

Properties and Mechanisms of Deletions, Insertions, and Substitutions in the Evolutionary History of SARS-CoV-2

 ,
, 
 , , and
, , and 
Abstract
:1. Introduction
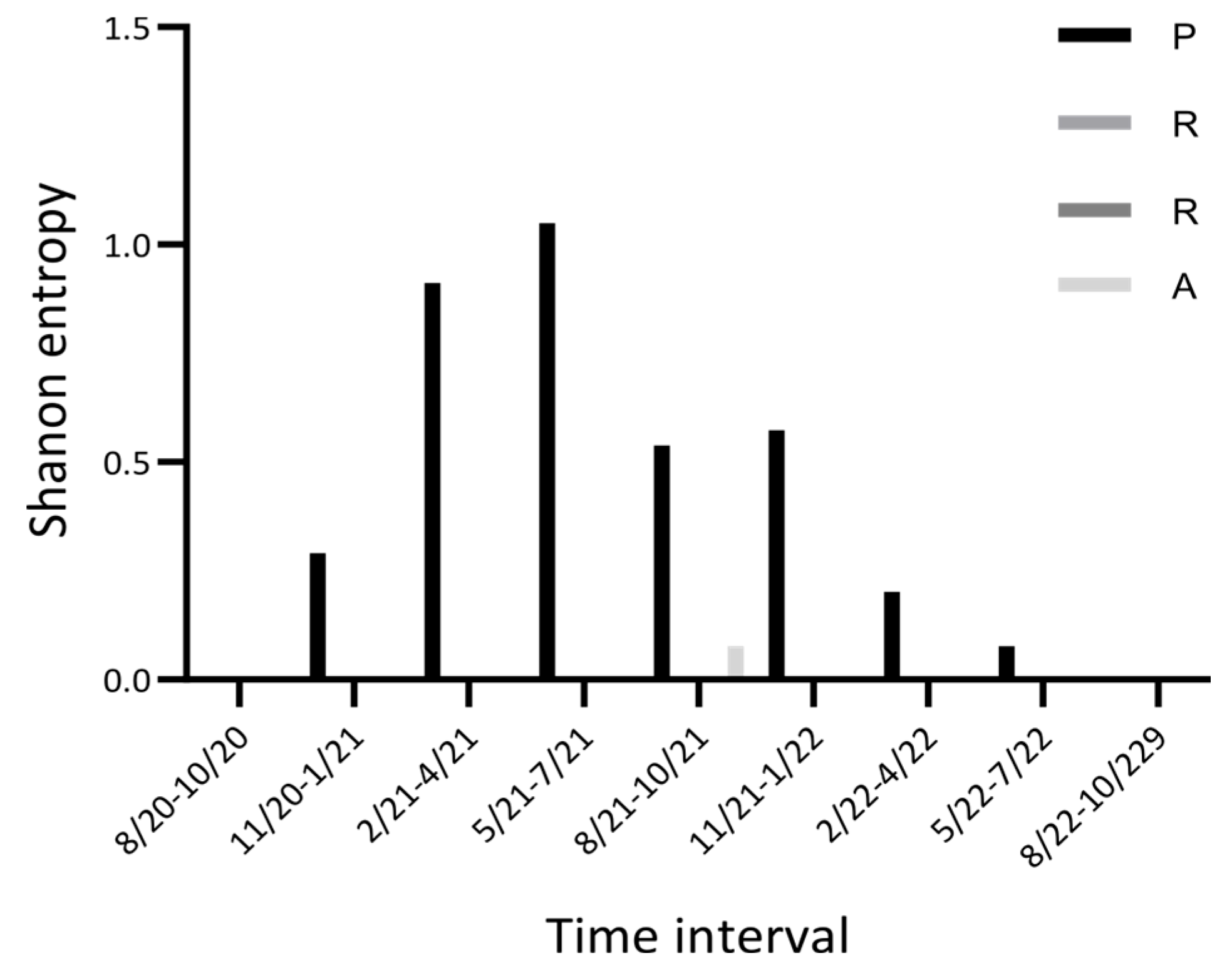
2. Results
2.1. SARS-CoV-2 Genome Structure and Replication
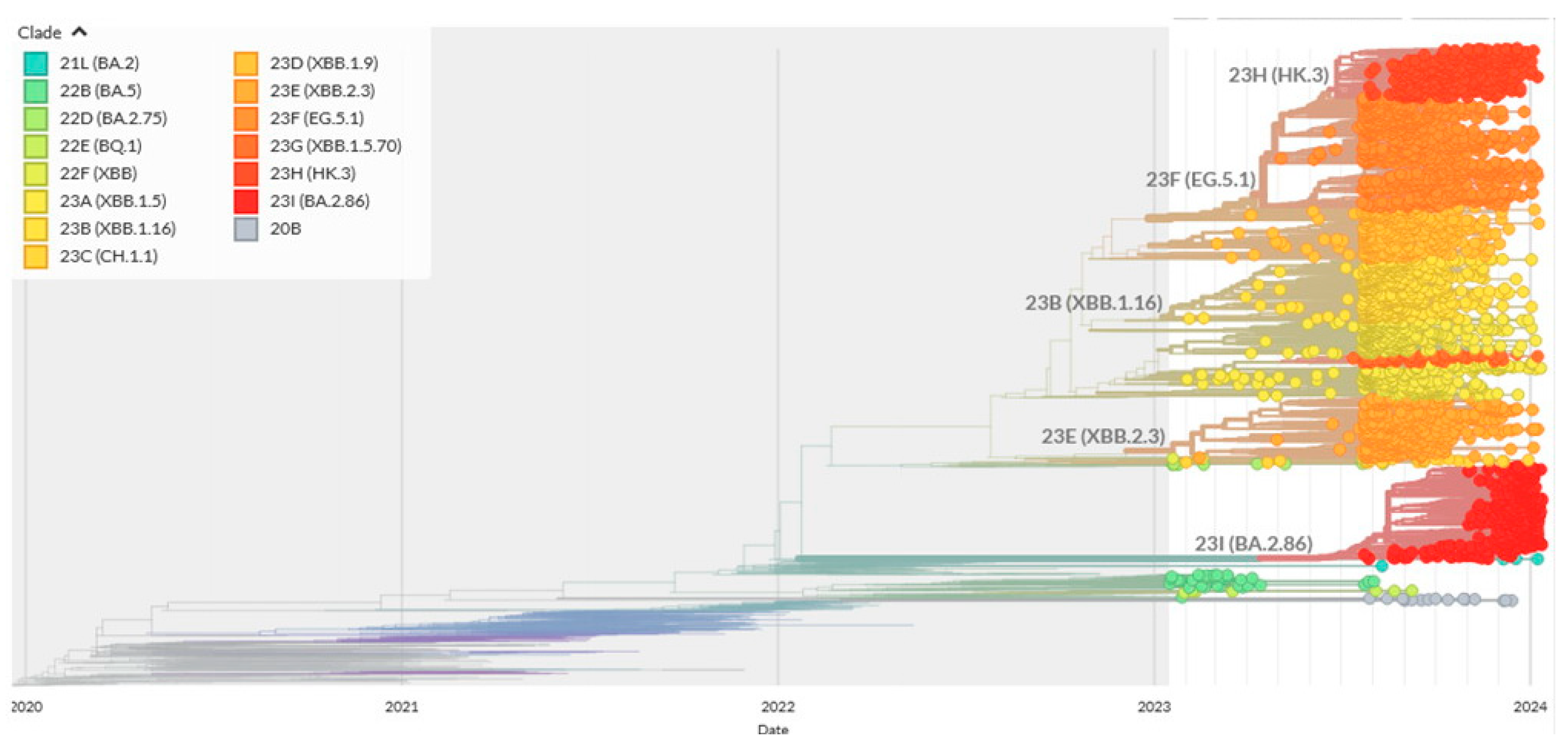
2.2. Reconstructions and Analyses of Mutation Spectra: Methodological Approaches
2.3. Molecular Mechanisms of Mutations
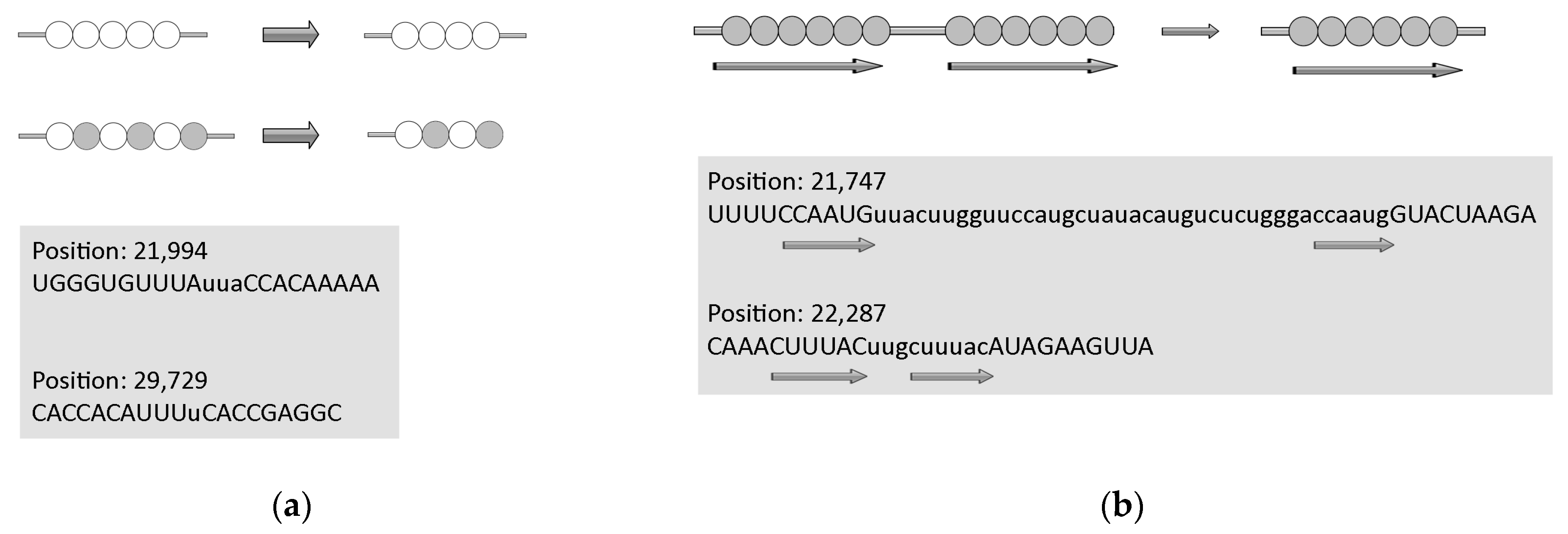
2.3.1. Deletions
2.3.2. Insertions
2.3.3. Substitutions
2.4. Natural Selection of Mutations
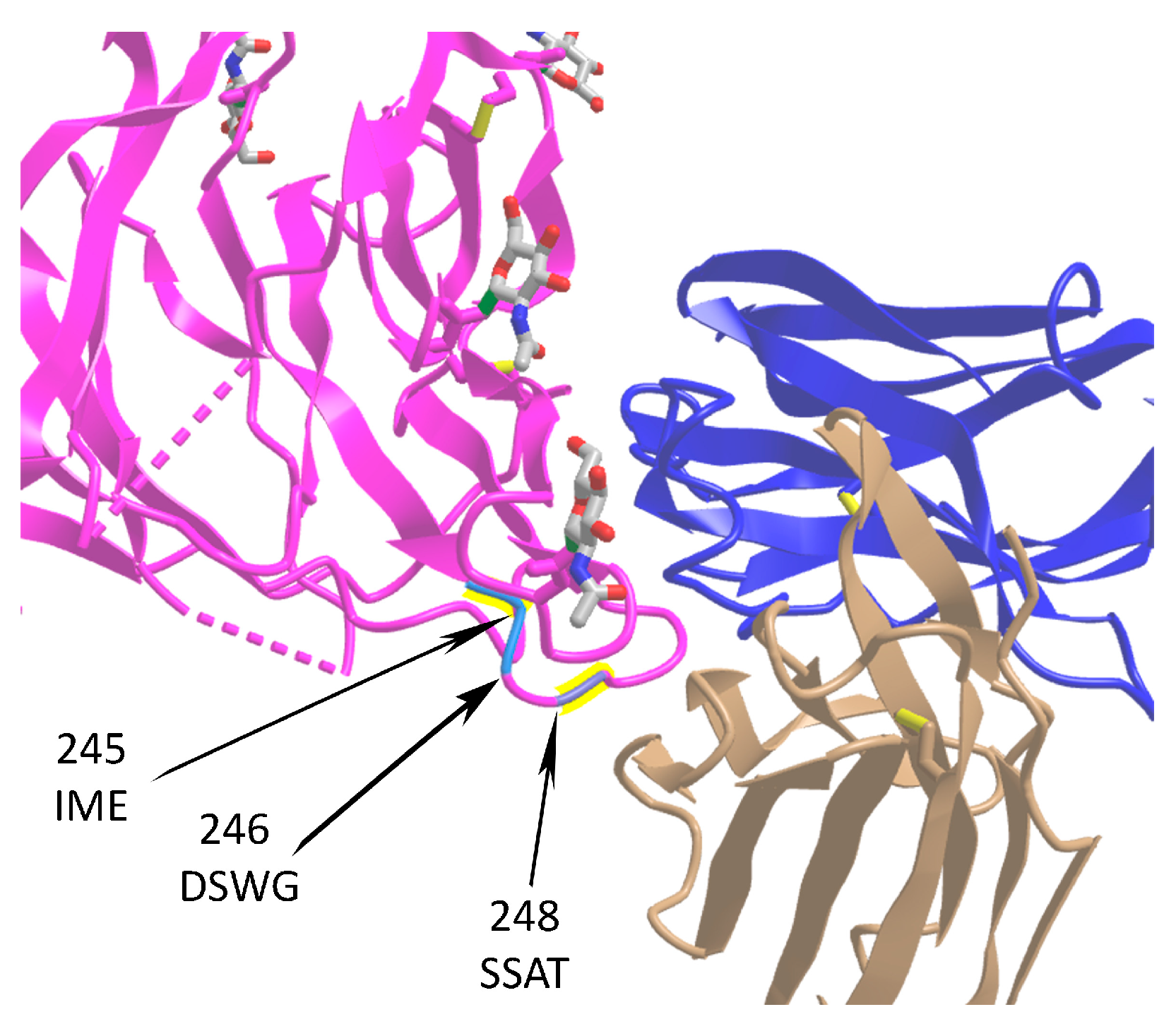
2.4.1. Selection of Deletions and Insertions
2.4.2. Selection of Substitutions
2.5. Interplay between Mutations and Selection
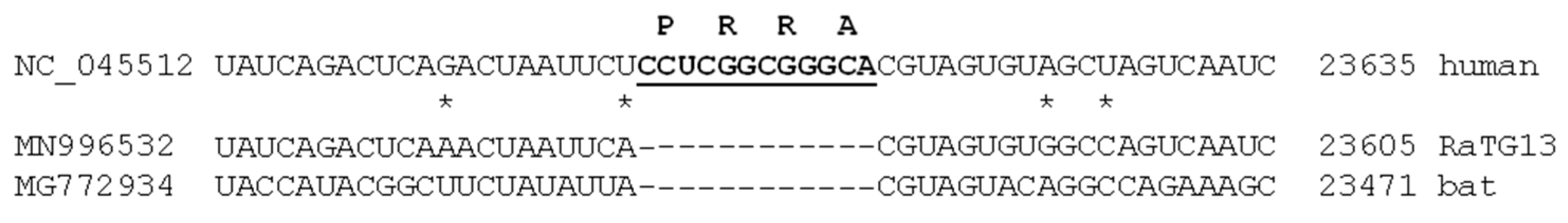
2.6. A Puzzle: Insertion and Recurrent Deletions of the -PRRA- Sequence
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drake, J.W.; Baltz, R.H. The biochemistry of mutagenesis. Annu. Rev. Biochem. 1976, 45, 11–37. [Google Scholar] [CrossRef] [PubMed]
- Maki, H. Origins of spontaneous mutations: Specificity and directionality of base-substitution, frameshift, and sequence-substitution mutageneses. Annu. Rev. Genet. 2002, 36, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Pavlov, Y.I. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat. Res. 2003, 544, 65–85. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Pavlov, Y.I.; Goncearenco, A.; De, S.; Lada, A.G.; Poliakov, E.; Panchenko, A.R.; Cooper, D.N. Mutational signatures and mutable motifs in cancer genomes. Brief. Bioinform. 2018, 19, 1085–1101. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Beichman, A.C.; Neher, R.A.; Harris, K. Evolution of the SARS-CoV-2 mutational spectrum. Mol. Biol. Evol. 2023, 40, msad085. [Google Scholar] [CrossRef] [PubMed]
- Saldivar-Espinoza, B.; Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Puigbo, P.; Cereto-Massague, A.; Pujadas, G.; Garcia-Vallve, S. Prediction of recurrent mutations in SARS-CoV-2 using artificial neural networks. Int. J. Mol. Sci. 2022, 23, 14683. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- McCullers, J.A.; Wang, G.C.; He, S.; Webster, R.G. Reassortment and insertion-deletion are strategies for the evolution of influenza B viruses in nature. J. Virol. 1999, 73, 7343–7348. [Google Scholar] [CrossRef]
- Taylor, K.Y.; Agu, I.; Jose, I.; Mantynen, S.; Campbell, A.J.; Mattson, C.; Chou, T.W.; Zhou, B.; Gresham, D.; Ghedin, E.; et al. Influenza A virus reassortment is strain dependent. PLoS Pathog. 2023, 19, e1011155. [Google Scholar] [CrossRef]
- Zdravkovic, M.; Berger-Estilita, J.; Zdravkovic, B.; Berger, D. Scientific quality of COVID-19 and SARS-CoV-2 publications in the highest impact medical journals during the early phase of the pandemic: A case control study. PLoS ONE 2020, 15, e0241826. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Holmes, E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Stadler, K.; Masignani, V.; Eickmann, M.; Becker, S.; Abrignani, S.; Klenk, H.D.; Rappuoli, R. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 2003, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Pancer, K.; Milewska, A.; Owczarek, K.; Dabrowska, A.; Kowalski, M.; Labaj, P.P.; Branicki, W.; Sanak, M.; Pyrc, K. The SARS-CoV-2 ORF10 is not essential in vitro or in vivo in humans. PLoS Pathog. 2020, 16, e1008959. [Google Scholar] [CrossRef]
- Mack, A.H.; Menzies, G.; Southgate, A.; Jones, D.D.; Connor, T.R. A proofreading mutation with an allosteric effect allows a cluster of SARS-CoV-2 viruses to rapidly evolve. Mol. Biol. Evol. 2023, 40, msad209. [Google Scholar] [CrossRef]
- Smith, E.C.; Blanc, H.; Surdel, M.C.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef]
- Long, S. SARS-CoV-2 subgenomic RNAs: Characterization, utility, and perspectives. Viruses 2021, 13, 1923. [Google Scholar] [CrossRef]
- Tang, M.E.; Ng, K.L.; Edslev, S.M.; Ellegaard, K.; Danish, C.-G.C.; Stegger, M.; Alexandersen, S. Comparative subgenomic mRNA profiles of SARS-CoV-2 Alpha, Delta and Omicron BA.1, BA.2 and BA.5 sub-lineages using Danish COVID-19 genomic surveillance data. EBioMedicine 2023, 93, 104669. [Google Scholar] [CrossRef]
- Chen, Z.; Ng, R.W.Y.; Lui, G.; Ling, L.; Chow, C.; Yeung, A.C.M.; Boon, S.S.; Wang, M.H.; Chan, K.C.C.; Chan, R.W.Y.; et al. Profiling of SARS-CoV-2 subgenomic rnas in clinical specimens. Microbiol. Spectr. 2022, 10, e0018222. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Template switching and duplications in SARS-CoV-2 genomes give rise to insertion variants that merit monitoring. Commun. Biol. 2021, 4, 1343. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between mutation spectra in hypermutated genomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Saura, A.; Bykova, A.; Brover, V.; Yurchenko, V. Deletions across the SARS-CoV-2 genome: Molecular mechanisms and putative functional consequences of deletions in accessory genes. Microorganisms 2023, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Bykova, A.; Saura, A.; Glazko, G.V.; Roche-Lima, A.; Yurchenko, V.; Rogozin, I.B. The 29-nucleotide deletion in SARS-CoV: Truncated versions of ORF8 are under purifying selection. BMC Genomics 2023, 24, 387. [Google Scholar] [CrossRef]
- Mavian, C.; Pond, S.K.; Marini, S.; Magalis, B.R.; Vandamme, A.M.; Dellicour, S.; Scarpino, S.V.; Houldcroft, C.; Villabona-Arenas, J.; Paisie, T.K.; et al. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-CoV-2 infections unreliable. Proc. Natl. Acad. Sci. USA 2020, 117, 12522–12523. [Google Scholar] [CrossRef]
- Turakhia, Y.; De Maio, N.; Thornlow, B.; Gozashti, L.; Lanfear, R.; Walker, C.R.; Hinrichs, A.S.; Fernandes, J.D.; Borges, R.; Slodkowicz, G.; et al. Stability of SARS-CoV-2 phylogenies. PLoS Genet. 2020, 16, e1009175. [Google Scholar] [CrossRef]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef]
- McBroome, J.; Thornlow, B.; Hinrichs, A.S.; Kramer, A.; De Maio, N.; Goldman, N.; Haussler, D.; Corbett-Detig, R.; Turakhia, Y. A daily-updated database and tools for comprehensive SARS-CoV-2 mutation-annotated trees. Mol. Biol. Evol. 2021, 38, 5819–5824. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferre, N.; Macip, G.; Martinez, R.; Puigbo, P.; Cereto-Massague, A.; Pujadas, G.; Garcia-Vallve, S. The mutational landscape of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 9072. [Google Scholar] [CrossRef]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and evolution of the SARS-CoV-2 Spike protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Scarpa, F.; Azzena, I.; Ciccozzi, A.; Giovanetti, M.; Locci, C.; Casu, M.; Fiori, P.L.; Borsetti, A.; Cella, E.; Quaranta, M.; et al. Integrative genome-based survey of the SARS-CoV-2 Omicron XBB.1.16 variant. Int. J. Mol. Sci. 2023, 24, 13573. [Google Scholar] [CrossRef]
- Zhu, X.; Mannar, D.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Saville, J.W.; Leopold, K.; Li, W.; Dimitrov, D.S.; Tuttle, K.S.; et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021, 19, e3001237. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977, 267, 275–276. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Koonin, E.V.; Rogozin, I.B. Getting positive about selection. Genome Biol. 2003, 4, 331. [Google Scholar] [CrossRef]
- Bukur, T.; Riesgo-Ferreiro, P.; Sorn, P.; Gudimella, R.; Hausmann, J.; Rosler, T.; Lower, M.; Schrors, B.; Sahin, U. CoVigator-a knowledge base for navigating SARS-CoV-2 genomic variants. Viruses 2023, 15, 1391. [Google Scholar] [CrossRef]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: Evidence for ongoing viral adaptation. J. Gen. Virol. 2021, 102, 001584. [Google Scholar] [CrossRef]
- Panzera, Y.; Calleros, L.; Goni, N.; Marandino, A.; Techera, C.; Grecco, S.; Ramos, N.; Frabasile, S.; Tomas, G.; Condon, E.; et al. Consecutive deletions in a unique Uruguayan SARS-CoV-2 lineage evidence the genetic variability potential of accessory genes. PLoS ONE 2022, 17, e0263563. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Charyyeva, A.; Sidorenko, I.A.; Babenko, V.N.; Yurchenko, V. Frequent recombination events in Leishmania donovani: Mining population data. Pathogens 2020, 9, 572. [Google Scholar] [CrossRef]
- Lovett, S.T.; Gluckman, T.J.; Simon, P.J.; Sutera, V.A., Jr.; Drapkin, P.T. Recombination between repeats in Escherichia coli by a recA-independent, proximity-sensitive mechanism. Mol. Gen. Genet. 1994, 245, 294–300. [Google Scholar] [CrossRef]
- Lovett, S.T. Encoded errors: Mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol. Microbiol. 2004, 52, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Bzymek, M.; Saveson, C.J.; Feschenko, V.V.; Lovett, S.T. Slipped misalignment mechanisms of deletion formation: In vivo susceptibility to nucleases. J. Bacteriol. 1999, 181, 477–482. [Google Scholar] [CrossRef]
- Hu, X.; Worton, R.G. Partial gene duplication as a cause of human disease. Hum. Mutat. 1992, 1, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Bard, J.D.; Triche, T.J.; Judkins, A.R.; Biegel, J.A.; Gai, X. Rapidly emerging SARS-CoV-2 B.1.1.7 sub-lineage in the United States of America with spike protein D178H and membrane protein V70L mutations. Emerg. Microbes Infect. 2021, 10, 1293–1299. [Google Scholar] [CrossRef]
- Akaishi, T.; Fujiwara, K.; Ishii, T. Insertion/deletion hotspots in the Nsp2, Nsp3, S1, and ORF8 genes of SARS-related coronaviruses. BMC Ecol. Evol. 2022, 22, 123. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Zheng, G.X.; Brankamp, R.G.; Allen, K.N. On the deletion of inverted repeated DNA in Escherichia coli: Effects of length, thermal stability, and cruciform formation in vivo. Genetics 1991, 129, 991–1005. [Google Scholar] [CrossRef]
- Gordenin, D.A.; Resnick, M.A. Yeast ARMs (DNA at-risk motifs) can reveal sources of genome instability. Mutat. Res. 1998, 400, 45–58. [Google Scholar] [CrossRef]
- Wakeley, J. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 1996, 11, 158–162. [Google Scholar] [CrossRef]
- Vogel, F.; Rohrborn, G. Amino-acid substitutions in haemoglobins and the mutation process. Nature 1966, 210, 116–117. [Google Scholar] [CrossRef]
- Fitch, W.M. Evidence suggesting a non-random character to nucleotide replacements in naturally occurring mutations. J. Mol. Biol. 1967, 26, 499–507. [Google Scholar] [CrossRef]
- Gojobori, T.; Li, W.H.; Graur, D. Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol. 1982, 18, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Wu, C.I.; Luo, C.C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984, 21, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Sankoff, D.; Morel, C.; Cedergren, R.J. Evolution of 5S RNA and the non-randomness of base replacement. Nat. New Biol. 1973, 245, 232–234. [Google Scholar] [CrossRef]
- Hixson, J.E.; Brown, W.M. A comparison of the small ribosomal RNA genes from the mitochondrial DNA of the great apes and humans: Sequence, structure, evolution, and phylogenetic implications. Mol. Biol. Evol. 1986, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Ansari, M.A. Extensive C->U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage- or host-mediated editing of viral RNA. PLoS Pathog. 2021, 17, e1009596. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Ode, H.; Kubota, M.; Kasahara, T.; Matsuoka, K.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res. 2023, 51, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Pecori, R.; Di Giorgio, S.; Paulo Lorenzo, J.; Nina Papavasiliou, F. Functions and consequences of AID/APOBEC-mediated DNA and RNA deamination. Nat. Rev. Genet. 2022, 23, 505–518. [Google Scholar] [CrossRef]
- Ooms, M.; Krikoni, A.; Kress, A.K.; Simon, V.; Munk, C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012, 86, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef]
- Kim, K.; Calabrese, P.; Wang, S.; Qin, C.; Rao, Y.; Feng, P.; Chen, X.S. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci. Rep. 2022, 12, 14972. [Google Scholar] [CrossRef]
- Song, Y.; He, X.; Yang, W.; Wu, Y.; Cui, J.; Tang, T.; Zhang, R. Virus-specific editing identification approach reveals the landscape of A-to-I editing and its impacts on SARS-CoV-2 characteristics and evolution. Nucleic Acids Res. 2022, 50, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Panchin, A.Y.; Panchin, Y.V. Excessive G-U transversions in novel allele variants in SARS-CoV-2 genomes. PeerJ 2020, 8, e9648. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, P.V.; Pavlov, Y.I.; Chilkova, O.; Rogozin, I.B.; Johansson, E.; Kunkel, T.A. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J. Biol. Chem. 2003, 278, 43770–43780. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef] [PubMed]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of oxidative stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) infection: A review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.D.W.; Magalis, B.R.; Kosakovsky Pond, S.L. Neutral theory and rapidly evolving viral pathogens. Mol. Biol. Evol. 2018, 35, 1348–1354. [Google Scholar] [CrossRef]
- Jackson, A.P.; Berry, A.; Aslett, M.; Allison, H.C.; Burton, P.; Vavrova-Anderson, J.; Brown, R.; Browne, H.; Corton, N.; Hauser, H.; et al. Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proc. Natl. Acad. Sci. USA 2012, 109, 3416–3421. [Google Scholar] [CrossRef]
- Bangs, J.D. Evolution of antigenic variation in african trypanosomes: Variant surface glycoprotein expression, structure, and function. Bioessays 2018, 40, e1800181. [Google Scholar] [CrossRef]
- Rehermann, B. Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. J. Clin. Invest. 2009, 119, 1745–1754. [Google Scholar] [CrossRef]
- Frost, S.D.; Wrin, T.; Smith, D.M.; Kosakovsky Pond, S.L.; Liu, Y.; Paxinos, E.; Chappey, C.; Galovich, J.; Beauchaine, J.; Petropoulos, C.J.; et al. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl. Acad. Sci. USA 2005, 102, 18514–18519. [Google Scholar] [CrossRef]
- Woelk, C.H.; Holmes, E.C. Reduced positive selection in vector-borne RNA viruses. Mol. Biol. Evol. 2002, 19, 2333–2336. [Google Scholar] [CrossRef]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012, 8, e1002529. [Google Scholar] [CrossRef]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Sepulcri, C.; Dentone, C.; Mikulska, M.; Bruzzone, B.; Lai, A.; Fenoglio, D.; Bozzano, F.; Bergna, A.; Parodi, A.; Altosole, T.; et al. The longest persistence of viable SARS-CoV-2 with recurrence of viremia and relapsing symptomatic COVID-19 in an immunocompromised patient—A case study. Open Forum Infect. Dis. 2021, 8, ofab217. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe 2021, 29, 819–833. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Computational inference of selection underlying the evolution of the novel coronavirus, severe acute respiratory syndrome coronavirus 2. J. Virol. 2020, 94, e00411–e00420. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, E275. [Google Scholar] [CrossRef]
- Maisnier-Patin, S.; Andersson, D.I. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004, 155, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.S.; Kemena, C.; Vlasov, P.K.; Notredame, C.; Kondrashov, F.A. Epistasis as the primary factor in molecular evolution. Nature 2012, 490, 535–538. [Google Scholar] [CrossRef]
- Kannan, S.; Shaik Syed Ali, P.; Sheeza, A. Omicron (B.1.1.529)—Variant of concern—Molecular profile and epidemiology: A mini review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8019–8022. [Google Scholar] [CrossRef] [PubMed]
- Muth, D.; Corman, V.M.; Roth, H.; Binger, T.; Dijkman, R.; Gottula, L.T.; Gloza-Rausch, F.; Balboni, A.; Battilani, M.; Rihtaric, D.; et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 2018, 8, 15177. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L. On the origins of SARS-CoV-2. Nat. Med. 2021, 27, 9. [Google Scholar] [CrossRef]
- Cyranoski, D. Profile of a killer: The complex biology powering the coronavirus pandemic. Nature 2020, 581, 22–26. [Google Scholar] [CrossRef]
- Postnikova, O.A.; Uppal, S.; Huang, W.; Kane, M.A.; Villasmil, R.; Rogozin, I.B.; Poliakov, E.; Redmond, T.M. The functional consequences of the novel ribosomal pausing site in SARS-CoV-2 spike glycoprotein RNA. Int. J. Mol. Sci. 2021, 22, 6490. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Tian, M.; Wu, N.; Yang, X.; Qi, J.; Ren, W.; Li, F.; Bian, H. SARS-CoV-2 and emerging variants: Unmasking structure, function, infection, and immune escape mechanisms. Front. Cell Infect. Microbiol. 2022, 12, 869832. [Google Scholar] [CrossRef]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Simas, M.C.C.; Costa, S.M.; Gomes, P.; Cruz, N.; Correa, I.A.; de Souza, M.R.M.; Dornelas-Ribeiro, M.; Nogueira, T.L.S.; Santos, C.; Hoffmann, L.; et al. Evaluation of SARS-CoV-2 ORF7a deletions from COVID-19-positive individuals and its impact on virus spread in cell culture. Viruses 2023, 15, 801. [Google Scholar] [CrossRef]
- Aroldi, A.; Angaroni, F.; D’Aliberti, D.; Spinelli, S.; Crespiatico, I.; Crippa, V.; Piazza, R.; Graudenzi, A.; Ramazzotti, D. Characterization of SARS-CoV-2 mutational signatures from 1.5+ million raw sequencing samples. Viruses 2022, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Eriksen, A.Z.; Moller, R.; Makovoz, B.; Uhl, S.A.; tenOever, B.R.; Blenkinsop, T.A. SARS-CoV-2 infects human adult donor eyes and hESC-derived ocular epithelium. Cell Stem Cell 2021, 28, 1205–1220. [Google Scholar] [CrossRef]
- Uppal, S.; Postnikova, O.; Villasmil, R.; Rogozin, I.B.; Bocharov, A.V.; Eggerman, T.L.; Poliakov, E.; Redmond, T.M. Low-Density Lipoprotein Receptor (LDLR) is involved in internalization of lentiviral particles pseudotyped with SARS-CoV-2 spike protein in ocular cells. Int. J. Mol. Sci. 2023, 24, 11860. [Google Scholar] [CrossRef]
- Widagdo, W.; Sooksawasdi Na Ayudhya, S.; Hundie, G.B.; Haagmans, B.L. Host determinants of MERS-CoV transmission and pathogenesis. Viruses 2019, 11, 280. [Google Scholar] [CrossRef]
- Cui, S.; Liu, Y.; Zhao, J.; Peng, X.; Lu, G.; Shi, W.; Pan, Y.; Zhang, D.; Yang, P.; Wang, Q. An updated review on SARS-CoV-2 infection in animals. Viruses 2022, 14, 1527. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Link |
|---|---|
| CDC strains | https://www.cdc.gov/coronavirus/2019-ncov/variants |
| NCBI | https://www.ncbi.nlm.nih.gov/activ |
| Nextstrain | https://nextstrain.org/ncov/gisaid/global/6m |
| GISAID | https://gisaid.org |
| SARS-CoV-2 mutation portal | http://sarscov2-mutation-portal.urv.cat |
| CoV-GLUE | https://cov-glue.cvr.gla.ac.uk |
| UShER | https://genome.ucsc.edu/cgi-bin/hgPhyloPlace |
| GESS | https://wan-bioinfo.shinyapps.io/GESS/ |
| CoVigator | https://github.com/TRON-bioinformatics/covigator |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogozin, I.B.; Saura, A.; Poliakov, E.; Bykova, A.; Roche-Lima, A.; Pavlov, Y.I.; Yurchenko, V. Properties and Mechanisms of Deletions, Insertions, and Substitutions in the Evolutionary History of SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 3696. https://doi.org/10.3390/ijms25073696
Rogozin IB, Saura A, Poliakov E, Bykova A, Roche-Lima A, Pavlov YI, Yurchenko V. Properties and Mechanisms of Deletions, Insertions, and Substitutions in the Evolutionary History of SARS-CoV-2. International Journal of Molecular Sciences. 2024; 25(7):3696. https://doi.org/10.3390/ijms25073696
Chicago/Turabian StyleRogozin, Igor B., Andreu Saura, Eugenia Poliakov, Anastassia Bykova, Abiel Roche-Lima, Youri I. Pavlov, and Vyacheslav Yurchenko. 2024. "Properties and Mechanisms of Deletions, Insertions, and Substitutions in the Evolutionary History of SARS-CoV-2" International Journal of Molecular Sciences 25, no. 7: 3696. https://doi.org/10.3390/ijms25073696
APA StyleRogozin, I. B., Saura, A., Poliakov, E., Bykova, A., Roche-Lima, A., Pavlov, Y. I., & Yurchenko, V. (2024). Properties and Mechanisms of Deletions, Insertions, and Substitutions in the Evolutionary History of SARS-CoV-2. International Journal of Molecular Sciences, 25(7), 3696. https://doi.org/10.3390/ijms25073696