance regulator activates aquaporin 3 in airway epithelial cells. J. Biol. Chem. 1999, 274, 11811–11816. [Google Scholar] [CrossRef]
- Boucher, R.C.; Stutts, M.J.; Knowles, M.R.; Cantley, L.; Gatzy, J.T. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J. Clin. Investig. 1986, 78, 1245–1252. [Google Scholar] [CrossRef]
- McNicholas, C.M.; Guggino, W.B.; Schwiebert, E.M.; Hebert, S.C.; Giebisch, G.; Egan, M.E. Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc. Natl. Acad. Sci. USA 1996, 93, 8083–8088. [Google Scholar] [CrossRef]
- Gabriel, S.E.; Clarke, L.L.; Boucher, R.C.; Stutts, M.J. CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature 1993, 363, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wigley, W.C.; Zeng, W.; Noel, L.E.; Marino, C.R.; Thomas, P.J.; Muallem, S. Regulation of Cl−/HCO3− exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J. Biol. Chem. 1999, 274, 3414–3421. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.-C.; Yeh, J.-T.; Zhang, J.; Yu, Y.-C.; Yeh, H.-I.; Destefano, S. Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol. 2018, 150, 539–570. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.F.; Kamis, A.B.; Aleksandrov, L.A.; Ford, R.C.; Riordan, J.R. Purification and crystallization of the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2004, 279, 39051–39057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597.e1589. [Google Scholar] [CrossRef]
- Mihályi, C.; Töröcsik, B.; Csanády, L. Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. Elife 2016, 5, e18164. [Google Scholar] [CrossRef]
- Mihályi, C.; Iordanov, I.; Töröcsik, B.; Csanády, L. Simple binding of protein kinase A prior to phosphorylation allows CFTR anion channels to be opened by nucleotides. Proc. Natl. Acad. Sci. USA 2020, 117, 21740–21746. [Google Scholar] [CrossRef]
- Naren, A.P.; Nelson, D.J.; Xie, W.; Jovov, B.; Pevsner, J.; Bennett, M.K.; Benos, D.J.; Quick, M.W.; Kirk, K.L. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature 1997, 390, 302–305. [Google Scholar] [CrossRef]
- Naren, A.P.; Cormet-Boyaka, E.; Fu, J.; Villain, M.; Blalock, J.E.; Quick, M.W.; Kirk, K.L. CFTR chloride channel regulation by an interdomain interaction. Science 1999, 286, 544–548. [Google Scholar] [CrossRef]
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Goor, F.V.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Okiyoneda, T. Peripheral protein quality control as a novel drug target for CFTR stabilizer. Front. Pharmacol. 2018, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discovery. 2018, 23, 111–121. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Bear, C.E. A therapy for most with cystic fibrosis. Cell 2020, 180, 211. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.B.; Yasothan, U.; Kirkpatrick, P. Ivacaftor. Nat. Rev. Drug Discov. 2012, 11, 349–351. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef]
- Sala, M.A.; Jain, M. Tezacaftor for the treatment of cystic fibrosis. Expert Rev. Respir. Med. 2018, 12, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Becq, F.; Mirval, S.; Carrez, T.; Lévêque, M.; Billet, A.; Coraux, C.; Sage, E.; Cantereau, A. The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. Eur. Respir. J. 2022, 59, 2100671. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Bell, S.C.; Barry, P.J.; De Boeck, K.; Drevinek, P.; Elborn, J.S.; Plant, B.J.; Minić, P.; Van Braeckel, E.; Verhulst, S.; Muller, K. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J. Cyst. Fibros. 2019, 18, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Sinha, C.; Zhang, W.; Moon, C.S.; Actis, M.; Yarlagadda, S.; Arora, K.; Woodroofe, K.; Clancy, J.P.; Lin, S.; Ziady, A.G.; et al. Capturing the Direct Binding of CFTR Correctors to CFTR by Using Click Chemistry. Chembiochem 2015, 16, 2017–2022. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-dependent but ATP-independent Manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Stauffer, B.B.; Imhoff, B.R.; Rab, A.; Hong, J.S.; Sorscher, E.J.; McCarty, N.A. VX-770-mediated potentiation of numerous human CFTR disease mutants is influenced by phosphorylation level. Sci. Rep. 2019, 9, 13460. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the major cystic fibrosis mutant interact through membrane-spanning domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef]
- Pedemonte, N.; Bertozzi, F.; Caci, E.; Sorana, F.; Di Fruscia, P.; Tomati, V.; Ferrera, L.; Rodríguez-Gimeno, A.; Berti, F.; Pesce, E. Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci. Adv. 2020, 6, eaay9669. [Google Scholar] [CrossRef]
- Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A.M.; Vortherms, T. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid(ABBV/GLPG-2222), a Potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for the Treatment of Cystic Fibrosis; ACS Publications: Washington, DC, USA, 2018. [Google Scholar]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Scanio, M.J.; Searle, X.B.; Liu, B.; Koenig, J.R.; Altenbach, R.; Gfesser, G.A.; Bogdan, A.; Greszler, S.; Zhao, G.; Singh, A. Discovery of ABBV/GLPG-3221, a Potent Corrector of CFTR for the Treatment of Cystic Fibrosis. ACS Med. Chem. Lett. 2019, 10, 1543–1548. [Google Scholar] [CrossRef]
- De Wilde, G.; Gees, M.; Musch, S.; Verdonck, K.; Jans, M.; Wesse, A.-S.; Singh, A.K.; Hwang, T.-C.; Christophe, T.; Pizzonero, M. Identification of GLPG/ABBV-2737, a novel class of corrector, which exerts functional synergy with other CFTR modulators. Front. Pharmacol. 2019, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Burgener, E.B.; Cornfield, D.N. Delivering a New Future for People with Cystic Fibrosis. Pediatrics 2023, 152, e2023062985. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.; Ramsey, B.W. New Therapeutic Approaches to Modulate and Correct Cystic Fibrosis Transmembrane Conductance Regulator. Pediatr. Clin. N. Am. 2016, 63, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Forge, A.; Schacht, J. Aminoglycoside Antibiotics. Audiol. Neurotol. 2000, 5, 3–22. [Google Scholar] [CrossRef]
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021, 12, 4358. [Google Scholar] [CrossRef]
- Konstan, M.W.; VanDevanter, D.R.; Rowe, S.M.; Wilschanski, M.; Kerem, E.; Sermet-Gaudelus, I.; DiMango, E.; Melotti, P.; McIntosh, J.; De Boeck, K. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J. Cyst. Fibros. 2020, 19, 595–601. [Google Scholar] [CrossRef]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef]
- Aksit, M.A.; Bowling, A.D.; Evans, T.A.; Joynt, A.T.; Osorio, D.; Patel, S.; West, N.; Merlo, C.; Sosnay, P.R.; Cutting, G.R.; et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J. Cyst. Fibros. 2019, 18, 606–613. [Google Scholar] [CrossRef]
- Lee, R.E.; Lewis, C.A.; He, L.; Bulik-Sullivan, E.C.; Gallant, S.C.; Mascenik, T.M.; Dang, H.; Cholon, D.M.; Gentzsch, M.; Morton, L.C.; et al. Small-molecule eRF3a degraders rescue CFTR nonsense mutations by promoting premature termination codon readthrough. J. Clin. Investig. 2022, 132, e154571. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, A.; Balgi, A.D.; Hosseini-Farahabadi, S.; Choi, K.; Has, C.; Roberge, M. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021, 49, 3692–3708. [Google Scholar] [CrossRef]
- Pedemonte, N.; Sonawane, N.D.; Taddei, A.; Hu, J.; Zegarra-Moran, O.; Suen, Y.F.; Robins, L.I.; Dicus, C.W.; Willenbring, D.; Nantz, M.H.; et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol. Pharmacol. 2005, 67, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Renda, M.; Barreca, M.; Borrelli, A.; Spanò, V.; Montalbano, A.; Raimondi, M.V.; Bivacqua, R.; Musante, I.; Scudieri, P.; Guidone, D.; et al. Novel tricyclic pyrrolo-quinolines as pharmacological correctors of the mutant CFTR chloride channel. Sci. Rep. 2023, 13, 7604. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Eckford, P.D.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional rescue of c. 3846G> A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J. Cyst. Fibros. 2020, 19, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021, 20, 106–119. [Google Scholar] [CrossRef]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-molecule drugs for cystic fibrosis: Where are we now? Pulm. Pharmacol. Ther. 2022, 72, 102098. [Google Scholar] [CrossRef]
- Mergiotti, M.; Murabito, A.; Prono, G.; Ghigo, A. CFTR Modulator Therapy for Rare CFTR Mutants. J. Respir. 2022, 2, 59–76. [Google Scholar] [CrossRef]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Pringle, I.; Hyde, S.; Gill, D. Non-viral vectors in cystic fibrosis gene therapy: Recent developments and future prospects. Expert Opin. Biol. Ther. 2009, 9, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.G.; Harbottle, R.P.; Lawrence, L.; Knapton, H.J.; Themis, M.; Coutelle, C. Enhancement of adenovirus-mediated gene transfer to the airways by DEAE dextran and sodium caprate in vivo. Mol. Ther. 2003, 7, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Scaria, A.; St. George, J.A.; Jiang, C.; Kaplan, J.M.; Wadsworth, S.C.; Gregory, R.J. Adenovirus-mediated persistent cystic fibrosis transmembrane conductance regulator expression in mouse airway epithelium. J. Virol. 1998, 72, 7302–7309. [Google Scholar] [CrossRef] [PubMed]
- Bellon, G.; Michel-Calemard, L.; Thouvenot, D.; Jagneaux, V.; Poitevin, F.; Malcus, C.; Accart, N.; Layani, M.P.; Aymard, M.; Bernon, H. Aerosol administration of a recombinant adenovirus expressing CFTR to cystic fibrosis patients: A phase I clinical trial. Hum. Gene Ther. 1997, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.L.; Harrison, P.T. Genetic therapies for cystic fibrosis lung disease. Curr. Opin. Pharmacol. 2017, 34, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.-Y.; Qiu, J. Human bocavirus type-1 capsid facilitates the transduction of ferret airways by adeno-associated virus genomes. Hum. Gene Ther. 2017, 28, 612–625. [Google Scholar] [CrossRef]
- Vidović, D.; Carlon, M.S.; Da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.; Ramalho, A.S.; Van den Haute, C.; Ferrante, M.; Baekelandt, V. rAAV-CFTRΔR rescues the cystic fibrosis phenotype in human intestinal organoids and cystic fibrosis mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef]
- Foldvari, M.; Chen, D.W.; Nafissi, N.; Calderon, D.; Narsineni, L.; Rafiee, A. Non-viral gene therapy: Gains and challenges of non-invasive administration methods. J. Control. Release 2016, 240, 165–190. [Google Scholar] [CrossRef]
- Catanese, D.; Fogg, J.; Schrock, D.; Gilbert, B.; Zechiedrich, L. Supercoiled Minivector DNA resists shear forces associated with gene therapy delivery. Gene Ther. 2012, 19, 94–100. [Google Scholar] [CrossRef]
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Bangel-Ruland, N.; Tomczak, K.; Fernández Fernández, E.; Leier, G.; Leciejewski, B.; Rudolph, C.; Rosenecker, J.; Weber, W.M. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: A novel alternative for cystic fibrosis gene therapy. J. Gene Med. 2013, 15, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Raychowdhury, M.K.; Tabatadze, D.R.; Cantiello, H.F. Reversal of cystic fibrosis phenotype in a cultured Δ508 cystic fibrosis transmembrane conductance regulator cell line by oligonucleotide insertion. Proc. Natl. Acad. Sci. USA 2004, 101, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Krainer, A.R. Antisense oligonucleotide therapeutics for cystic fibrosis: Recent developments and perspectives. Mol. Cells 2023, 46, 10–20. [Google Scholar] [CrossRef]
- Chiba-Falek, O.; Kerem, E.; Shoshani, T.; Aviram, M.; Augarten, A.; Bentur, L.; Tal, A.; Tullis, E.; Rahat, A.; Kerem, B. The molecular basis of disease variability among cystic fibrosis patients carrying the 3849 + 10 kb C → T mutation. Genomics 1998, 53, 276–283. [Google Scholar] [CrossRef]
- Friedman, K.J.; Kole, J.; Cohn, J.A.; Knowles, M.R.; Silverman, L.M.; Kole, R. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem. 1999, 274, 36193–36199. [Google Scholar] [CrossRef]
- Michaels, W.E.; Pena-Rasgado, C.; Kotaria, R.; Bridges, R.J.; Hastings, M.L. Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2114886119. [Google Scholar] [CrossRef]
- Zaher, A.; ElSaygh, J.; Elsori, D.; ElSaygh, H.; Sanni, A.; Elsaygh, H.; Sanni, A. A review of Trikafta: Triple cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy. Cureus 2021, 13, e16144. [Google Scholar] [CrossRef]
- Oren, Y.S.; Sinai, M.I.-T.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.R.; Zhao, C.; Jiang, C.; Bai, D.; Katz, M.; Greenlee, S.; Kawabe, H.; McCaleb, M.; Rotin, D.; Guo, S. Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros. 2017, 16, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Karoubi, G.; Duchesneau, P.; Shutova, M.V.; Sung, H.-K.; Tonge, P.; Bear, C.; Rogers, I.; Nagy, A.; Waddell, T.K. Generation of induced progenitor-like cells from mature epithelial cells using interrupted reprogramming. Stem Cell Rep. 2017, 9, 1780–1795. [Google Scholar] [CrossRef]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef]
- Pollard, B.S.; Pollard, H.B. Induced pluripotent stem cells for treating cystic fibrosis: State of the science. Pediatr. Pulmonol. 2018, 53, S12–S29. [Google Scholar] [CrossRef]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharmacol. Adv. Appl. 2016, 8, 127–140. [Google Scholar]
- Simsek, S.; Zhou, T.; Robinson, C.L.; Tsai, S.-Y.; Crespo, M.; Amin, S.; Lin, X.; Hon, J.; Evans, T.; Chen, S. Modeling cystic fibrosis using pluripotent stem cell-derived human pancreatic ductal epithelial cells. Stem Cells Transl. Med. 2016, 5, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed]
- Reid, L.M.; Jones, R. Mucous membrane of respiratory epithelium. Environ. Health Perspect. 1980, 35, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Meyrick, B.; Sturgess, J.M.; Reid, L. A reconstruction of the duct system and secretory tubules of the human bronchial submucosal gland. Thorax 1969, 24, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Moninger, T.O.; Thurman, A.L.; Xie, Y.; Jain, A.; Zarei, K.; Powers, L.S.; Pezzulo, A.A.; Stoltz, D.A.; Welsh, M.J. Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs. Proc. Natl. Acad. Sci. USA 2022, 119, e2119759119. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef]
- Hogg, J.C.; Macklem, P.T.; Thurlbeck, W. Site and nature of airway obstruction in chronic obstructive lung disease. N. Engl. J. Med. 1968, 278, 1355–1360. [Google Scholar] [CrossRef]
- Hyde, D.M.; Hamid, Q.; Irvin, C.G. Anatomy, pathology, and physiology of the tracheobronchial tree: Emphasis on the distal airways. J. Allergy Clin. Immunol. 2009, 124, S72–S77. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.F.; Altisheh, R.; Zitt, M. Small airways disease and severe asthma. World Allergy Organ. J. 2017, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Paré, P.D.; Hackett, T.-L. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol. Rev. 2017, 97, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090. [Google Scholar] [CrossRef]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef]
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018, 19, 54. [Google Scholar] [CrossRef]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef]
- Yuan, F.; Gasser, G.N.; Lemire, E.; Montoro, D.T.; Jagadeesh, K.; Zhang, Y.; Duan, Y.; Ievlev, V.; Wells, K.L.; Rotti, P.G.; et al. Transgenic ferret models define pulmonary ionocyte diversity and function. Nature 2023, 621, 857–867. [Google Scholar] [CrossRef]
- Sato, Y.; Kim, D.; Turner, M.J.; Luo, Y.; Zaidi, S.S.Z.; Thomas, D.Y.; Hanrahan, J.W. Ionocyte-Specific Regulation of Cystic Fibrosis Transmembrane Conductance Regulator. Am. J. Respir. Cell Mol. Biol. 2023, 69, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Luo, M.; Tang, Y.; Yu, M.; Yuan, F.; Gasser, G.N.; Liu, X.; Engelhardt, J.F. Sonic Hedgehog Signaling Is Essential for Pulmonary Ionocyte Specification in Human and Ferret Airway Epithelia. Am. J. Respir. Cell Mol. Biol. 2023, 69, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Wang, F.; Rollins, S.; Bawa, P.S.; Lu, J.; Tang, Y.; Beermann, M.L.; Schlaeger, T.; et al. De Novo Generation of Pulmonary Ionocytes from Normal and Cystic Fibrosis Human Induced Pluripotent Stem Cells. Am. J. Respir. Crit. Care Med. 2023, 207, 1249–1253. [Google Scholar] [CrossRef]
- Jeffery, P.; Li, D. Airway mucosa: Secretory cells, mucus and mucin genes. Eur. Respir. J. 1997, 10, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Joo, N.S.; Reitz, B.; Wine, J.J.; Verkman, A.S. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc. Natl. Acad. Sci. USA 2001, 98, 8119–8123. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Morley, M.P.; Jiang, C.; Wu, Y.; Li, G.; Du, Y.; Zhao, S.; Wagner, A.; Cakar, A.C.; Kouril, M.; et al. Guided construction of single cell reference for human and mouse lung. Nat. Commun. 2023, 14, 4566. [Google Scholar] [CrossRef]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef]
- Carraro, G.; Langerman, J.; Sabri, S.; Lorenzana, Z.; Purkayastha, A.; Zhang, G.; Konda, B.; Aros, C.J.; Calvert, B.A.; Szymaniak, A. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med. 2021, 27, 806–814. [Google Scholar] [CrossRef]
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single-Cell Reconstruction of Human Basal Cell Diversity in Normal and Idiopathic Pulmonary Fibrosis Lungs. Am. J. Respir. Crit. Care Med. 2020, 202, 1540–1550. [Google Scholar] [CrossRef]
- Beppu, A.K.; Zhao, J.; Yao, C.; Carraro, G.; Israely, E.; Coelho, A.L.; Drake, K.; Hogaboam, C.M.; Parks, W.C.; Kolls, J.K.; et al. Epithelial plasticity and innate immune activation promote lung tissue remodeling following respiratory viral infection. Nat. Commun. 2023, 14, 5814. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Mustafina, K.R.; Luo, Y.; Martini, C.; Thomas, D.Y.; Wiseman, P.W.; Hanrahan, J.W. Nonspecific binding of common anti-CFTR antibodies in ciliated cells of human airway epithelium. Sci. Rep. 2021, 11, 23256. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Lotti, V.; Merigo, F.; Lagni, A.; Di Clemente, A.; Ligozzi, M.; Bernardi, P.; Rossini, G.; Concia, E.; Plebani, R.; Romano, M.; et al. CFTR Modulation Reduces SARS-CoV-2 Infection in Human Bronchial Epithelial Cells. Cells 2022, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Januska, M.N.; Walsh, M.J. Single-Cell RNA Sequencing Reveals New Basic and Translational Insights in the Cystic Fibrosis Lung. Am. J. Respir. Cell Mol. Biol. 2023, 68, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.C.; Khanal, S.; Gomez, J.L.; Sauler, M.; Adams, T.S.; Chupp, G.L.; Yan, X.; Poli, S.; Zhao, Y.; Montgomery, R.R. Single-cell transcriptional archetypes of airway inflammation in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2020, 202, 1419–1429. [Google Scholar] [CrossRef]
- Leir, S.-H.; Yin, S.; Kerschner, J.L.; Cosme, W.; Harris, A. An atlas of human proximal epididymis reveals cell-specific functions and distinct roles for CFTR. Life Sci. Alliance 2020, 3, e202000744. [Google Scholar] [CrossRef]
- Busslinger, G.A.; Weusten, B.L.; Bogte, A.; Begthel, H.; Brosens, L.A.; Clevers, H. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep. 2021, 34, 108819. [Google Scholar] [CrossRef]
- Burclaff, J.; Bliton, R.J.; Breau, K.A.; Ok, M.T.; Gomez-Martinez, I.; Ranek, J.S.; Bhatt, A.P.; Purvis, J.E.; Woosley, J.T.; Magness, S.T. A proximal-to-distal survey of healthy adult human small intestine and colon epithelium by single-cell transcriptomics. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1554–1589. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct binding of the corrector VX-809 to human CFTR NBD1: Evidence of an allosteric coupling between the binding site and the NBD1: CL4 interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef]
- Krainer, G.; Treff, A.; Hartmann, A.; Stone, T.A.; Schenkel, M.; Keller, S.; Deber, C.M.; Schlierf, M. A minimal helical-hairpin motif provides molecular-level insights into misfolding and pharmacological rescue of CFTR. Commun. Biol. 2018, 1, 154. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Corrector VX-809 promotes interactions between cytoplasmic loop one and the first nucleotide-binding domain of CFTR. Biochem. Pharmacol. 2017, 136, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Moon, C.; Zhang, W.; Yarlagadda, S.; Penmatsa, H.; Ren, A.; Sinha, C.; Naren, A.P. Stabilizing rescued surface-localized δf508 CFTR by potentiation of its interaction with Na(+)/H(+) exchanger regulatory factor 1. Biochemistry 2014, 53, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martínez-Bartolomé, S.; Lavallée-Adam, M.; Balch, W.E.; Yates, J.R., III. ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Yates, J.R., 3rd. A posttranslational modification code for CFTR maturation is altered in cystic fibrosis. Sci. Signal 2019, 12, eaan7984. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019, 48, 44–54. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Chevalier, B.; Baatallah, N.; Najm, M.; Castanier, S.; Jung, V.; Pranke, I.; Golec, A.; Stoven, V.; Marullo, S.; Antigny, F.; et al. Differential CFTR-Interactome Proximity Labeling Procedures Identify Enrichment in Multiple SLC Transporters. Int. J. Mol. Sci. 2022, 23, 8937. [Google Scholar] [CrossRef]
- Hutt, D.M.; Loguercio, S.; Campos, A.R.; Balch, W.E. A proteomic variant approach (ProVarA) for personalized medicine of inherited and somatic disease. J. Mol. Biol. 2018, 430, 2951–2973. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Bear, C.; Auerbach, W.; Reyes, E.; Jensen, T.; Kartner, N.; Riordan, J.R.; Buchwald, M. Regulation of CFTR expression and function during differentiation of intestinal epithelial cells. EMBO J. 1992, 11, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125 Pt 5, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Edelman, A. Cytoskeleton and CFTR. Int. J. Biochem. Cell Biol. 2014, 52, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Chanoux, R.A.; Rubenstein, R.C. Molecular Chaperones as Targets to Circumvent the CFTR Defect in Cystic Fibrosis. Front. Pharmacol. 2012, 3, 137. [Google Scholar] [CrossRef] [PubMed]
- Ameen, N.; Silvis, M.; Bradbury, N.A. Endocytic trafficking of CFTR in health and disease. J. Cyst. Fibros. 2007, 6, 1–14. [Google Scholar] [CrossRef]
- Sun, F.; Hug, M.J.; Bradbury, N.A.; Frizzell, R.A. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 2000, 275, 14360–14366. [Google Scholar] [CrossRef]
- Klein, H.; Abu-Arish, A.; Trinh, N.T.; Luo, Y.; Wiseman, P.W.; Hanrahan, J.W.; Brochiero, E.; Sauvé, R. Investigating CFTR and KCa3.1 Protein/Protein Interactions. PLoS ONE 2016, 11, e0153665. [Google Scholar] [CrossRef]
- Bertrand, C.A.; Mitra, S.; Mishra, S.K.; Wang, X.; Zhao, Y.; Pilewski, J.M.; Madden, D.R.; Frizzell, R.A. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L912–L925. [Google Scholar] [CrossRef]
- Davezac, N.; Tondelier, D.; Lipecka, J.; Fanen, P.; Demaugre, F.; Debski, J.; Dadlez, M.; Schrattenholz, A.; Cahill, M.A.; Edelman, A. Global proteomic approach unmasks involvement of keratins 8 and 18 in the delivery of cystic fibrosis transmembrane conductance regulator (CFTR)/ΔF508-CFTR to the plasma membrane. Proteomics 2004, 4, 3833–3844. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, S.S.; Silva, I.A.; Amaral, M.D.; Farinha, C.M. Rare trafficking CFTR mutations involve distinct cellular retention machineries and require different rescuing strategies. Int. J. Mol. Sci. 2021, 23, 24. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiong, X.; Helm, A.; Kimani, K.; Bragin, A.; Skach, W.R. Co-and posttranslational translocation mechanisms direct cystic fibrosis transmembrane conductance regulator N terminus transmembrane assembly. J. Biol. Chem. 1998, 273, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.T. Membrane insertion, processing, and topology of cystic fibrosis transmembrane conductance regulator (CFTR) in microsomal membranes. Mol. Membr. Biol. 1996, 13, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Pitonzo, D.; Yang, Z.; Matsumura, Y.; Johnson, A.E.; Skach, W.R. Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol. Biol. Cell 2009, 20, 685–698. [Google Scholar] [CrossRef]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef]
- Kleizen, B.; van Vlijmen, T.; de Jonge, H.R.; Braakman, I. Folding of CFTR is predominantly cotranslational. Mol. Cell 2005, 20, 277–287. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef]
- Pranke, I.M.; Sermet-Gaudelus, I. Biosynthesis of cystic fibrosis transmembrane conductance regulator. Int. J. Biochem. Cell Biol. 2014, 52, 26–38. [Google Scholar] [CrossRef]
- Estabrooks, S.; Brodsky, J.L. Regulation of CFTR biogenesis by the proteostatic network and pharmacological modulators. Int. J. Mol. Sci. 2020, 21, 452. [Google Scholar] [CrossRef]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar] [PubMed]
- Loo, M.A.; Jensen, T.J.; Cui, L.; Hou, Y.-X.; Chang, X.-B.; Riordan, J.R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998, 17, 6879–6887. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; David, L.L.; Skach, W.R. Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum–associated degradation. Mol. Biol. Cell 2011, 22, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Bohse, K.; Arndt, V.; Schmitz, A.; Hohfeld, J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 4003–4010. [Google Scholar] [CrossRef] [PubMed]
- Grove, D.E.; Fan, C.-Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum–associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3–dependent degradation of nascent CFTRΔF508. Mol. Biol. Cell 2011, 22, 301–314. [Google Scholar] [CrossRef]
- Yamamoto, Y.-h.; Kimura, T.; Momohara, S.; Takeuchi, M.; Tani, T.; Kimata, Y.; Kadokura, H.; Kohno, K. A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 2010, 35, 107–116. [Google Scholar] [CrossRef]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002, 366, 797–806. [Google Scholar] [CrossRef]
- Zhang, H.; Schmidt, B.Z.; Sun, F.; Condliffe, S.B.; Butterworth, M.B.; Youker, R.T.; Brodsky, J.L.; Aridor, M.; Frizzell, R.A. Cysteine string protein monitors late steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2006, 281, 11312–11321. [Google Scholar] [CrossRef]
- Huang, Y.; Arora, K.; Mun, K.S.; Yang, F.; Moon, C.; Yarlagadda, S.; Jegga, A.; Weaver, T.; Naren, A.P. Targeting DNAJB9, a novel ER luminal co-chaperone, to rescue ΔF508-CFTR. Sci. Rep. 2019, 9, 9808. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Weixel, K.M.; Bradbury, N.A. The carboxyl terminus of the cystic fibrosis transmembrane conductance regulator binds to AP-2 clathrin adaptors. J. Biol. Chem. 2000, 275, 3655–3660. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Rab, A.; Tang, L.P.; Rowe, S.M.; Bebok, Z.; Collawn, J.F. Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 2012, 441, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, M.; Chang, X.-B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and ΔF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Moyer, B.D.; Milewski, M.; Loffing, J.; Ikeda, M.; Mickle, J.E.; Cutting, G.R.; Li, M.; Stanton, B.A.; Guggino, W.B. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J. Biol. Chem. 2002, 277, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.W.; Qi, J.; Watkins, S.C.; Frizzell, R.A. Syntaxin 1A inhibits regulated CFTR trafficking in Xenopus oocytes. Am. J. Physiol. Cell Physiol. 1999, 277, C174–C180. [Google Scholar] [CrossRef]
- Arora, K.; Liyanage, P.; Zhong, Q.; Naren, A.P. A SNARE protein Syntaxin 17 captures CFTR to potentiate autophagosomal clearance under stress. FASEB J. 2021, 35, e21185. [Google Scholar] [CrossRef]
- Assani, K.; Tazi, M.F.; Amer, A.O.; Kopp, B.T. IFN-γ stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS ONE 2014, 9, e96681. [Google Scholar] [CrossRef]
- Yuan, K.; Huang, C.; Fox, J.; Laturnus, D.; Carlson, E.; Zhang, B.; Yin, Q.; Gao, H.; Wu, M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J. Cell Sci. 2012, 125, 507–515. [Google Scholar] [CrossRef]
- Della Sala, A.; Prono, G.; Hirsch, E.; Ghigo, A. Role of protein kinase A-mediated phosphorylation in CFTR channel activity regulation. Front. Physiol. 2021, 12, 690247. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.; Hung, M.; Bear, C.E. Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell. Mol. Life Sci. 2017, 74, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Seavilleklein, G.; Amer, N.; Evagelidis, A.; Chappe, F.; Irvine, T.; Hanrahan, J.W.; Chappe, V. PKC phosphorylation modulates PKA-dependent binding of the R domain to other domains of CFTR. Am. J. Physiol. Cell Physiol. 2008, 295, C1366–C1375. [Google Scholar] [CrossRef] [PubMed]
- Chappe, V.; Hinkson, D.A.; Howell, L.D.; Evagelidis, A.; Liao, J.; Chang, X.-B.; Riordan, J.R.; Hanrahan, J.W. Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 2004, 101, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Billet, A.; Luo, Y.; Balghi, H.; Hanrahan, J.W. Role of tyrosine phosphorylation in the muscarinic activation of the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013, 288, 21815–21823. [Google Scholar] [CrossRef]
- Ren, A.; Zhang, W.; Yarlagadda, S.; Sinha, C.; Arora, K.; Moon, C.S.; Naren, A.P. MAST205 competes with cystic fibrosis transmembrane conductance regulator (CFTR)-associated ligand for binding to CFTR to regulate CFTR-mediated fluid transport. J. Biol. Chem. 2013, 288, 12325–12334. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Zhang, Y.; Naren, A.P. CFTR-NHERF2-LPA2 complex in the airway and gut epithelia. Int. J. Mol. Sci. 2017, 18, 1896. [Google Scholar] [CrossRef]
- Naren, A.P.; Cobb, B.; Li, C.; Roy, K.; Nelson, D.; Heda, G.D.; Liao, J.; Kirk, K.L.; Sorscher, E.J.; Hanrahan, J. A macromolecular complex of β2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA 2003, 100, 342–346. [Google Scholar] [CrossRef]
- Li, C.; Dandridge, K.S.; Di, A.; Marrs, K.L.; Harris, E.L.; Roy, K.; Jackson, J.S.; Makarova, N.V.; Fujiwara, Y.; Farrar, P.L. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 2005, 202, 975–986. [Google Scholar] [CrossRef]
- Li, C.; Krishnamurthy, P.C.; Penmatsa, H.; Marrs, K.L.; Wang, X.Q.; Zaccolo, M.; Jalink, K.; Li, M.; Nelson, D.J.; Schuetz, J.D. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 2007, 131, 940–951. [Google Scholar] [CrossRef]
- Arora, K.; Sinha, C.; Zhang, W.; Moon, C.S.; Ren, A.; Yarlagadda, S.; Dostmann, W.R.; Adebiyi, A.; Haberman, Y.; Denson, L.A. Altered cGMP dynamics at the plasma membrane contribute to diarrhea in ulcerative colitis. Am. J. Pathol. 2015, 185, 2790–2804. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Hug, M.J.; Lewarchik, C.M.; Yun, C.-H.C.; Bradbury, N.A.; Frizzell, R.A. E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells. J. Biol. Chem. 2000, 275, 29539–29546. [Google Scholar] [CrossRef]
- Sadana, R.; Dessauer, C.W. Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, R.A.; Hanrahan, J.W. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb. Perspect. Med. 2012, 2, a009563. [Google Scholar] [CrossRef] [PubMed]
- Field, M. Intestinal secretion: Effect of cyclic AMP and its role in cholera. N. Engl. J. Med. 1971, 284, 1137–1144. [Google Scholar]
- Bartlett, S. Water, sanitation and urban children: The need to go beyond “improved” provision. Environ. Urban. 2003, 15, 57–70. [Google Scholar]
- Demissie, G.D.; Yeshaw, Y.; Aleminew, W.; Akalu, Y. Diarrhea and associated factors among under five children in sub-Saharan Africa: Evidence from demographic and health surveys of 34 sub-Saharan countries. PLoS ONE 2021, 16, e0257522. [Google Scholar] [CrossRef]
- Kimberg, D.V.; Field, M.; Johnson, J.; Henderson, A.; Gershon, E. Stimulation of intestinal mucosal adenyl cyclase by cholera enterotoxin and prostaglandins. J. Clin. Investig. 1971, 50, 1218–1230. [Google Scholar] [CrossRef]
- Kantor, H.S. Enterotoxins of Escherichia coli and Vibrio cholerae: Tools for the molecular biologist. J. Infect. Dis. 1975, 131, s22–s32. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Bot, A.G.; Ruth, P.; Pfeifer, A.; Hofmann, F.; De Jonge, H.R. Differential role of cyclic GMP–dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 2000, 118, 108–114. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Smolenski, A.; Tilly, B.C.; Houtsmuller, A.B.; Ehlert, E.M.; Bot, A.G.; Edixhoven, M.; Boomaars, W.E.; Lohmann, S.M.; De Jonge, H.R. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl− channel activation. Proc. Natl. Acad. Sci. USA 1998, 95, 1466–1471. [Google Scholar] [CrossRef]
- Francis, S.H.; Turko, I.V.; Corbin, J.D. Cyclic nucleotide phosphodiesterases: Relating structure and function. Prog. Nucleic Acid. Res. Mol. Biol. 2000, 65, 1–52. [Google Scholar]
- Penmatsa, H.; Zhang, W.; Yarlagadda, S.; Li, C.; Conoley, V.G.; Yue, J.; Bahouth, S.W.; Buddington, R.K.; Zhang, G.; Nelson, D.J. Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol. Cell 2010, 21, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Ramananda, Y.; Mun, K.; Naren, A.P.; Arora, K. AC6 is the major adenylate cyclase forming a diarrheagenic protein complex with cystic fibrosis transmembrane conductance regulator in cholera. J. Biol. Chem. 2018, 293, 12949–12959. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.A.; Murali, S.K.; Kaji, I.; Akiba, Y.; Kaunitz, J.D.; Kristensen, T.B.; Poulsen, S.B.; Dominguez Rieg, J.A.; Rieg, T. Adenylyl Cyclase 6 Expression Is Essential for Cholera Toxin-Induced Diarrhea. J. Infect. Dis. 2019, 220, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Sinha, C.; Zhang, W.; Ren, A.; Moon, C.S.; Yarlagadda, S.; Naren, A.P. Compartmentalization of cyclic nucleotide signaling: A question of when, where, and why? Pflügers Arch. Eur. J. Physiol. 2013, 465, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Lund, J.R.; Naren, N.A.; Zingarelli, B.; Naren, A.P. AC6 regulates the microtubule-depolymerizing kinesin KIF19A to control ciliary length in mammals. J. Biol. Chem. 2020, 295, 14250–14259. [Google Scholar] [CrossRef]
- Basu, N.; Arshad, N.; Visweswariah, S.S. Receptor guanylyl cyclase C (GC-C): Regulation and signal transduction. Mol. Cell. Biochem. 2010, 334, 67–80. [Google Scholar] [CrossRef]
- Navaneethan, U.; Giannella, R.A. Mechanisms of infectious diarrhea. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 637–647. [Google Scholar] [CrossRef]
- Schulz, S.; Green, C.K.; Yuen, P.S.; Garbers, D.L. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 1990, 63, 941–948. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; Arshad, N.; Haukanes, B.I.; Tronstad, R.R.; Pham, K.D.-C.; Johansson, S.; Håvik, B.; Tønder, S.L.; Levy, S.E.; Brackman, D. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 2012, 366, 1586–1595. [Google Scholar] [CrossRef]
- Romi, H.; Cohen, I.; Landau, D.; Alkrinawi, S.; Yerushalmi, B.; Hershkovitz, R.; Newman-Heiman, N.; Cutting, G.R.; Ofir, R.; Sivan, S. Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am. J. Hum. Genet. 2012, 90, 893–899. [Google Scholar] [CrossRef]
- Corsetti, M.; Tack, J. Linaclotide: A new drug for the treatment of chronic constipation and irritable bowel syndrome with constipation. United Eur. Gastroenterol. J. 2013, 1, 7–20. [Google Scholar] [CrossRef]
- Arora, K.; Huang, Y.; Mun, K.; Yarlagadda, S.; Sundaram, N.; Kessler, M.M.; Hannig, G.; Kurtz, C.B.; Silos-Santiago, I.; Helmrath, M.; et al. Guanylate cyclase 2C agonism corrects CFTR mutants. JCI Insight 2017, 2, 93686. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramananda, Y.; Naren, A.P.; Arora, K. Functional Consequences of CFTR Interactions in Cystic Fibrosis. Int. J. Mol. Sci. 2024, 25, 3384. https://doi.org/10.3390/ijms25063384
Ramananda Y, Naren AP, Arora K. Functional Consequences of CFTR Interactions in Cystic Fibrosis. International Journal of Molecular Sciences. 2024; 25(6):3384. https://doi.org/10.3390/ijms25063384
Chicago/Turabian StyleRamananda, Yashaswini, Anjaparavanda P. Naren, and Kavisha Arora. 2024. "Functional Consequences of CFTR Interactions in Cystic Fibrosis" International Journal of Molecular Sciences 25, no. 6: 3384. https://doi.org/10.3390/ijms25063384
APA StyleRamananda, Y., Naren, A. P., & Arora, K. (2024). Functional Consequences of CFTR Interactions in Cystic Fibrosis. International Journal of Molecular Sciences, 25(6), 3384. https://doi.org/10.3390/ijms25063384