
Multiomics-Based Feature Extraction and Selection for the Prediction of Lung Cancer Survival



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
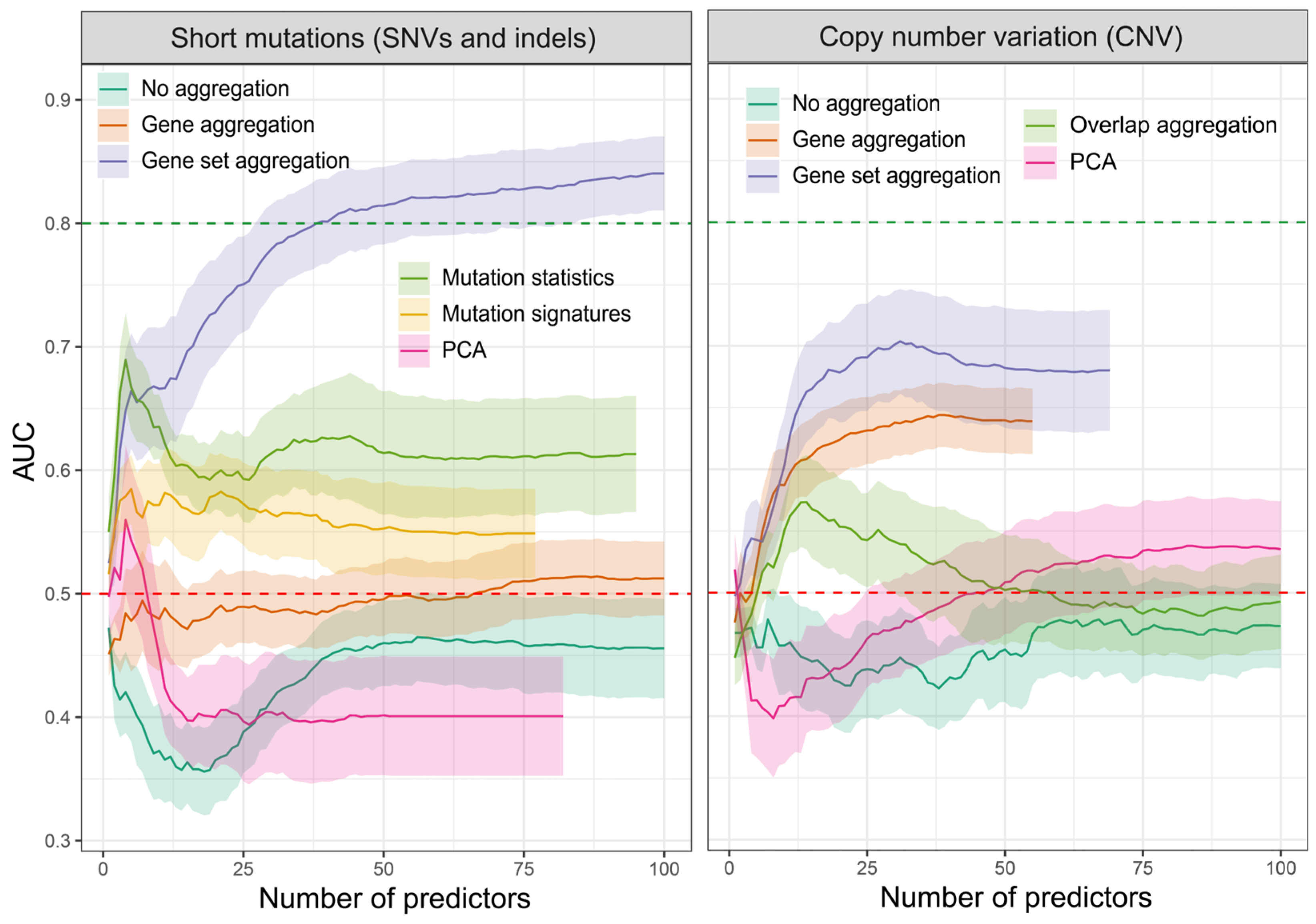
2.1. Feature Evaluation Strategy
2.2. Feature Extraction
- Region overlap aggregation—Combining altered regions based on a partial overlap. This is achieved by dividing the genomic regions representing alterations into smaller sections that are shared by at least two samples. Since the mutation set contains primarily single nucleotide variants (SNVs), this method is applicable only to the CNV set. Each smaller region is assigned a distinct copy number value, represented as a segment mean variable defined with log2(CN/2), where CN represents the absolute copy number. It should be emphasized that although this method involves segmenting the genome across all samples, not every genomic region is assigned a numerical value. Sections lacking alterations in at least one sample are omitted;
- Gene level aggregation—Aggregating mutations and copy number changes at the gene level by counting the total number of mutations associated with each gene. This approach is similar to the region overlap; however, the genome is divided based on gene coordinates, irrespective of the size of the altered regions. We then assign 1 to each gene that contains at least one protein-altering mutation and 0 if no such changes were detected. In the case of CNV, we assign the CN to the region in which the gene is located. We use the altered copy number value even if the region does not include the entire gene. Unfortunately, under certain conditions, particularly when the total length of copy number-altered regions is minimal, this method can also yield a sparse feature matrix. Additionally, it is important to recognize that genes situated in close proximity are likely to show a high correlation with the assigned values;
- Gene set aggregation—Grouping genes into biological pathways and aggregating mutations at the pathway level. This method provides a higher-level view of the functional impact of mutations and CNVs. This is achieved by further aggregating mutations and CNV statistics obtained for individual genes across specific gene sets that represent genes involved in the same signaling process, having similar functions, or showing altered expression levels as a result of specific stimuli. By adopting this method, both high-correlation and low-variance issues can be avoided. Furthermore, it has the potential to decrease the overall number of features, especially in scenarios with an exceptionally high number of aggregated regions;
- Mutation statistics—Calculating the number of specific mutation types, which are aggregated using three methods: SBS, DBS, and ID. In SBS (single-base substitutions), we calculate the number of variants depending on the reference and observed nucleotide and its context, e.g., G[T>C]T indicates a T-to-C mutation in a context of G and T on both sides (there are 96 types in total). DBS are the frequencies of double-base substitutions generated concurrently modifying two consecutive nucleotide bases, e.g., AC>GA (78 types). IDs are small indels (insertions and deletions) aggregated into 83 different types, based on their length, number and length of repeat units, and length of microhomologous sequences (sequence found at the edge of the deleted sequence and in the reference genome right next to it);
- Mutation signatures—Exploring the previous mutation statistics to identify mutational signatures associated with specific biological processes or exposures. Each of the mutation statistic groups (SBS, DBS, and ID) was used to calculate the association of each sample with individual signatures from each group;
- PCA—Principal component analysis (PCA) constitutes a distinct category within the spectrum of feature extraction methods, allowing the condensation of the feature space to match the number of tested samples. PCA is widely used in machine learning to maximize the variance or dispersion along the newly delineated variables.
2.3. Evaluation of Prediction Capabilities of Each Dataset
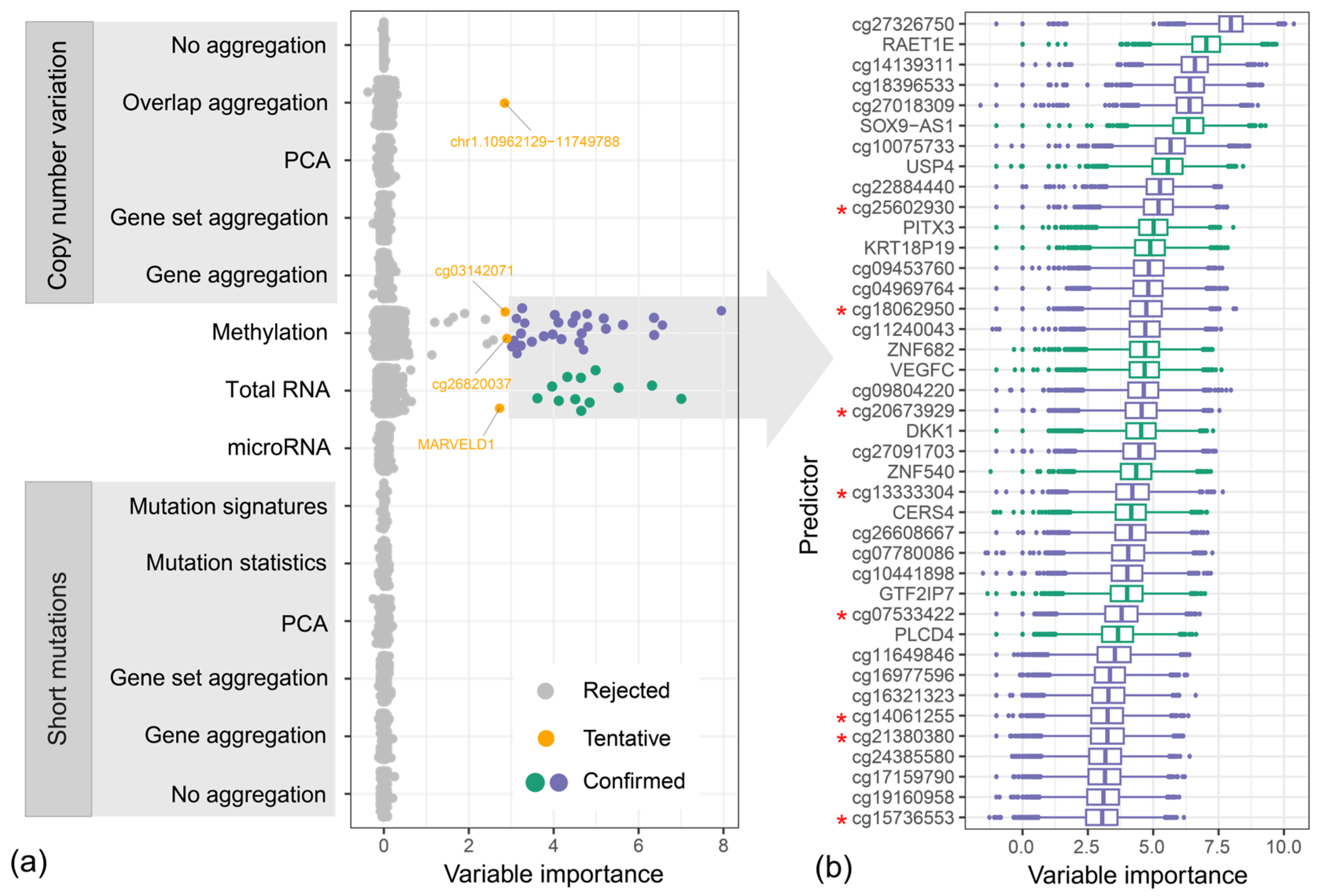
2.4. Variable Importance Assessment
3. Discussion
4. Materials and Methods
4.1. Multiomics Data
- Gene expression levels derived from total RNA sequencing (RNA-seq) in the form of raw read counts assigned to individual genes;
- microRNA expression levels derived from RNA sequencing (miRNA-seq) in the form of raw read counts assigned to individual miRNAs;
- Locations of somatic mutations (single nucleotide variants and short indels) obtained in a whole-exome sequencing experiment (WES) in the form of genomic coordinates (for the GRCh38 genome version) with supporting annotation data;
- DNA methylation levels acquired via Illumina Infinium HumanMethylation450 (TCGA) or Methylation Epic (CPTAC-3) BeadChip microarrays, expressed as values within a range of 0 to 1;
- Copy number variation (CNV) data acquired using Affymetrix SNP 6.0 microarrays, presented as genomic intervals (GRCh38 reference genome), along with segment mean statistics (log2(CN/2), where CN represents the observed copy number of a particular region).
4.2. Feature Definition
4.3. Variable Importance Study
4.4. Classification of Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Toonkel, R.L.; Powell, C.A. Genomics of lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 152–158. [Google Scholar] [CrossRef]
- Xiong, Y.; Feng, Y.; Qiao, T.; Han, Y. Identifying prognostic biomarkers of non-small cell lung cancer by transcriptome analysis. Cancer Biomark. Sect. A Dis. Markers 2020, 27, 243–250. [Google Scholar] [CrossRef]
- Cheung, C.H.Y.; Juan, H.F. Quantitative proteomics in lung cancer. J. Biomed. Sci. 2017, 24, 37. [Google Scholar] [CrossRef]
- Qi, S.A.; Wu, Q.; Chen, Z.; Zhang, W.; Zhou, Y.; Mao, K.; Li, J.; Li, Y.; Chen, J.; Huang, Y.; et al. High-resolution metabolomic biomarkers for lung cancer diagnosis and prognosis. Sci. Rep. 2021, 11, 11805. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.D.; Jia, P.; Aldrich, M.C.; Zhao, Z. Lung Cancer: One Disease or Many. Hum. Hered. 2018, 83, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, M.; Liu, B. Exploring and comparing of the gene expression and methylation differences between lung adenocarcinoma and squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 4454–4459. [Google Scholar] [CrossRef] [PubMed]
- Relli, V.; Trerotola, M.; Guerra, E.; Alberti, S. Distinct lung cancer subtypes associate to distinct drivers of tumor progression. Oncotarget 2018, 9, 35528–35540. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.A.; Wishart, D.S. Applications of machine learning in cancer prediction and prognosis. Cancer Inform. 2007, 2, 59–77. [Google Scholar] [CrossRef]
- Simes, R.J. Treatment selection for cancer patients: Application of statistical decision theory to the treatment of advanced ovarian cancer. J. Chronic Dis. 1985, 38, 171–186. [Google Scholar] [CrossRef]
- Astion, M.L.; Wilding, P. Application of neural networks to the interpretation of laboratory data in cancer diagnosis. Clin. Chem. 1992, 38, 34–38. [Google Scholar] [CrossRef]
- Sesen, M.B.; Nicholson, A.E.; Banares-Alcantara, R.; Kadir, T.; Brady, M. Bayesian networks for clinical decision support in lung cancer care. PLoS ONE 2013, 8, e82349. [Google Scholar] [CrossRef]
- Guo, S.; Yan, F.; Xu, J.; Bao, Y.; Zhu, J.; Wang, X.; Wu, J.; Li, Y.; Pu, W.; Liu, Y.; et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin. Epigenetics 2015, 7, 3. [Google Scholar] [CrossRef]
- Wang, L. Deep Learning Techniques to Diagnose Lung Cancer. Cancers 2022, 14, 5569. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, X.; Sun, D. A narrative review of prognosis prediction models for non-small cell lung cancer: What kind of predictors should be selected and how to improve models? Ann. Transl. Med. 2021, 9, 1597. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Woerl, A.C.; Jungmann, F.; Glasner, C.; Stenzel, P.; Strobl, S.; Fernandez, A.; Wagner, D.C.; Haferkamp, A.; Mildenberger, P.; et al. Multimodal Deep Learning for Prognosis Prediction in Renal Cancer. Front. Oncol. 2021, 11, 788740. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xie, L.; Han, J.; Guo, X. The Application of Deep Learning in Cancer Prognosis Prediction. Cancers 2020, 12, 603. [Google Scholar] [CrossRef] [PubMed]
- Ten Haaf, K.; Jeon, J.; Tammemagi, M.C.; Han, S.S.; Kong, C.Y.; Plevritis, S.K.; Feuer, E.J.; de Koning, H.J.; Steyerberg, E.W.; Meza, R. Risk prediction models for selection of lung cancer screening candidates: A retrospective validation study. PLoS Med. 2017, 14, e1002277. [Google Scholar] [CrossRef] [PubMed]
- Ten Haaf, K.; van der Aalst, C.M.; de Koning, H.J.; Kaaks, R.; Tammemagi, M.C. Personalising lung cancer screening: An overview of risk-stratification opportunities and challenges. Int. J. Cancer 2021, 149, 250–263. [Google Scholar] [CrossRef]
- Yeo, Y.; Shin, D.W.; Han, K.; Park, S.H.; Jeon, K.H.; Lee, J.; Kim, J.; Shin, A. Individual 5-Year Lung Cancer Risk Prediction Model in Korea Using a Nationwide Representative Database. Cancers 2021, 13, 3496. [Google Scholar] [CrossRef]
- Tufail, A.B.; Ma, Y.K.; Kaabar, M.K.A.; Martinez, F.; Junejo, A.R.; Ullah, I.; Khan, R. Deep Learning in Cancer Diagnosis and Prognosis Prediction: A Minireview on Challenges, Recent Trends, and Future Directions. Comput. Math. Methods Med. 2021, 2021, 9025470. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, R.; Lyu, Q. Multiomics and machine learning in lung cancer prognosis. J. Thorac. Dis. 2020, 12, 4531–4535. [Google Scholar] [CrossRef]
- Laios, A.; Katsenou, A.; Tan, Y.S.; Johnson, R.; Otify, M.; Kaufmann, A.; Munot, S.; Thangavelu, A.; Hutson, R.; Broadhead, T.; et al. Feature Selection is Critical for 2-Year Prognosis in Advanced Stage High Grade Serous Ovarian Cancer by Using Machine Learning. Cancer Control J. Moffitt Cancer Cent. 2021, 28, 10732748211044678. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, H.; Zhang, S.; Luo, W. A practical framework RNMF for exploring the association between mutational signatures and genes using gene cumulative contribution abundance. Cancer Med. 2022, 11, 4053–4069. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.A.; Diaz-Gay, M.; Wu, Y.; Barnes, M.; Vangara, R.; Bergstrom, E.N.; He, Y.; Vella, M.; Wang, J.; Teague, J.W.; et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2022, 2, 100179. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef] [PubMed]
- Sanjaya, P.; Maljanen, K.; Katainen, R.; Waszak, S.M.; Genomics England Research Consortium; Aaltonen, L.A.; Stegle, O.; Korbel, J.O.; Pitkanen, E. Mutation-Attention (MuAt): Deep representation learning of somatic mutations for tumour typing and subtyping. Genome Med. 2023, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, W.S.; Rashid, M. A review of deep learning applications in human genomics using next-generation sequencing data. Hum. Genom. 2022, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Piernik, M.; Brzezinski, D.; Sztromwasser, P.; Pacewicz, K.; Majer-Burman, W.; Gniot, M.; Sielski, D.; Bryzghalov, O.; Wozna, A.; Zawadzki, P. DBFE: Distribution-based feature extraction from structural variants in whole-genome data. Bioinformatics 2022, 38, 4466–4473. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, J.; Wagner, J. STR-based feature extraction and selection for genetic feature discovery in neurological disease genes. Sci. Rep. 2023, 13, 2480. [Google Scholar] [CrossRef] [PubMed]
- Pancotti, C.; Rollo, C.; Birolo, G.; Benevenuta, S.; Fariselli, P.; Sanavia, T. Unravelling the instability of mutational signatures extraction via archetypal analysis. Front. Genet. 2022, 13, 1049501. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, Y.; Jiang, N.; Boot, A.; Rozen, S.G. mSigHdp: Hierarchical Dirichlet process mixture modeling for mutational signature discovery. NAR Genom. Bioinform. 2023, 5, lqad005. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. TIG 2008, 24, 529–538. [Google Scholar] [CrossRef]
- How, J.A.; Jazaeri, A.A.; Fellman, B.; Daniels, M.S.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.S.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef]
- Jaksik, R.; Śmieja, J. Prediction of Lung Cancer Survival Based on Multiomic Data. In Proceedings of the Intelligent Information and Database Systems, Ho Chi Minh City, Vietnam, 28–30 November 2022; pp. 116–127. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 4768–4777. [Google Scholar]
- McGilvray, R.W.; Eagle, R.A.; Rolland, P.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int. J. Cancer 2010, 127, 1412–1420. [Google Scholar] [CrossRef]
- Bowden, M.; Nadal, R.; Zhou, C.W.; Werner, L.; Barletta, J.; Juanpere, N.; Lloreta, J.; Hernandez-Llodra, S.; Morote, J.; de Torres, I.; et al. Transcriptomic analysis of micropapillary high grade T1 urothelial bladder cancer. Sci. Rep. 2020, 10, 20135. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, L.; Zong, J.; Ye, Y.; Chen, G.; Chen, Y.; Liao, X.; Guo, Q.; Qiu, S.; Lin, S.; et al. Decreased expression of the NKG2D ligand ULBP4 may be an indicator of poor prognosis in patients with nasopharyngeal carcinoma. Oncotarget 2017, 8, 42007–42019. [Google Scholar] [CrossRef]
- Wu, L.; Yang, J.; Ke, R.S.; Liu, Y.; Guo, P.; Feng, L.; Li, Z. Impact of lncRNA SOX9-AS1 overexpression on the prognosis and progression of intrahepatic cholangiocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101999. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Y.; Hou, B.; Wang, Y.; Deng, D.; Fu, Z.; Xu, Z. A SOX9-AS1/miR-5590-3p/SOX9 positive feedback loop drives tumor growth and metastasis in hepatocellular carcinoma through the Wnt/beta-catenin pathway. Mol. Oncol. 2019, 13, 2194–2210. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Herrero, J.F.; Pluvinet, R.; Luna de Haro, A.; Sumoy, L. Paired-end small RNA sequencing reveals a possible overestimation in the isomiR sequence repertoire previously reported from conventional single read data analysis. BMC Bioinform. 2021, 22, 215. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.Q.; Varmus, H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. eLife 2015, 4, e06907. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-H.; Chen, W.-N.; Hsu, T.-C.; Lin, C.; Tsao, Y.; Wu, S. Overall survival prediction of non-small cell lung cancer by integrating microarray and clinical data with deep learning. Sci. Rep. 2020, 10, 4679. [Google Scholar] [CrossRef]
- Emaminejad, N.; Qian, W.; Guan, Y.; Tan, M.; Qiu, Y.; Liu, H.; Zheng, B. Fusion of Quantitative Image and Genomic Biomarkers to Improve Prognosis Assessment of Early Stage Lung Cancer Patients. IEEE Trans. Bio-Med. Eng. 2016, 63, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Zhou, W.; Yan, H.; Wong, M.; Lee, V. Personalized prediction of EGFR mutation-induced drug resistance in lung cancer. Sci. Rep. 2013, 3, 2855. [Google Scholar] [CrossRef]
- Matsubara, T.; Ochiai, T.; Hayashida, M.; Akutsu, T.; Nacher, J.C. Convolutional neural network approach to lung cancer classification integrating protein interaction network and gene expression profiles. J. Bioinform. Comput. Biol. 2019, 17, 1940007. [Google Scholar] [CrossRef]
- Lee, N.S.-Y.; Shafiq, J.; Field, M.; Fiddler, C.; Varadarajan, S.; Gandhidasan, S.; Hau, E.; Vinod, S.K. Predicting 2-year survival in stage I-III non-small cell lung cancer: The development and validation of a scoring system from an Australian cohort. Radiat. Oncol. 2022, 17, 74. [Google Scholar] [CrossRef]
- Herrmann, M.; Probst, P.; Hornung, R.; Jurinovic, V.; Boulesteix, A.-L. Large-scale benchmark study of survival prediction methods using multi-omics data. Brief. Bioinform. 2021, 22, bbaa167. [Google Scholar] [CrossRef]
- Giang, T.T.; Nguyen, T.P.; Tran, D.H. Stratifying patients using fast multiple kernel learning framework: Case studies of Alzheimer’s disease and cancers. BMC Med. Inf. Decis. Mak. 2020, 20, 108. [Google Scholar] [CrossRef]
- Jayasurya, K.; Fung, G.; Yu, S.; Dehing-Oberije, C.; De Ruysscher, D.; Hope, A.; De Neve, W.; Lievens, Y.; Lambin, P.; Dekker, A.L. Comparison of Bayesian network and support vector machine models for two-year survival prediction in lung cancer patients treated with radiotherapy. Med. Phys. 2010, 37, 1401–1407. [Google Scholar] [CrossRef]
- Sun, T.; Wang, J.; Li, X.; Lv, P.; Liu, F.; Luo, Y.; Gao, Q.; Zhu, H.; Guo, X. Comparative evaluation of support vector machines for computer aided diagnosis of lung cancer in CT based on a multi-dimensional data set. Comput. Methods Programs Biomed. 2013, 111, 519–524. [Google Scholar] [CrossRef]
- Hyun, S.H.; Ahn, M.S.; Koh, Y.W.; Lee, S.J. A Machine-Learning Approach Using PET-Based Radiomics to Predict the Histological Subtypes of Lung Cancer. Clin. Nucl. Med. 2019, 44, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Briley, P.; Bolin, L.P.; Kitko, L.; Ellis, C. Disparities in Comorbidities in Lung Cancer: Findings From the Behavioral Risk Factor Surveillance System. Cancer Nurs. 2022, 45, E883–E889. [Google Scholar] [CrossRef]
- Sigel, K.; Yin Kong, C.; Leiter, A.; Kale, M.; Mhango, G.; Huang, B.; Gould, M.K.; Wisnivesky, J. Assessment of treatment strategies for stage I non-small cell lung cancer in patients with comorbidities. Lung Cancer 2022, 170, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Stabellini, N.; Bruno, D.S.; Dmukauskas, M.; Barda, A.J.; Cao, L.; Shanahan, J.; Waite, K.; Montero, A.J.; Barnholtz-Sloan, J.S. Sex Differences in Lung Cancer Treatment and Outcomes at a Large Hybrid Academic-Community Practice. JTO Clin. Res. Rep. 2022, 3, 100307. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, R.J.; Zhang, X.; Switchenko, J.M.; Patel, P.R.; Shelton, J.W.; Tian, S.; Nanda, R.H.; Steuer, C.E.; Pillai, R.N.; Owonikoko, T.K.; et al. Health care disparities among octogenarians and nonagenarians with stage III lung cancer. Cancer 2018, 124, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Tufman, A.; Holle, R.; Schwarzkopf, L. “Age matters”-German claims data indicate disparities in lung cancer care between elderly and young patients. PLoS ONE 2019, 14, e0217434. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.; Conron, M.; Wright, G.; Mitchell, P.; Ball, D.; Philip, J.; Brand, M.; Zalcberg, J.; Stirling, R.G. Excess mortality and undertreatment in elderly lung cancer patients: Treatment nihilism in the modern era? ERJ Open Res. 2021, 7, 00393–2020. [Google Scholar] [CrossRef]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front. Oncol. 2018, 8, 621. [Google Scholar] [CrossRef]
- Lee, M.; Napier, C.E.; Yang, S.F.; Arthur, J.W.; Reddel, R.R.; Pickett, H.A. Comparative analysis of whole genome sequencing-based telomere length measurement techniques. Methods 2017, 114, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kursa, M.; Rudnicki, W. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Lewis, M.J.; Spiliopoulou, A.; Goldmann, K.; Pitzalis, C.; McKeigue, P.; Barnes, M.R. nestedcv: An R package for fast implementation of nested cross-validation with embedded feature selection designed for transcriptomics and high-dimensional data. Bioinform. Adv. 2023, 3, vbad048. [Google Scholar] [CrossRef]
- Jethani, M.S.N.; Covert, S.I.; Lee, R. Ranganath. FastSHAP: Real-Time Shapley Value Estimation. arXiv 2022, arXiv:2107.07436. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaksik, R.; Szumała, K.; Dinh, K.N.; Śmieja, J. Multiomics-Based Feature Extraction and Selection for the Prediction of Lung Cancer Survival. Int. J. Mol. Sci. 2024, 25, 3661. https://doi.org/10.3390/ijms25073661
Jaksik R, Szumała K, Dinh KN, Śmieja J. Multiomics-Based Feature Extraction and Selection for the Prediction of Lung Cancer Survival. International Journal of Molecular Sciences. 2024; 25(7):3661. https://doi.org/10.3390/ijms25073661
Chicago/Turabian StyleJaksik, Roman, Kamila Szumała, Khanh Ngoc Dinh, and Jarosław Śmieja. 2024. "Multiomics-Based Feature Extraction and Selection for the Prediction of Lung Cancer Survival" International Journal of Molecular Sciences 25, no. 7: 3661. https://doi.org/10.3390/ijms25073661
APA StyleJaksik, R., Szumała, K., Dinh, K. N., & Śmieja, J. (2024). Multiomics-Based Feature Extraction and Selection for the Prediction of Lung Cancer Survival. International Journal of Molecular Sciences, 25(7), 3661. https://doi.org/10.3390/ijms25073661