
Hydrogenolysis of Glycerol over NiCeZr Catalyst Modified with Mg, Cu, and Sn at the Surface Level
Abstract
1. Introduction
2. Results and Discussion
2.1. Textural Properties and Bulk Chemical Composition
2.2. Structural Characterization
2.3. Redox Properties from the H2-TPR Analysis and Oxygen Storage Capacity
2.4. Characterization of Acid and Metallic Sites
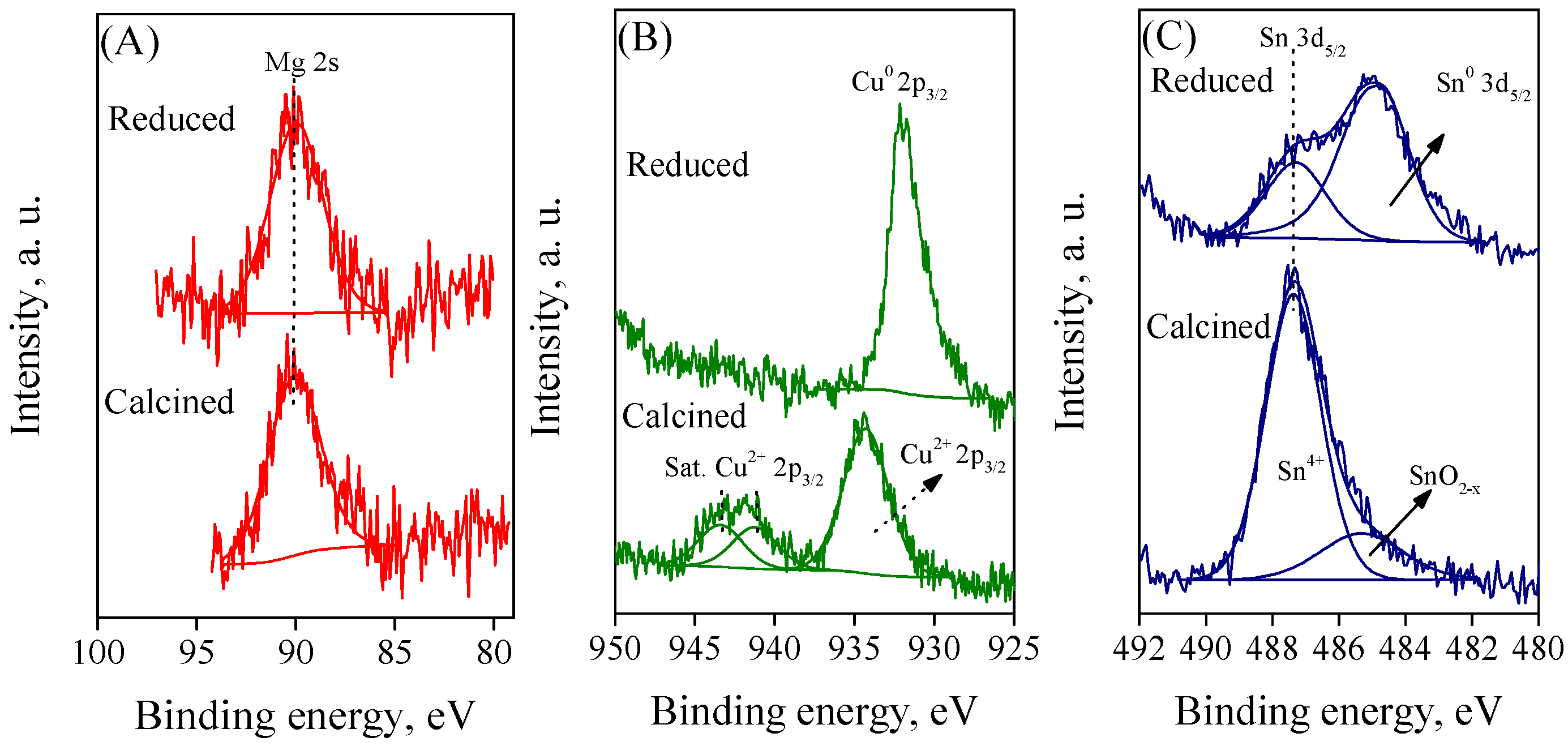
2.5. Surface Characterization Using XPS Analysis
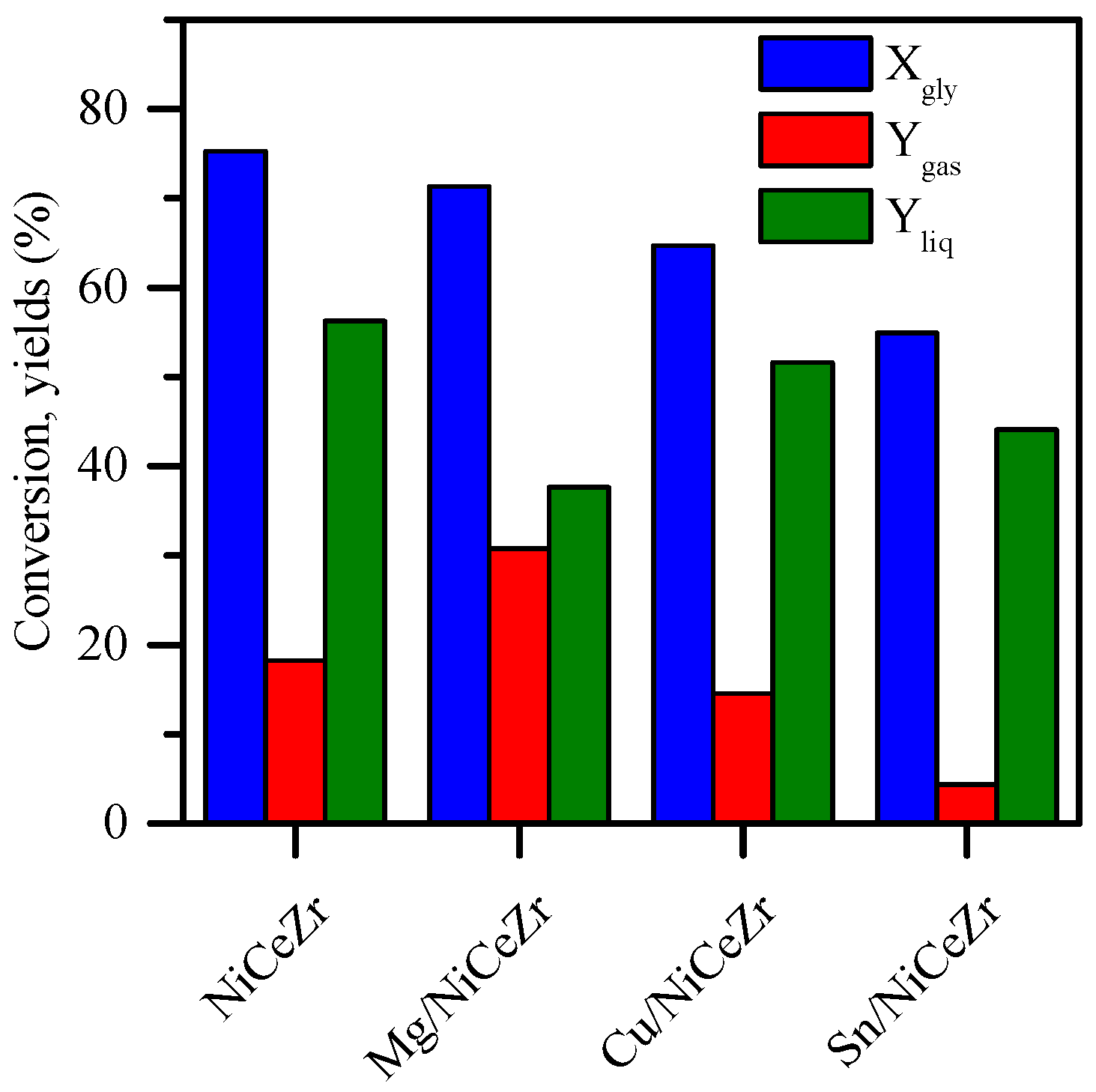
2.6. Hydrogenolysis of Glycerol
2.6.1. Gas Products
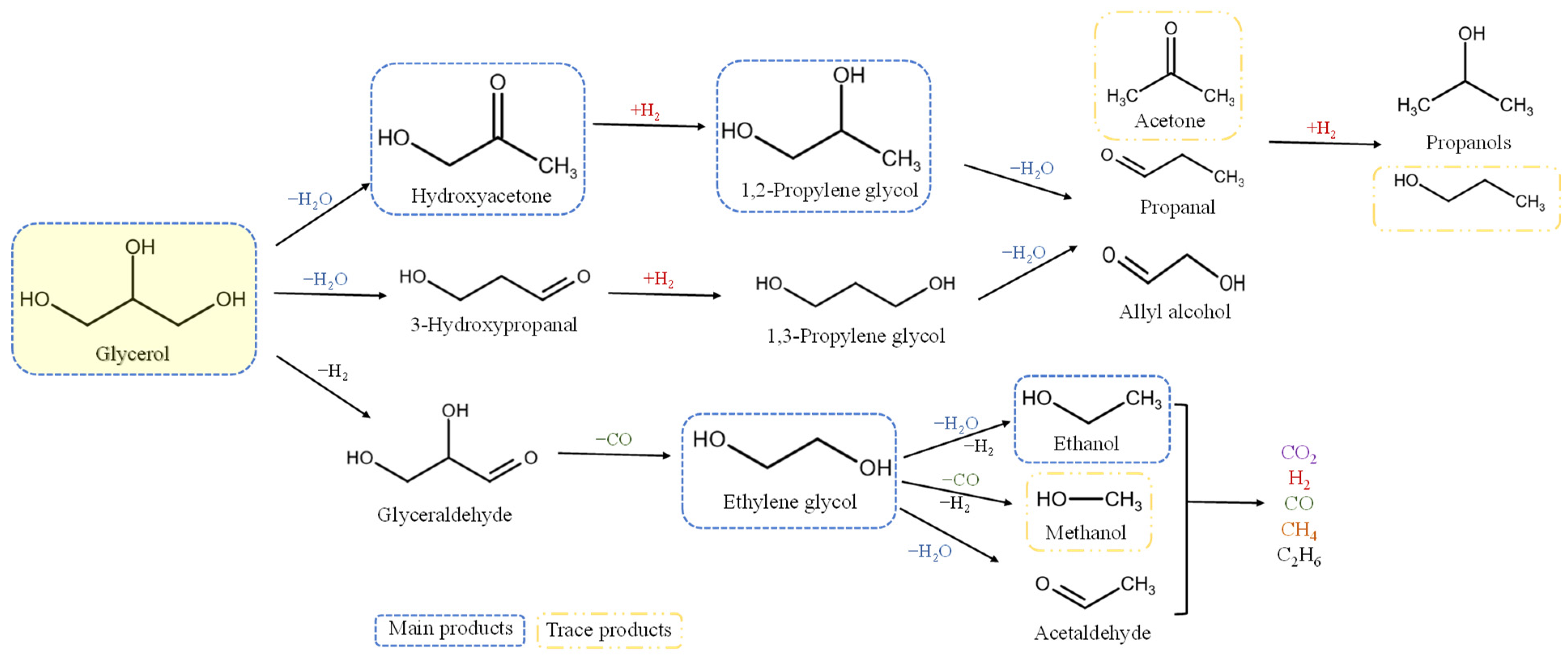
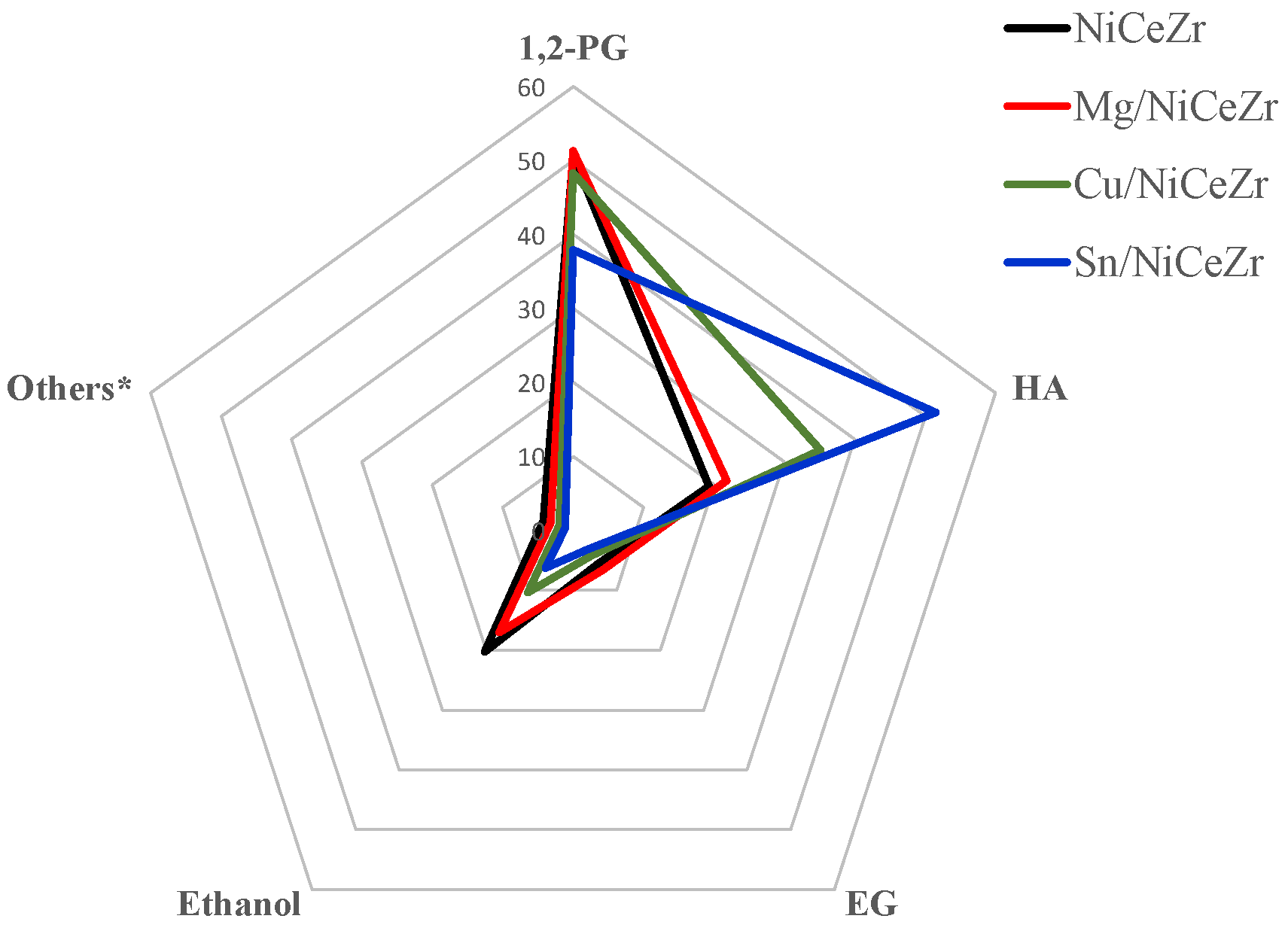
2.6.2. Liquid Products
2.7. Characterization of Spent Catalysts
3. Materials and Methods
3.1. Materials
3.1.1. Chemicals
3.1.2. Synthesis of Catalysts
3.2. Characterization of the Catalysts
3.3. Catalytic Tests
3.4. Equations Used for Calculations for Catalytic Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Wild, P.J.; den Uil, H.; Reith, J.H.; Kiel, J.H.A.; Heeres, H.J. Biomass Valorisation by Staged Degasification. A New Pyrolysis-Based Thermochemical Conversion Option to Produce Value-Added Chemicals from Lignocellulosic Biomass. J. Anal. Appl. Pyrolysis 2009, 85, 124–133. [Google Scholar] [CrossRef]
- Mishra, S.; Upadhyay, R.K. Review on Biomass Gasification: Gasifiers, Gasifying Mediums, and Operational Parameters. Mater. Sci. Energy Technol. 2021, 4, 329–340. [Google Scholar] [CrossRef]
- Gao, Y.; Remón, J.; Matharu, A.S. Microwave-Assisted Hydrothermal Treatments for Biomass Valorisation: A Critical Review. Green Chem. 2021, 23, 3502–3525. [Google Scholar] [CrossRef]
- Nzeteu, C.; Coelho, F.; Davis, E.; Trego, A.; O’Flaherty, V. Current Trends in Biological Valorization of Waste-Derived Biomass: The Critical Role of VFAs to Fuel a Biorefinery. Fermentation 2022, 8, 445. [Google Scholar] [CrossRef]
- Seo, M.W.; Lee, S.H.; Nam, H.; Lee, D.; Tokmurzin, D.; Wang, S.; Park, Y.K. Recent Advances of Thermochemical Conversion Processes for Biorefinery. Bioresour. Technol. 2022, 343, 126109. [Google Scholar] [CrossRef]
- Awasthi, M.K.; Sarsaiya, S.; Patel, A.; Juneja, A.; Singh, R.P.; Yan, B.; Awasthi, S.K.; Jain, A.; Liu, T.; Duan, Y.; et al. Refining Biomass Residues for Sustainable Energy and Bio-Products: An Assessment of Technology, Its Importance, and Strategic Applications in Circular Bio-Economy. Renew. Sustain. Energy Rev. 2020, 127, 109876. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Martin-Martinez, F.J. Biorefineries: Achievements and Challenges for a Bio-Based Economy. Front. Chem. 2022, 10, 973417. [Google Scholar] [CrossRef] [PubMed]
- Checa, M.; Nogales-Delgado, S.; Montes, V.; Encinar, J.M. Recent Advances in Glycerol Catalytic Valorization: A Review. Catalysts 2020, 10, 1279. [Google Scholar] [CrossRef]
- Wan, W.; Ammal, S.C.; Lin, Z.; You, K.E.; Heyden, A.; Chen, J.G. Controlling Reaction Pathways of Selective C–O Bond Cleavage of Glycerol. Nat. Commun. 2018, 9, 4612. [Google Scholar] [CrossRef]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards Understanding the Bifunctional Hydrodeoxygenation and Aqueous Phase Reforming of Glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Dou, B.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Ni/Y2O3–ZrO2 Catalyst for Hydrogen Production through the Glycerol Steam Reforming Reaction. Int. J. Hydrogen Energy 2020, 45, 10442–10460. [Google Scholar] [CrossRef]
- Dahdah, E.; Estephane, J.; Gennequin, C.; Aboukaïs, A.; Aouad, S.; Abi-Aad, E. Effect of La Promotion on Ni/Mg-Al Hydrotalcite Derived Catalysts for Glycerol Steam Reforming. J. Environ. Chem. Eng. 2020, 8, 104228. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Papageridis, K.N.; Baklavaridis, A.; Tzounis, L.; Polychronopoulou, K.; Goula, M.A. Hydrogen Production via the Glycerol Steam Reforming Reaction over Nickel Supported on Alumina and Lanthana-Alumina Catalysts. Int. J. Hydrogen Energy 2017, 42, 13039–13060. [Google Scholar] [CrossRef]
- Gatti, M.N.; Nichio, N.N.; Pompeo, F. Advances for Biorefineries: Glycerol Hydrogenolysis to 1,3-Propylene Glycol. Reactions 2022, 3, 451–498. [Google Scholar] [CrossRef]
- Martin, A.; Armbruster, U.; Gandarias, I.; Arias, P.L. Glycerol Hydrogenolysis into Propanediols Using in Situ Generated Hydrogen—A Critical Review. Eur. J. Lipid Sci. Technol. 2013, 115, 9–27. [Google Scholar] [CrossRef]
- Nanda, M.R.; Yuan, Z.; Qin, W.; Xu, C. (Charles) Recent Advancements in Catalytic Conversion of Glycerol into Propylene Glycol: A Review. Catal. Rev. 2016, 58, 309–336. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tomishige, K. Heterogeneous Catalysis of the Glycerol Hydrogenolysis. Catal. Sci. Technol. 2011, 1, 179–190. [Google Scholar] [CrossRef]
- Vasiliadou, E.S.; Lemonidou, A.A. Kinetic Study of Liquid-Phase Glycerol Hydrogenolysis over Cu/SiO2 Catalyst. Chem. Eng. J. 2013, 231, 103–112. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, J.; Guo, X. Catalytic Hydrogenolysis of Glycerol to Propanediols: A Review. RSC Adv. 2015, 5, 74611–74628. [Google Scholar] [CrossRef]
- Zhou, J.; Guo, L.; Guo, X.; Mao, J.; Zhang, S. Selective Hydrogenolysis of Glycerol to Propanediols on Supported Cu-Containing Bimetallic Catalysts. Green Chem. 2010, 12, 1835–1843. [Google Scholar] [CrossRef]
- Maris, E.P.; Ketchie, W.C.; Murayama, M.; Davis, R.J. Glycerol Hydrogenolysis on Carbon-Supported PtRu and AuRu Bimetallic Catalysts. J. Catal. 2007, 251, 281–294. [Google Scholar] [CrossRef]
- Pipitone, G.; Zoppi, G.; Pirone, R.; Bensaid, S. A Critical Review on Catalyst Design for Aqueous Phase Reforming. Int. J. Hydrogen Energy 2022, 47, 151–180. [Google Scholar] [CrossRef]
- Dieuzeide, M.L.; Laborde, M.; Amadeo, N.; Cannilla, C.; Bonura, G.; Frusteri, F. Hydrogen Production by Glycerol Steam Reforming: How Mg Doping Affects the Catalytic Behaviour of Ni/Al2O3 Catalysts. Int. J. Hydrogen Energy 2016, 41, 157–166. [Google Scholar] [CrossRef]
- Wu, K.; Dou, B.; Zhang, H.; Liu, D.; Chen, H.; Xu, Y. Aqueous Phase Reforming of Biodiesel Byproduct Glycerol over Mesoporous Ni-Cu/CeO2 for Renewable Hydrogen Production. Fuel 2022, 308, 122014. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Dumesic, J.A. Aqueous-Phase Reforming of Oxygenated Hydrocarbons over Sn-Modified Ni Catalysts. J. Catal. 2004, 222, 180–191. [Google Scholar] [CrossRef]
- Vera-Hincapie, N.; Iriarte-Velasco, U.; Ayastuy, J.L.; Gutiérrez-Ortiz, M.A. Effect of the Synthesis Method on the Performance of Ni-CeO2-ZrO2 Catalysts for the Hydrogenolysis of Glycerol with Endogenous Hydrogen. J. Environ. Chem. Eng. 2023, 11, 111072. [Google Scholar] [CrossRef]
- Cangiano, M.D.L.A.; Ojeda, M.W.; Carreras, A.C.; González, J.A.; Ruiz, M.D.C. A Study of the Composition and Microstructure of Nanodispersed Cu-Ni Alloys Obtained by Different Routes from Copper and Nickel Oxides. Mater. Charact. 2010, 61, 1135–1146. [Google Scholar] [CrossRef]
- Dhanapal, K.; Narayanan, V.; Stephen, A. Influence of Sn on the Magnetic Ordering of Ni-Sn Alloy Synthesized Using Chemical Reduction Method. J. Magn. Magn. Mater. 2016, 406, 103–109. [Google Scholar] [CrossRef]
- Berwal, U.; Singh, V.; Sharma, R. Effect of Ce4+→Ce3+ Conversion on the Structural and Luminescence Properties of Ce4+ Doped Gd2Ti2O7 Pyrochlore Oxide. J. Lumin. 2023, 257, 119687. [Google Scholar] [CrossRef]
- Loridant, S. Raman Spectroscopy as a Powerful Tool to Characterize Ceria-Based Catalysts. Catal. Today 2021, 373, 98–111. [Google Scholar] [CrossRef]
- Sartoretti, E.; Novara, C.; Giorgis, F.; Piumetti, M.; Bensaid, S.; Russo, N.; Fino, D. In Situ Raman Analyses of the Soot Oxidation Reaction over Nanostructured Ceria-Based Catalysts. Sci. Rep. 2019, 9, 3875. [Google Scholar] [CrossRef]
- Shan, W.; Luo, M.; Ying, P.; Shen, W.; Li, C. Reduction Property and Catalytic Activity of Ce1-XNiXO2 Mixed Oxide Catalysts for CH4 Oxidation. Appl. Catal. A Gen. 2003, 246, 1–9. [Google Scholar] [CrossRef]
- Yisup, N.; Cao, Y.; Feng, W.L.; Dai, W.L.; Fan, K.N. Catalytic Oxidation of Methane over Novel Ce-Ni-O Mixed Oxide Catalysts Prepared by Oxalate Gel-Coprecipitation. Catal. Lett. 2005, 99, 207–213. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, C.; Chen, W. Hydrogen Production from Steam Reforming of Ethanol over Ni/MgO-CeO2 Catalyst at Low Temperature. J. Rare Earths 2009, 27, 948–954. [Google Scholar] [CrossRef]
- Siakavelas, G.I.; Charisiou, N.D.; AlKhoori, S.; AlKhoori, A.A.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Yentekakis, I.V.; Polychronopoulou, K.; Goula, M.A. Highly Selective and Stable Nickel Catalysts Supported on Ceria Promoted with Sm2O3, Pr2O3 and MgO for the CO2 Methanation Reaction. Appl. Catal. B Environ. 2021, 282, 119562. [Google Scholar] [CrossRef]
- Saw, E.T.; Oemar, U.; Ang, M.L.; Kus, H.; Kawi, S. High-Temperature Water Gas Shift Reaction on Ni-Cu/CeO2 Catalysts: Effect of Ceria Nanocrystal Size on Carboxylate Formation. Catal. Sci. Technol. 2016, 6, 5336–5349. [Google Scholar] [CrossRef]
- Saw, E.T.; Oemar, U.; Tan, X.R.; Du, Y.; Borgna, A.; Hidajat, K.; Kawi, S. Bimetallic Ni-Cu Catalyst Supported on CeO2 for High-Temperature Water-Gas Shift Reaction: Methane Suppression via Enhanced CO Adsorption. J. Catal. 2014, 314, 32–46. [Google Scholar] [CrossRef]
- Courtois, X.; Perrichon, V.; Courtois, X. Distinct Roles of Copper in Bimetallic Copper-Rhodium Three-Way-Catalysts Deposited on Redox Supports. Distinct Roles of Copper in Bimetallic Copper-Rhodium Three-Way-Catalysts Deposited on Redox Supports. Appl. Catal. B Environ. 2005, 57, 63–72. [Google Scholar] [CrossRef]
- Solsona, B.; Concepción, P.; Demicol, B.; Hernández, S.; Delgado, J.J.; Calvino, J.J.; López Nieto, J.M. Selective Oxidative Dehydrogenation of Ethane over SnO2-Promoted NiO Catalysts. J. Catal. 2012, 295, 104–114. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.; Kang, M. Hydrogen Production from Ethanol Steam Reforming over SnO2-K2O/Zeolite y Catalyst. Bull. Korean Chem. Soc. 2011, 32, 1912–1920. [Google Scholar] [CrossRef]
- Parmaliana, A.; Arena, F.; Frusteri, F.; Giordano, N. Temperature-Programmed Reduction Study of NiO–MgO Interactions in Magnesia-Supported Ni Catalysts and NiO–MgO Physical Mixture. J. Chem. Soc. Faraday Trans. 1990, 86, 2663–2669. [Google Scholar] [CrossRef]
- Lin, F.; Chen, Z.; Gong, H.; Wang, X.; Chen, L.; Yu, H. Oxygen Vacancy Induced Strong Metal-Support Interactions on Ni/Ce0.8Zr0.2O2 Nanorod Catalysts for Promoting Steam Reforming of Toluene: Experimental and Computational Studies. Langmuir 2023, 39, 4495–4506. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, N.; Zhou, Z. Synergetic Effect of Cu Active Sites and Oxygen Vacancies in Cu/CeO2-ZrO2 for the Water-Gas Shift Reaction. Catal. Sci. Technol. 2021, 11, 2518–2528. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.; Li, Y.; Schwank, J.W. A Review on Oxygen Storage Capacity of CeO2 -Based Materials: Influence Factors, Measurement Techniques, and Applications in Reactions Related to Catalytic Automotive Emissions Control. Catal. Today 2019, 327, 90–115. [Google Scholar] [CrossRef]
- Kaminski, P.; Ziolek, M. Mobility of Gold, Copper and Cerium Species in Au, Cu/Ce, Zr-Oxides and Its Impact on Total Oxidation of Methanol. Appl. Catal. B Environ. 2016, 187, 328–341. [Google Scholar] [CrossRef]
- Fang, C.; Shi, L.; Li, H.; Huang, L.; Zhang, J.; Zhang, D. Creating Hierarchically Macro-/Mesoporous Sn/CeO2 for the Selective Catalytic Reduction of NO with NH3. RSC Adv. 2016, 6, 78727–78736. [Google Scholar] [CrossRef]
- Caballero, A.; Holgado, J.P.; Gonzalez-Delacruz, V.M.; Habas, S.E.; Herranz, T.; Salmeron, M. In Situ Spectroscopic Detection of SMSI Effect in a Ni/CeO2 System: Hydrogen-Induced Burial and Dig out of Metallic Nickel. Chem. Commun. 2010, 46, 1097–1099. [Google Scholar] [CrossRef]
- Jin, B.; Li, S.; Liang, X. Enhanced Activity and Stability of MgO-Promoted Ni/Al2O3 Catalyst for Dry Reforming of Methane: Role of MgO. Fuel 2021, 284, 119082. [Google Scholar] [CrossRef]
- Gutta, N.; Velisoju, V.K.; Chatla, A.; Boosa, V.; Tardio, J.; Patel, J.; Akula, V. Promotional Effect of Cu and Influence of Surface Ni-Cu Alloy for Enhanced H2 Yields from CH4 Decomposition over Cu-Modified Ni Supported on MCM-41 Catalyst. Energy Fuels 2018, 32, 4008–4015. [Google Scholar] [CrossRef]
- Miranda, B.C.; Chimentão, R.J.; Szanyi, J.; Braga, A.H.; Santos, J.B.O.; Gispert-Guirado, F.; Llorca, J.; Medina, F. Influence of Copper on Nickel-Based Catalysts in the Conversion of Glycerol. Appl. Catal. B Environ. 2015, 166–167, 166–180. [Google Scholar] [CrossRef]
- Khan, M.E.; Khan, M.M.; Cho, M.H. Ce3+-Ion, Surface Oxygen Vacancy, and Visible Light-Induced Photocatalytic Dye Degradation and Photocapacitive Performance of CeO2-Graphene Nanostructures. Sci. Rep. 2017, 7, 5928. [Google Scholar] [CrossRef]
- Weidler, N.; Schuch, J.; Knaus, F.; Stenner, P.; Hoch, S.; Maljusch, A.; Schäfer, R.; Kaiser, B.; Jaegermann, W. X-Ray Photoelectron Spectroscopic Investigation of Plasma-Enhanced Chemical Vapor Deposited NiOx, NiOx(OH)y, and CoNiOx(OH)y: Influence of the Chemical Composition on the Catalytic Activity for the Oxygen Evolution Reaction. J. Phys. Chem. C 2017, 121, 6455–6463. [Google Scholar] [CrossRef]
- Shen, Y.; Lua, A.C. Sol-Gel Synthesis of Ni and Ni Supported Catalysts for Hydrogen Production by Methane Decomposition. RSC Adv. 2014, 4, 42159–42167. [Google Scholar] [CrossRef]
- Casillas-Trujillo, L.; Osinger, B.; Lindblad, R.; Karlsson, D.; Abrikosov, A.I.; Fritze, S.; Von Fieandt, K.; Alling, B.; Hotz, I.; Jansson, U.; et al. Experimental and Theoretical Evidence of Charge Transfer in Multi-Component Alloys-How Chemical Interactions Reduce Atomic Size Mismatch. Mater. Chem. Front. 2021, 5, 5746–5759. [Google Scholar] [CrossRef]
- Azdad, Z.; Marot, L.; Moser, L.; Steiner, R.; Meyer, E. Valence Band Behaviour of Zirconium Oxide, Photoelectron and Auger Spectroscopy Study. Sci. Rep. 2018, 8, 16251. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Park, H.R.; Lee, Y.L.; Ahn, S.Y.; Kim, K.J.; Hong, G.R.; Roh, H.S. Customized Ni-MgO-ZrO2catalysts for the Dry Reforming of Methane Using Coke Oven Gas: Optimizing the MgO Content. J. CO2 Util. 2023, 68, 102379. [Google Scholar] [CrossRef]
- Asencios, Y.J.O.; Yigit, N.; Wicht, T.; Stöger-Pollach, M.; Lucrédio, A.F.; Marcos, F.C.F.; Assaf, E.M.; Rupprechter, G. Partial Oxidation of Bio-Methane over Nickel Supported on MgO–ZrO2 Solid Solutions. Top. Catal. 2023, 66, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Aghamohammadi, S.; Li, D.; Li, K.; Farrauto, R. Structure Dependence of Nb2O5−X Supported Manganese Oxide for Catalytic Oxidation of Propane: Enhanced Oxidation Activity for MnOx on a Low Surface Area Nb2O5−X. Appl. Catal. B Environ. 2019, 244, 438–447. [Google Scholar] [CrossRef]
- Deng, J.; Zhou, Y.; Cui, Y.; Lan, L.; Wang, J.; Yuan, S.; Chen, Y. The Influence of H2O2 on the Properties of CeO2-ZrO2 Mixed Oxides. J. Mater. Sci. 2017, 52, 5242–5255. [Google Scholar] [CrossRef]
- Sádaba, I.; Ojeda, M.; Mariscal, R.; Fierro, J.L.G.; Granados, M.L. Catalytic and Structural Properties of Co-Precipitated Mg-Zr Mixed Oxides for Furfural Valorization via Aqueous Aldol Condensation with Acetone. Appl. Catal. B Environ. 2011, 101, 638–648. [Google Scholar] [CrossRef]
- Zhuang, H.D.; Bai, S.F.; Liu, X.M.; Yan, Z.F. Structure and Performance of Cu/ZrO2 Catalyst for the Synthesis of Methanol from CO2 Hydrogenation. Ranliao Huaxue Xuebao/J. Fuel Chem. Technol. 2010, 38, 462–467. [Google Scholar] [CrossRef]
- Vasquez, R.P. CuO by XPS. Surf. Sci. Spectra 1998, 5, 262–266. [Google Scholar] [CrossRef]
- Ghodselahi, T.; Vesaghi, M.A.; Shafiekhani, A.; Baghizadeh, A.; Lameii, M. XPS Study of the Cu@Cu2O Core-Shell Nanoparticles. Appl. Surf. Sci. 2008, 255, 2730–2734. [Google Scholar] [CrossRef]
- Hengne, A.M.; Samal, A.K.; Enakonda, L.R.; Harb, M.; Gevers, L.E.; Anjum, D.H.; Hedhili, M.N.; Saih, Y.; Huang, K.W.; Basset, J.M. Ni-Sn-Supported ZrO2 Catalysts Modified by Indium for Selective CO2 Hydrogenation to Methanol. ACS Omega 2018, 3, 3688–3701. [Google Scholar] [CrossRef] [PubMed]
- Degerman, D.; Lömker, P.; Goodwin, C.M.; Shipilin, M.; García-Martínez, F.; Schlueter, C.; Nilsson, A.; Amann, P. State of the Surface During CO Hydrogenation over Ni(111) and Ni(211) Probed by Operando X-Ray Photoelectron Spectroscopy. J. Phys. Chem. C 2023, 127, 4021–4032. [Google Scholar] [CrossRef]
- Concepción, P.; Boronat, M.; García-García, S.; Fernández, E.; Corma, A. Enhanced Stability of Cu Clusters of Low Atomicity against Oxidation. Effect on the Catalytic Redox Process. ACS Catal. 2017, 7, 3560–3568. [Google Scholar] [CrossRef]
- Fan, J.; Weng, D.; Wu, X.; Wu, X.; Ran, R. Modification of CeO2-ZrO2 Mixed Oxides by Coprecipitated/Impregnated Sr: Effect on the Microstructure and Oxygen Storage Capacity. J. Catal. 2008, 258, 177–186. [Google Scholar] [CrossRef]
- Serrano-Lotina, A.; Cruz, K.; Bañares, M.A.; Daturi, M.; Ávila, P. Bimetallic MnO2-Supported Catalysts for Selective Reduction of NO with NH3. Operando IR Studies. Appl. Surf. Sci. 2023, 610, 155550. [Google Scholar] [CrossRef]
- Popescu, I.; Piumetti, M.; Bensaid, S.; Marcu, I.C. Study of Ce-Cu Mixed Oxide Catalysts by: In Situ Electrical Conductivity Measurements. Phys. Chem. Chem. Phys. 2017, 19, 31929–31939. [Google Scholar] [CrossRef]
- Liang, P.; Zhang, Y.; Dong, X.; Zhang, J.; Li, B.; Zhang, Y.; Jiao, T.; Zhang, W.; Li, X. Effect of Sn(II)/Sn(IV) Additive on the Catalytic Combustion of Diethylamine and N2 Selectivity over CeZrOx Catalyst. Fuel 2024, 358, 130164. [Google Scholar] [CrossRef]
- Li, S.; Deng, J.; Wang, J.; Chen, Y.; Li, Y. Effects of Thermal Treatment Conditions on Redox Properties of Ceria-Zirconia Materials. J. Rare Earths 2023, 41, 1969–1975. [Google Scholar] [CrossRef]
- Coronado, I.; Stekrova, M.; Reinikainen, M.; Simell, P.; Lefferts, L.; Lehtonen, J. A Review of Catalytic Aqueous-Phase Reforming of Oxygenated Hydrocarbons Derived from Biorefinery Water Fractions. Int. J. Hydrogen Energy 2016, 41, 11003–11032. [Google Scholar] [CrossRef]
- Larimi, A.S.; Kazemeini, M.; Khorasheh, F. Highly Selective Doped Pt-MgO Nano-Sheets for Renewable Hydrogen Production from APR of Glycerol. Int. J. Hydrogen Energy 2016, 41, 17390–17398. [Google Scholar] [CrossRef]
- Guo, Y.; Azmat, M.U.; Liu, X.; Wang, Y.; Lu, G. Effect of Support’s Basic Properties on Hydrogen Production in Aqueous-Phase Reforming of Glycerol and Correlation between WGS and APR. Appl. Energy 2012, 92, 218–223. [Google Scholar] [CrossRef]
- Wen, G.; Xu, Y.; Ma, H.; Xu, Z.; Tian, Z. Production of Hydrogen by Aqueous-Phase Reforming of Glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
- Robinson, A.M.; Hensley, J.E.; Will Medlin, J. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Q.; Zhou, Z.; Li, Z.; Meng, K.; Fang, T.; You, Z.; Zhang, G.; Yin, B.; Shen, J.; et al. Hydrogenolysis of Glycerol to 1,3-Propanediol: Are Spatial and Electronic Configuration of “Metal-Solid Acid” Interface Key for Active and Durable Catalysts? ChemCatChem 2022, 14, 1–21. [Google Scholar] [CrossRef]
- Pendem, C.; Sarkar, B.; Siddiqui, N.; Konathala, L.N.S.; Baskar, C.; Bal, R. K-Promoted Pt-Hydrotalcite Catalyst for Production of H2 by Aqueous Phase Reforming of Glycerol. ACS Sustain. Chem. Eng. 2018, 6, 2122–2131. [Google Scholar] [CrossRef]
- Manfro, R.L.; Pires, T.P.; Ribeiro, N.F.P.; Souza, M.M. Aqueous-Phase Reforming of Glycerol Using Ni-Cu Catalysts Prepared from Hydrotalcite-like Precursors. Catal. Sci. Technol. 2013, 3, 1278–1287. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Simonetti, D.A.; Cortright, R.D.; Dumesic, J.A. Sn-Modified Ni Catalysts for Aqueous-Phase Reforming: Characterization and Deactivation Studies. J. Catal. 2005, 231, 67–76. [Google Scholar] [CrossRef]
- Zhang, C.C.; Hartlaub, S.; Petrovic, I.; Yilmaz, B. Raman Spectroscopy Characterization of Amorphous Coke Generated in Industrial Processes. ACS Omega 2022, 7, 2565–2570. [Google Scholar] [CrossRef]
- Rosli, S.N.A.; Abidin, S.Z.; Osazuwa, O.U.; Fan, X.; Jiao, Y. The Effect of Oxygen Mobility/Vacancy on Carbon Gasification in Nano Catalytic Dry Reforming of Methane: A Review. J. CO2 Util. 2022, 63, 102109. [Google Scholar] [CrossRef]
- Perosa, A.; Tundo, P. Selective Hydrogenolysis of Glycerol with Raney Nickel. Ind. Eng. Chem. Res. 2005, 44, 8535–8537. [Google Scholar] [CrossRef]
- Yu, W.; Xu, J.; Ma, H.; Chen, C.; Zhao, J.; Miao, H.; Song, Q. A Remarkable Enhancement of Catalytic Activity for KBH4 Treating the Carbothermal Reduced Ni/AC Catalyst in Glycerol Hydrogenolysis. Catal. Commun. 2010, 11, 493–497. [Google Scholar] [CrossRef]
- Marinoiu, A.; Ionita, G.; Gáspár, C.L.; Cobzaru, C.; Oprea, S. Glycerol Hydrogenolysis to Propylene Glycol. React. Kinet. Catal. Lett. 2009, 97, 315–320. [Google Scholar] [CrossRef]
- Syuhada, A.; Ameen, M.; Azizan, M.T.; Aqsha, A.; Yusoff, M.H.M.; Ramli, A.; Alnarabiji, M.S.; Sher, F. In-Situ Hydrogenolysis of Glycerol Using Hydrogen Produced via Aqueous Phase Reforming of Glycerol over Sonochemically Synthesized Nickel-Based Nano-Catalyst. Mol. Catal. 2021, 514, 111860. [Google Scholar] [CrossRef]
- Freitas, I.C.; Manfro, R.L.; Souza, M.M.V.M. Hydrogenolysis of Glycerol to Propylene Glycol in Continuous System without Hydrogen Addition over Cu-Ni Catalysts. Appl. Catal. B Environ. 2018, 220, 31–41. [Google Scholar] [CrossRef]
- Raso, R.; García, L.; Ruiz, J.; Oliva, M.; Arauzo, J. Aqueous Phase Hydrogenolysis of Glycerol over Ni/Al-Fe Catalysts without External Hydrogen Addition. Appl. Catal. B Environ. 2021, 283, 119598. [Google Scholar] [CrossRef]
- Cai, F.; Pan, D.; Ibrahim, J.J.; Zhang, J.; Xiao, G. Hydrogenolysis of Glycerol over Supported Bimetallic Ni/Cu Catalysts with and without External Hydrogen Addition in a Fixed-Bed Flow Reactor. Appl. Catal. A Gen. 2018, 564, 172–182. [Google Scholar] [CrossRef]
- Gallego-García, D.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A.; Ayastuy, J.L. Nickel Aluminate Spinel-Derived Catalysts for Aqueous-Phase Hydrogenolysis of Glycerol with in-Situ Hydrogen Production: Effect of Molybdenum Doping. Appl. Catal. B Environ. 2024, 344, 123671. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Me (wt.%) | Ni (wt.%) | Ce/Zr (at./at.) | SBET (m2/g) | dNiCeZr.XRD (nm) | DA a | Lattice * (Å) | dNi0 (nm) | Ni0 Sites (×10−19) (Sites/gcat) b |
|---|---|---|---|---|---|---|---|---|---|
| NiCeZr | 0 | 9.2 | 14.1/85.9 | 235 (190) | 2.7 (2.9) | 1.6 | a = 3.6094 (3.6559) c = 5.2539 (5.2705) | 11.4 | 4.2 |
| Mg/NiCeZr | 1.05 | 9.2 | 13.6/86.4 | 226 (197) | 2.8 (3.3) | 1.6 | a = 3.6241 (3.6615) c = 5.2315 (5.2181) | 10.9 | 2.9 |
| Cu/NiCeZr | 1.06 | 9.2 | 15.7/84.3 | 224 (202) | 2.3 (2.9) | 1.9 | a = 3.6452 (3.6698) c = 5.2014 (5.2232) | 5.0 | 0.7 |
| Sn/NiCeZr | 0.82 | 9.2 | 14.6/85.4 | 200 (199) | 2.5 (3.0) | 2.0 | a = 3.6366 (3.6720) b = 5.2244 (5.2239) | 20.1 | 0.5 |
| Catalyst | H2 Uptake (mmolH2/g) | OSC (mmolo/gcat) | Sites Density (Sites/nm2) | Metal/Acid (Sites/Sites) | ||||
|---|---|---|---|---|---|---|---|---|
| Overall a | Peak | Metal b | Acid c | |||||
| α | β | γ | ||||||
| NiCeZr | 2.20 (106) | 0.11 | 0.44 | 1.65 | 63 | 0.22 | 0.36 | 0.62 |
| Mg/NiCeZr | 1.62 (78) | 0.03 | 0.25 | 1.34 | 60 | 0.15 | 0.30 | 0.49 |
| Cu/NiCeZr | 2.28 (101) | 0.10 | 0.62 | 1.56 | 65 | 0.03 | 0.42 | 0.08 |
| Sn/NiCeZr | 2.28 (103) | 0.04 | 0.46 | 1.78 | 43 | 0.02 | 0.51 | 0.05 |
| Catalyst | Ce/Zr | Ni/(Ce + Zr) | Ce3+/(Ce4+ + Ce3+) | Bulk Me/ (Ni + Ce + Zr) | Me/(Ni + Ce + Zr) | Ni0/NiTotal |
|---|---|---|---|---|---|---|
| NiCeZr | 0.13 (0.17) | 0.17 (0.08) | 0.16 (1.0) | 0 | 0 (0) | 0.0 (0.70) |
| Mg/NiCeZr | 0.18 (0.21) | 0.33 (0.10) | 0.27 (1.0) | 0.05 | 0.11 (0.10) | 0.0 (0.79) |
| Cu/NiCeZr | 0.17 (0.22) | 0.39 (0.10) | 0.19 (1.0) | 0.02 | 0.06 (0.05) | 0.0 (0.89) |
| Sn/NiCeZr | 0.16 (0.23) | 0.31 (0.10) | 0.19 (1.0) | 0.01 | 0.03 (0.02) | 0.0 (0.86) |
| Catalyst | Yield (%) | Selectivity (%) | H2/CO2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CH4 | CO | CO2 | C2+ | H2 | CH4 | CO | CO2 | C2+ | ||
| NiCeZr | 2.3 | 4.30 | 0.65 | 11.15 | 2.15 | 32.2 | 23.5 | 3.6 | 61.1 | 11.8 | 0.48 |
| Mg/NiCeZr | 3.2 | 9.21 | 0.76 | 17.55 | 3.30 | 24.8 | 29.9 | 2.5 | 56.9 | 10.7 | 0.43 |
| Cu/NiCeZr | 1.8 | 2.46 | 0.99 | 8.07 | 3.01 | 32.5 | 16.9 | 6.8 | 55.5 | 20.8 | 0.51 |
| Sn/NiCeZr | 0.6 | 0.69 | 0.43 | 1.60 | 1.61 | 29.5 | 16.0 | 10.1 | 36.9 | 37.0 | 0.87 |
| Catalyst | Primary Products | Secondary Products | C–C | C–O | O/C | Y1,2-PG |
|---|---|---|---|---|---|---|
| NiCeZr | 75.5 | 24.5 | 6.6 | 93.4 | 0.65 | 28.5 |
| Mg/NiCeZr | 79.6 | 20.4 | 7.9 | 92.1 | 0.66 | 19.3 |
| Cu/NiCeZr | 87.9 | 12.1 | 5.1 | 94.9 | 0.66 | 25.0 |
| Sn/NiCeZr | 92.6 | 7.4 | 3.9 | 96.1 | 0.67 | 16.7 |
| Catalyst | SBET (m2/g) | ΔVpore (%) | Δdpore (%) | dNiº (nm) | Leaching (wt.%) | |||
|---|---|---|---|---|---|---|---|---|
| Ni | Me | Ce | Zr | |||||
| NiCeZr | 218 (+15%) | +21 | +7 | 21.0 | 3.9 | 0.0 | 0.9 | 0.0 |
| Mg/NiCeZr | 211 (−7%) | −21 | −15 | 8.9 | 2.1 | 40.3 | 0.1 | 0.0 |
| Cu/NiCeZr | 279 (+25%) | +28 | +16 | 7.8 | 0.9 | 0.0 | 0.1 | 0.0 |
| Sn/NiCeZr | 246 (+23%) | +35 | −7 | 18.2 | 0.6 | 0.0 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera-Hincapie, N.; Iriarte-Velasco, U.; Ayastuy, J.L.; Gutiérrez-Ortiz, M.Á. Hydrogenolysis of Glycerol over NiCeZr Catalyst Modified with Mg, Cu, and Sn at the Surface Level. Int. J. Mol. Sci. 2024, 25, 3484. https://doi.org/10.3390/ijms25063484
Vera-Hincapie N, Iriarte-Velasco U, Ayastuy JL, Gutiérrez-Ortiz MÁ. Hydrogenolysis of Glycerol over NiCeZr Catalyst Modified with Mg, Cu, and Sn at the Surface Level. International Journal of Molecular Sciences. 2024; 25(6):3484. https://doi.org/10.3390/ijms25063484
Chicago/Turabian StyleVera-Hincapie, Norberto, Unai Iriarte-Velasco, Jose Luis Ayastuy, and Miguel Ángel Gutiérrez-Ortiz. 2024. "Hydrogenolysis of Glycerol over NiCeZr Catalyst Modified with Mg, Cu, and Sn at the Surface Level" International Journal of Molecular Sciences 25, no. 6: 3484. https://doi.org/10.3390/ijms25063484
APA StyleVera-Hincapie, N., Iriarte-Velasco, U., Ayastuy, J. L., & Gutiérrez-Ortiz, M. Á. (2024). Hydrogenolysis of Glycerol over NiCeZr Catalyst Modified with Mg, Cu, and Sn at the Surface Level. International Journal of Molecular Sciences, 25(6), 3484. https://doi.org/10.3390/ijms25063484