
Immunological Signatures in Blood and Urine in 80 Individuals Hospitalized during the Initial Phase of COVID-19 Pandemic with Quantified Nicotine Exposure

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
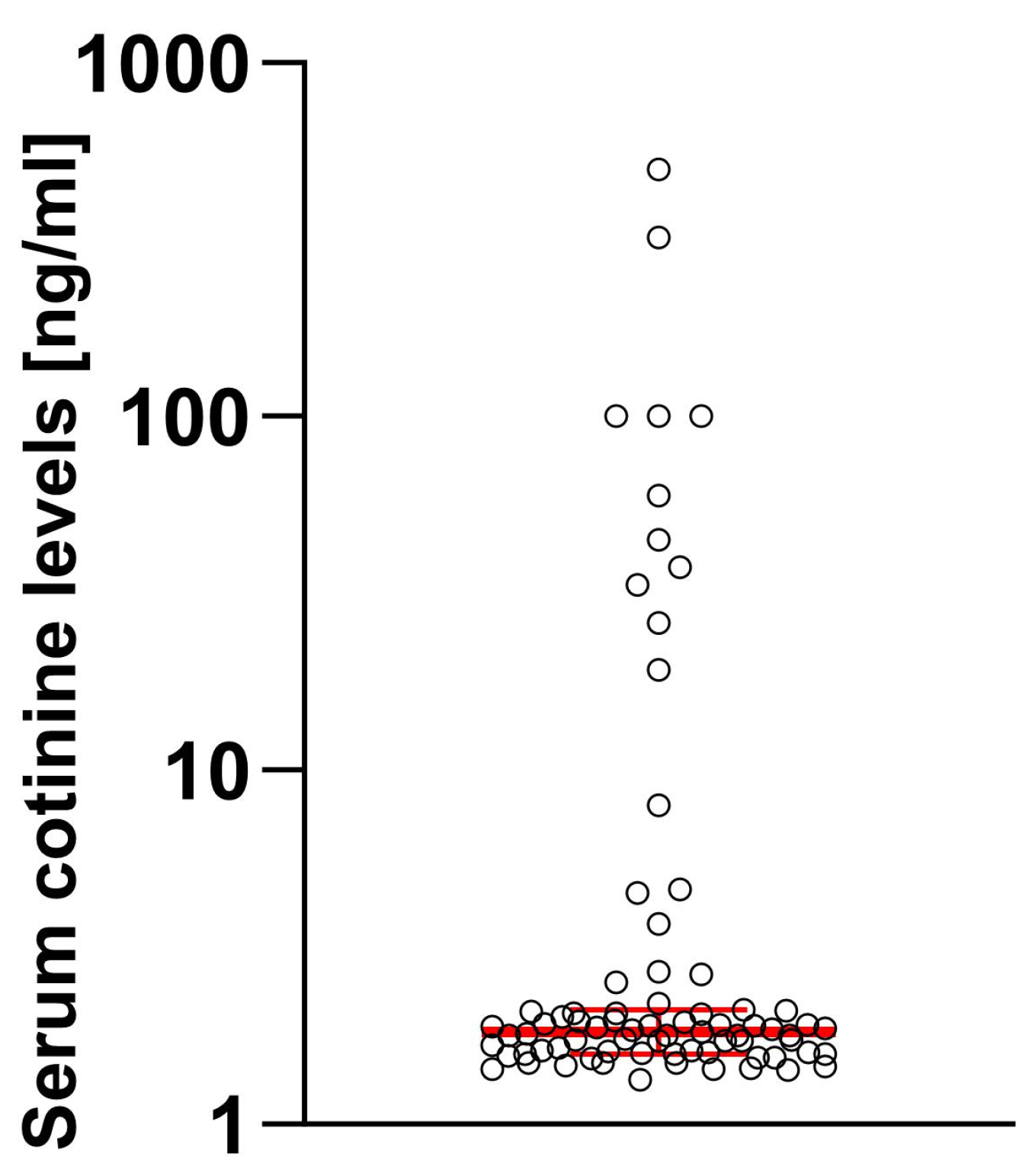
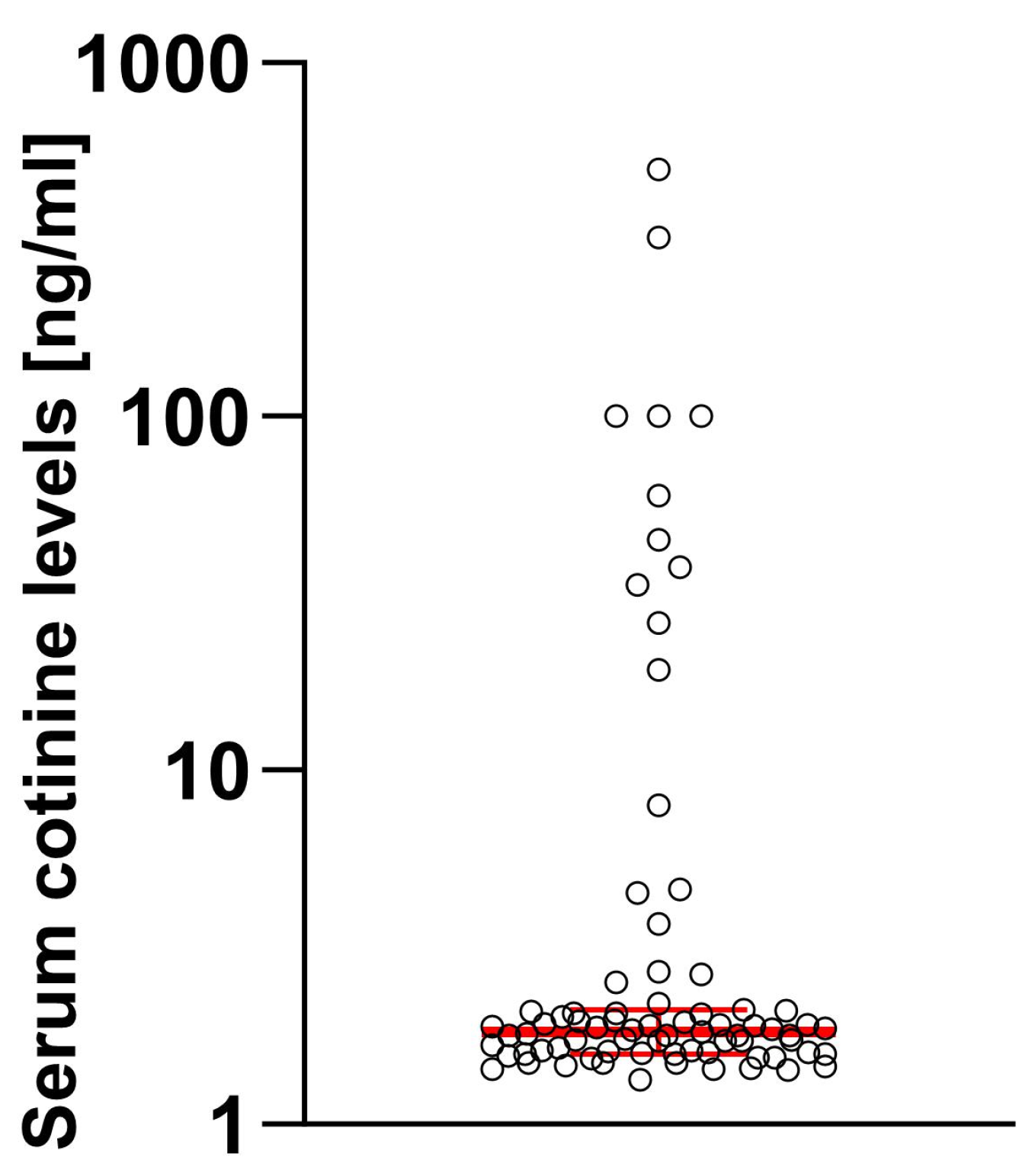
2.1. Characteristics of the Sample and Prevalence of EHR-Reported and Serum-Tested Cotinine Levels
2.2. Effect of Smoking on Demographical Variables of the Patients
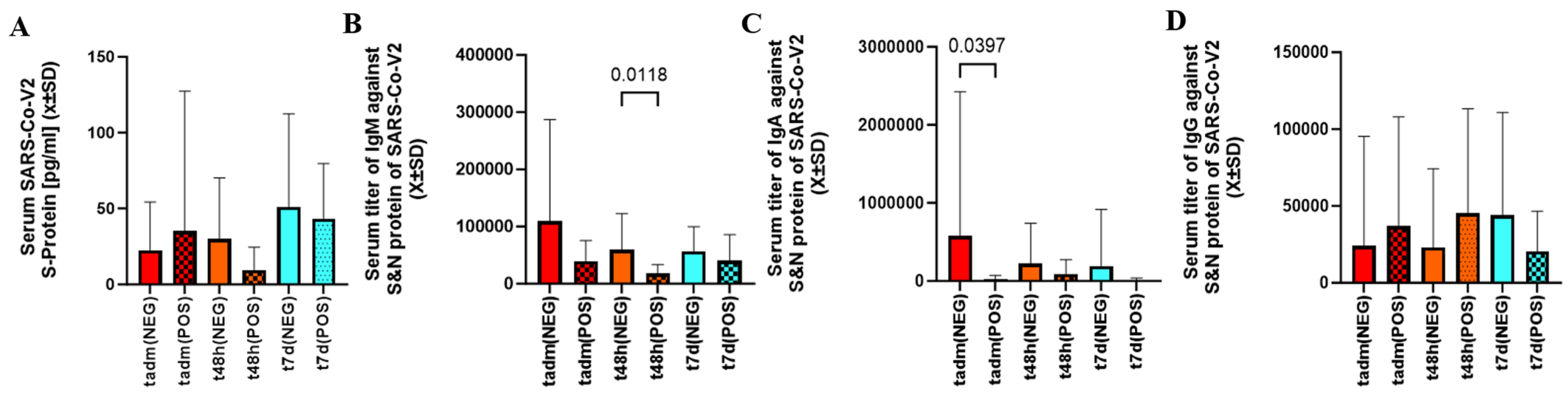
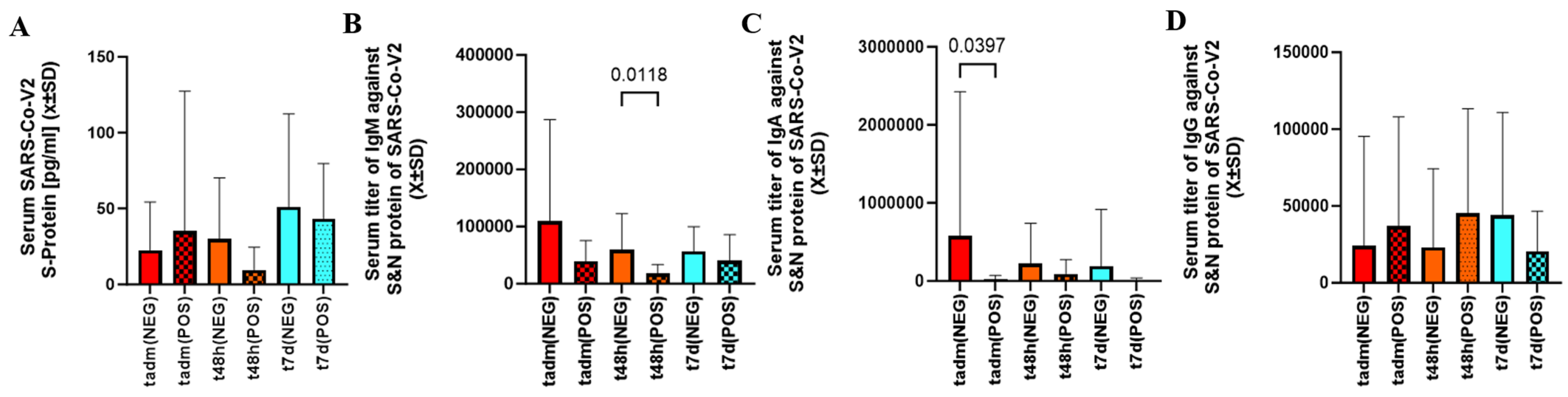
2.3. Viral Load, Immunoglobulin Titers, Level of Tissue Necrosis, and Non-Specific Inflammatory Markers Assessed in Patients with Cotinine Levels in SARS-CoV-2 Infection
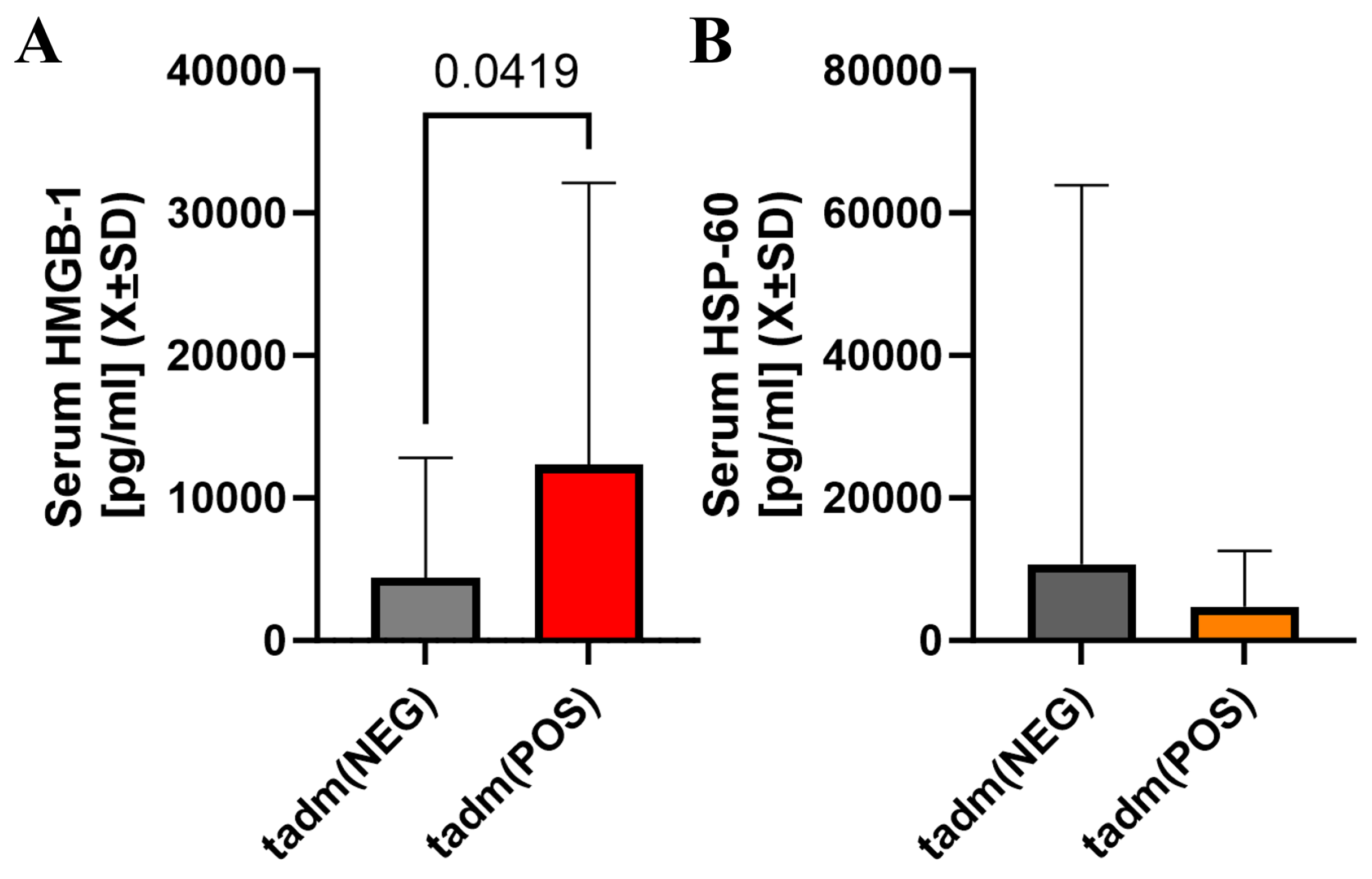
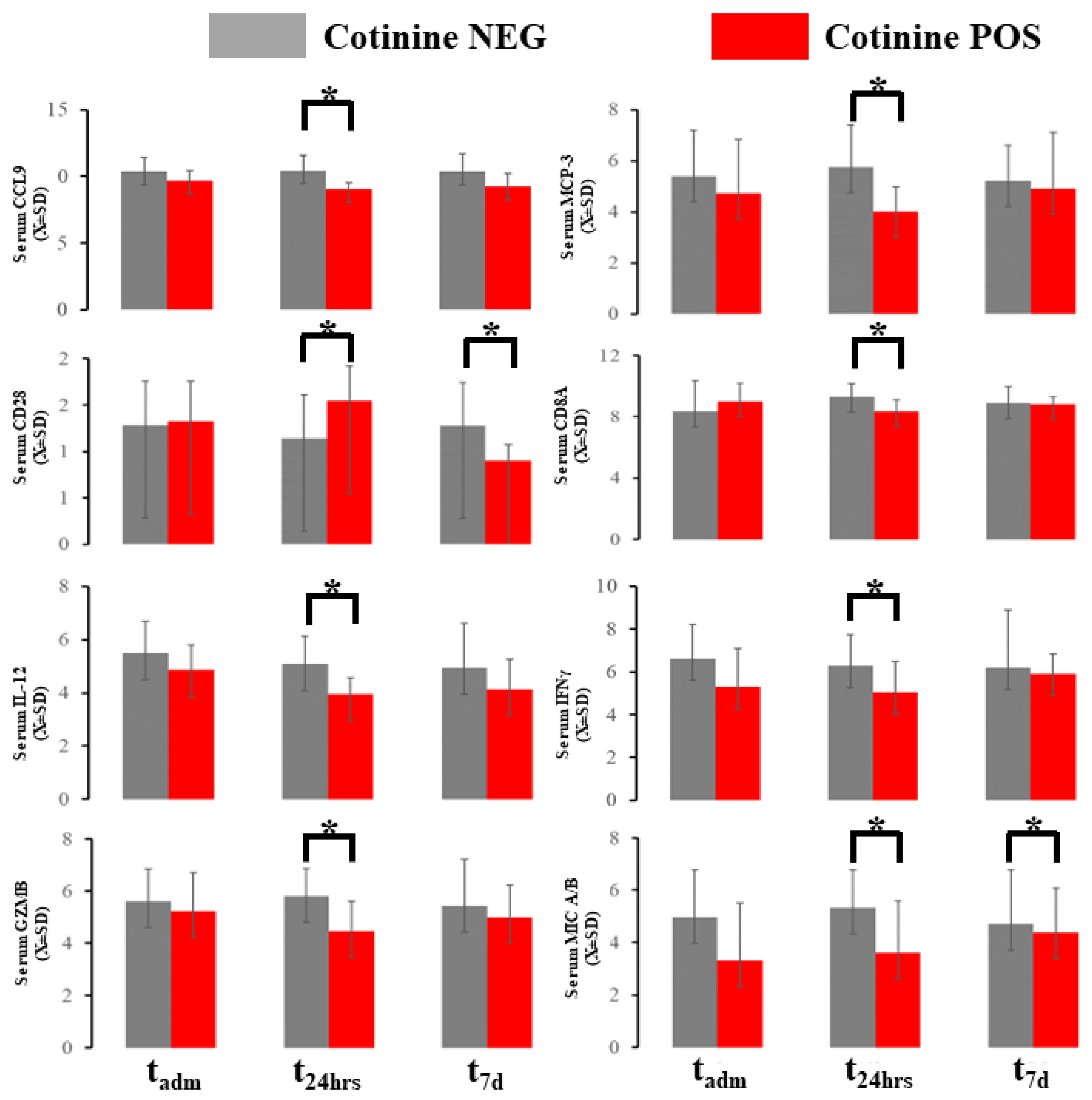
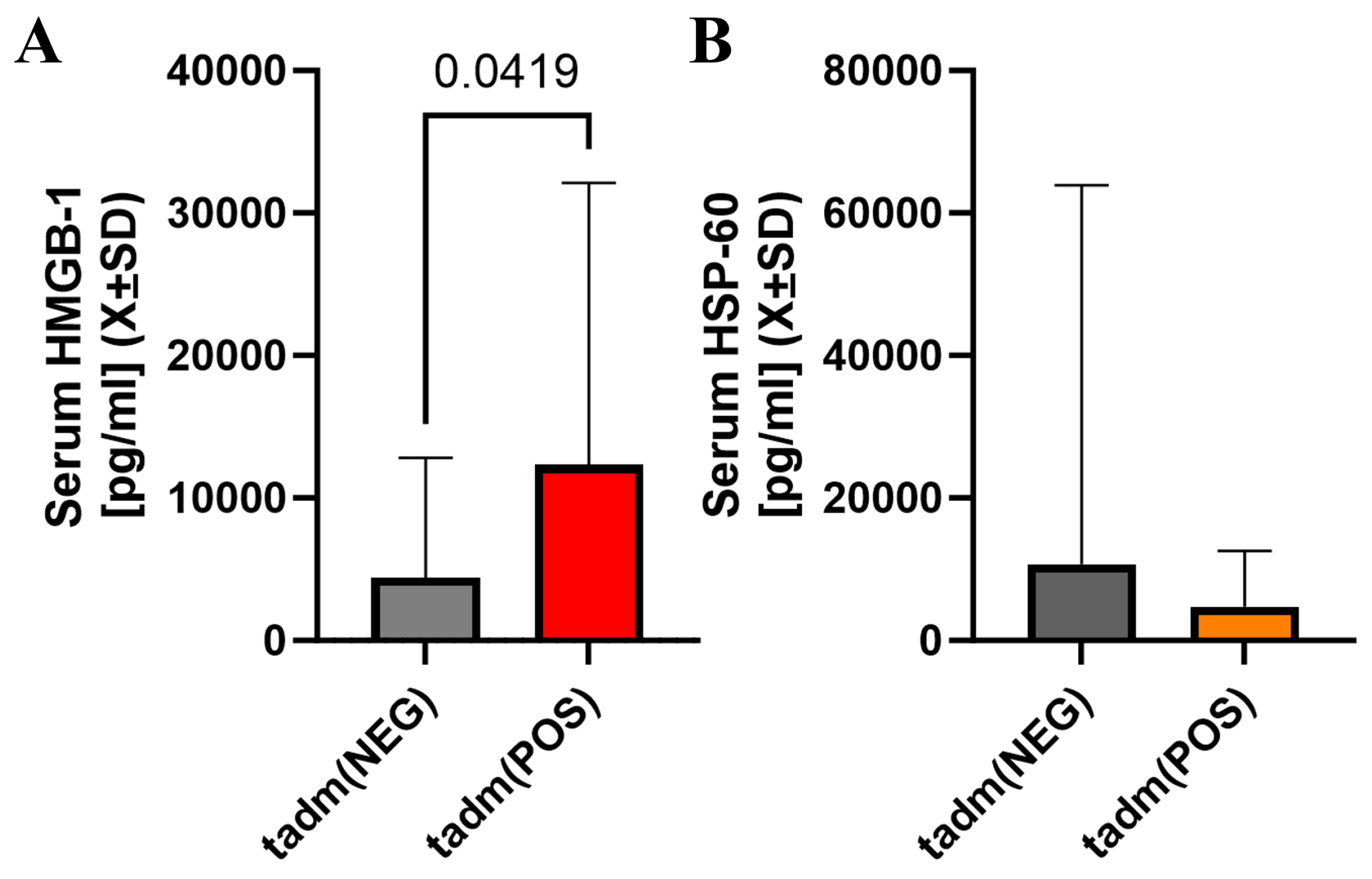
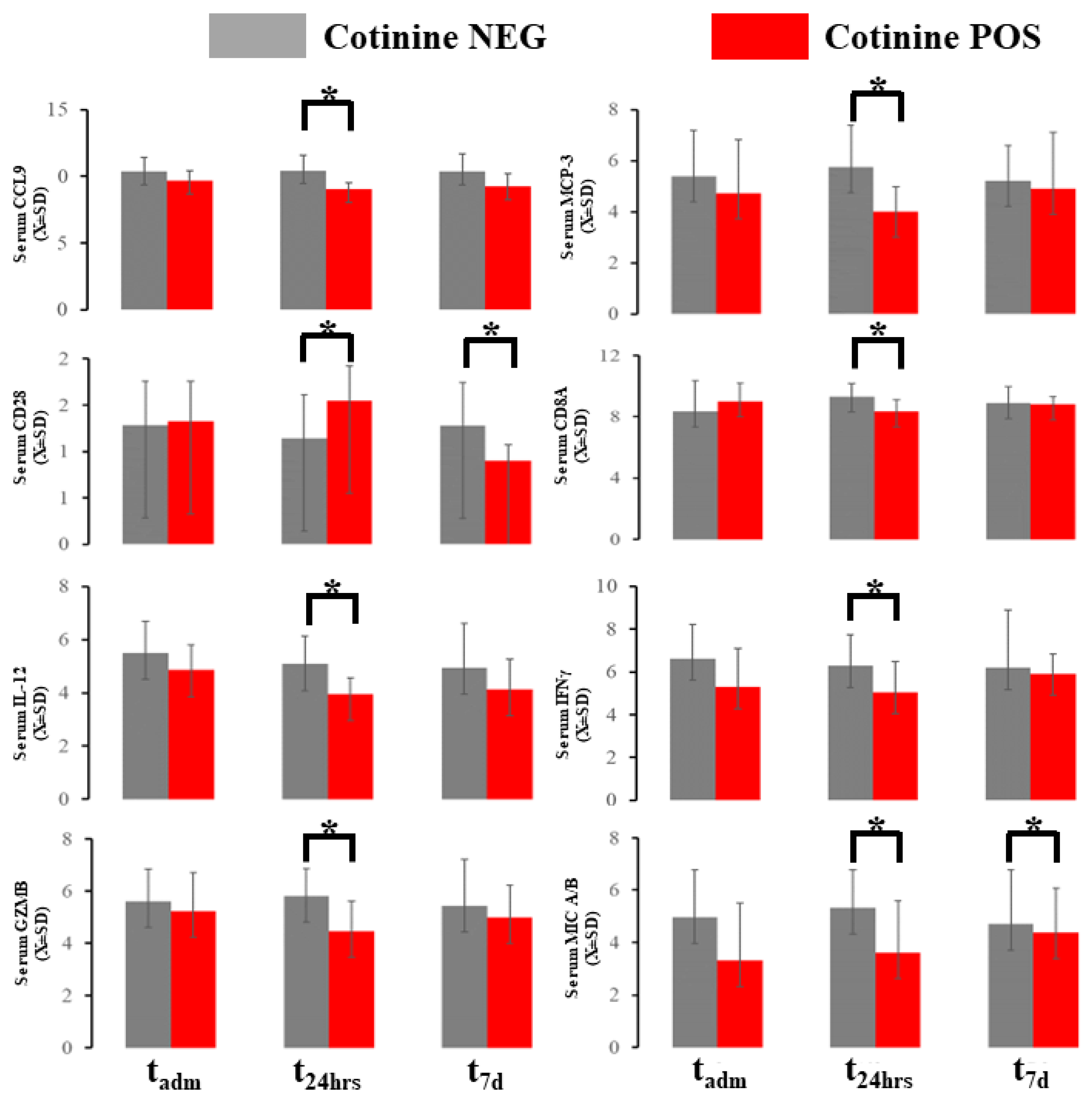
2.4. Acute Immunological Blood of Blood Serum in Cotinine-Positive Individuals at Admission
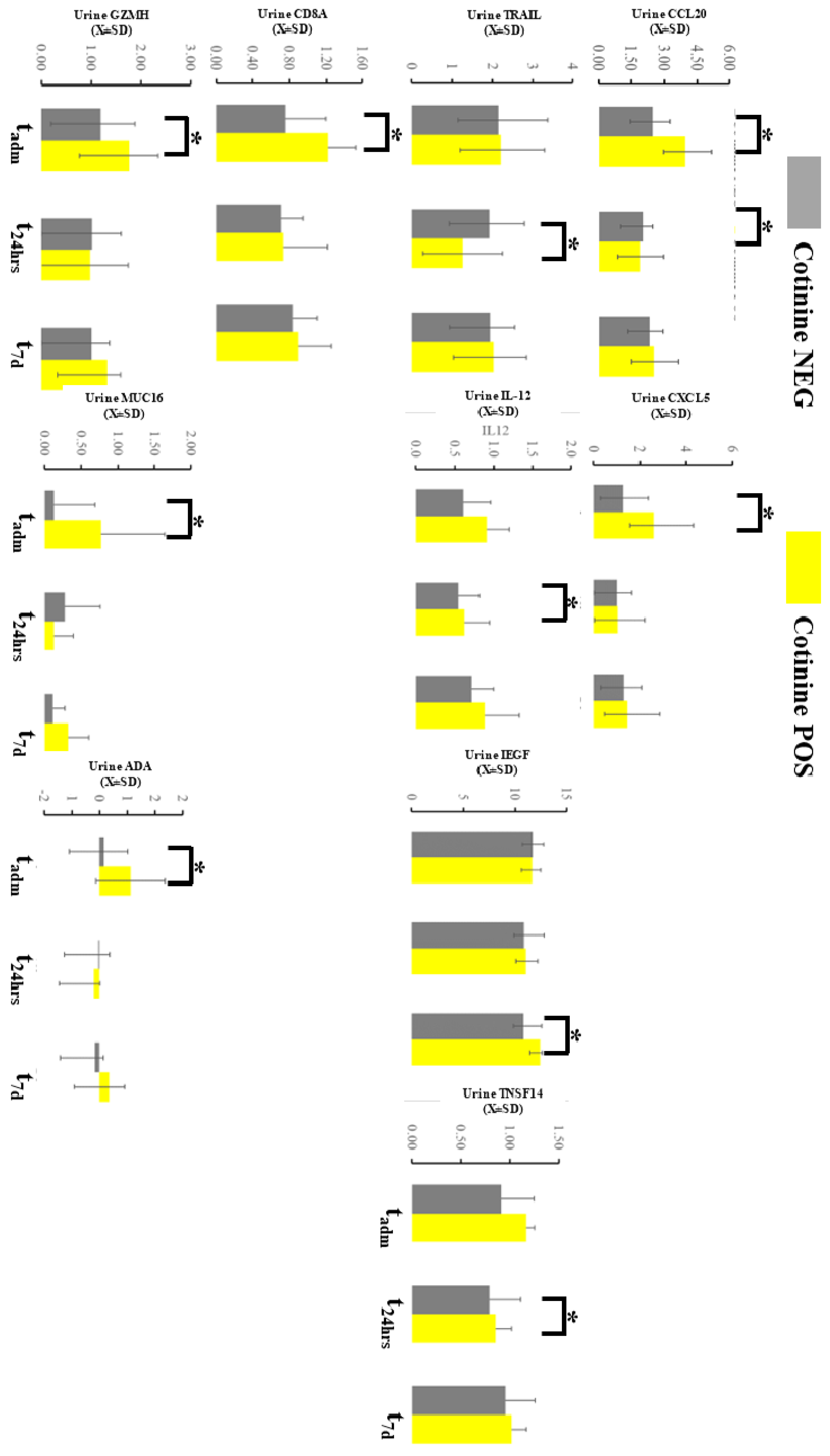
2.5. Urine Profile during COVID-19
3. Discussion
4. Materials and Methods
4.1. Ethical Concerns
4.2. Study Cohort and Clinical Data Extraction
4.3. Study Procedure and Sample Collection
4.4. Assessment of the Viral Load, the Release of Danger-Associated Molecular Patterns, and Non-Specific Immune Response
4.5. Immune Markers Testing
4.6. Immunological Profiling with Olink
4.7. Details of Tobacco Exposure
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santoro, A.; Tomino, C.; Prinzi, G.; Lamonaca, P.; Cardaci, V.; Fini, M.; Russo, P. Tobacco smoking: Risk to develop addiction, chronic obstructive pulmonary disease, and lung cancer. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 39–52. [Google Scholar] [CrossRef]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef]
- Lee, K.M.; Renne, R.A.; Harbo, S.J.; Clark, M.L.; Johnson, R.E.; Gideon, K.M. 3-week inhalation exposure to cigarette smoke and/or lipopolysaccharide in AKR/J mice. Inhal. Toxicol. 2007, 19, 23–35. [Google Scholar] [CrossRef]
- Vernooy, J.H.; Bracke, K.R.; Drummen, N.E.; Pauwels, N.S.; Zabeau, L.; van Suylen, R.J.; Tavernier, J.; Joos, G.F.; Wouters, E.F.; Brusselle, G.G. Leptin modulates innate and adaptive immune cell recruitment after cigarette smoke exposure in mice. J. Immunol. 2010, 184, 7169–7177. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Parrish, W.R.; Rosas-Ballina, M.; Ochani, M.; Puerta, M.; Ochani, K.; Chavan, S.; Al-Abed, Y.; Tracey, K.J. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav. Immun. 2009, 23, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Pena, G.; Cai, B.; Ramos, L.; Vida, G.; Deitch, E.A.; Ulloa, L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J. Immunol. 2011, 187, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Fink, B.; Thürmann, L.; Eszlinger, M.; Herberth, G.; Lehmann, I. Tobacco smoking differently influences cell types of the innate and adaptive immune system—Indications from CpG site methylation. Clin. Epigenet. 2016, 8, 83. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Samet, J.; Soleimanpour, N.; Chaffee, B.W. Biomarkers of Improved Health Outcomes after Smoking Cessation. Addict. Neurosci. 2022, 5, 100054. [Google Scholar] [CrossRef]
- Huttunen, R.; Heikkinen, T.; Syrjänen, J. Smoking and the outcome of infection. J. Intern. Med. 2011, 269, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, V.; Murray, R.L.; Hunter, A.; Lim, W.S.; McKeever, T.M. Effect of tobacco smoking on the risk of developing community acquired pneumonia: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0220204. [Google Scholar] [CrossRef] [PubMed]
- Bello, S.; Menéndez, R.; Antoni, T.; Reyes, S.; Zalacain, R.; Capelastegui, A.; Aspa, J.; Borderías, L.; Martin-Villasclaras, J.J.; Alfageme, I. Tobacco smoking increases the risk for death from pneumococcal pneumonia. Chest 2014, 146, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Oldridge, D.A.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, 1210. [Google Scholar] [CrossRef]
- Marik, P.E.; Iglesias, J.; Varon, J.; Kory, P. A scoping review of the pathophysiology of COVID-19. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211048026. [Google Scholar] [CrossRef]
- Albini, A.; Di Guardo, G.; Noonan, D.M.; Lombardo, M. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: Implications for ACE-inhibitor- and angiotensin II receptor blocker-based cardiovascular therapies. Intern. Emerg. Med. 2020, 15, 759–766. [Google Scholar] [CrossRef]
- Jain, V.; Yuan, J.-M. Predictive symptoms and comorbidities for severe COVID-19 and intensive care unit admission: A systematic review and meta-analysis. Int. J. Public Health 2020, 65, 533–546. [Google Scholar] [CrossRef]
- Eketunde, A.O.; Mellacheruvu, S.P.; Oreoluwa, P. A Review of Postmortem Findings in Patients With COVID-19. Cureus 2020, 12, e9438. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Magro, G. SARS-CoV-2 and COVID-19: Is interleukin-6 (IL-6) the ‘culprit lesion’ of ARDS onset? What is there besides Tocilizumab? SGP130Fc. Cytokine X 2020, 2, 100029. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef] [PubMed]
- Nidadavolu, L.; Walston, J. Underlying Vulnerabilities to the Cytokine Storm and Adverse COVID-19 Outcomes in the Aging Immune System. J. Gerontology. Ser. A Biol. Sci. Med. Sci. 2020, 76, e13–e18. [Google Scholar] [CrossRef] [PubMed]
- Remy, K.E.; Mazer, M.; Striker, D.A.; Ellebedy, A.H.; Walton, A.H.; Unsinger, J.; Blood, T.M.; Mudd, P.A.; Yi, D.J.; Mannion, D.A.; et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 2020, 5, e140329. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Froushani, S.M.A.; Ajami, M.; Mahmoudzadeh, M. Effect of nicotine on immune system function. Adv. Pharm. Bull. 2023, 13, 69. [Google Scholar]
- Maggi, F.; Rosellini, A.; Spezia, P.G.; Focosi, D.; Macera, L.; Lai, M.; Pistello, M.; de Iure, A.; Tomino, C.; Bonassi, S.; et al. Nicotine upregulates ACE2 expression and increases competence for SARS-CoV-2 in human pneumocytes. ERJ Open Res. 2021, 7, 00713-2020. [Google Scholar] [CrossRef]
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. How nicotine can inhibit cytokine storm in the lungs and prevent or lessen the severity of COVID-19 infection? Immunol. Lett. 2020, 224, 28–29. [Google Scholar] [CrossRef]
- Oakes, J.M.; Fuchs, R.M.; Gardner, J.D.; Lazartigues, E.; Yue, X. Nicotine and the renin-angiotensin system. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2018, 315, R895–R906. [Google Scholar] [CrossRef]
- Samet, J.M. Tobacco Products and the Risks of SARS-CoV-2 Infection and COVID-19. Nicotine Tob. Res. 2020, 22, S93–S95. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Rodda, L.B.; Netland, J.; Shehata, L.; Pruner, K.B.; Morawski, P.M.; Thouvenel, C.; Takehara, K.K.; Eggenberger, J.; Hemann, E.A.; Waterman, H.R.; et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Yamaguchi, N.H. Smoking, immunity, and DNA damage. Transl. Lung Cancer Res. 2019, 8, S3–S6. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Krishnan, N.; Abroms, L.C.; Berg, C.J. The Impact of Tobacco Use on COVID-19 Outcomes: A Systematic Review. J. Smok. Cessat. 2022, 2022, 5474397. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, M.S.; Zagà, V.; Gallus, S.; D’Argenio, P.; Gorini, G. Tobacco smoking and COVID-19 pandemic: Old and new issues. A summary of the evidence from the scientific literature. Acta Biomed 2020, 91, 106–112. [Google Scholar] [CrossRef]
- Warner, D.O. Tobacco dependence in surgical patients. Curr. Opin. Anaesthesiol. 2007, 20, 279–283. [Google Scholar] [CrossRef]
- Klebanoff, M.A.; Levine, R.J.; Clemens, J.D.; DerSimonian, R.; Wilkins, D.G. Serum Cotinine Concentration and Self-reported Smoking during Pregnancy. Am. J. Epidemiol. 1998, 148, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Laudanski, K.; Okeke, T.; Hajj, J.; Siddiq, K.; Rader, D.J.; Wu, J.; Susztak, K. Longitudinal urinary biomarkers of immunological activation in COVID-19 patients without clinically apparent kidney disease versus acute and chronic failure. Sci. Rep. 2021, 11, 19675. [Google Scholar] [CrossRef]
- Xu, K.; Shang, N.; Levitman, A.; Corker, A.; Kudose, S.; Yaeh, A.; Neupane, U.; Stevens, J.; Mohan, S.; Sampogna, R.; et al. Urine test predicts kidney injury and death in COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Perezstable, E.J.; Benowitz, N.L.; Marin, G. Is serum cotinine a better measure of cigarette-smoking than self-report? Prev. Med. 1995, 24, 171–179. [Google Scholar] [CrossRef]
- Benowitz, N.L. Cotinine as a biomarker of environmental tobacco smoke exposure. Epidemiol. Rev. 1996, 18, 188–204. [Google Scholar] [CrossRef]
- Gorber, S.C.; Schofield-Hurwitz, S.; Hardt, J.; Levasseur, G.; Tremblay, M. The accuracy of self-reported smoking: A systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tob. Res. 2009, 11, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.; Garg, A.; Yadav, P.; Jha, K.; Handa, S. Diagnostic Methods for Detection of Cotinine Level in Tobacco Users: A Review. J. Clin. Diagn. Res. JCDR 2016, 10, ZE04–ZE06. [Google Scholar] [CrossRef]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium; Donlin, L.T.; Raychaudhuri, S. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med. 2021, 13, 64. [Google Scholar] [CrossRef]
- Ellis, G.T.; Davidson, S.; Crotta, S.; Branzk, N.; Papayannopoulos, V.; Wack, A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. 2015, 16, 1203–1218. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Araya, P.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; Baxter, R.; Jordan, K.R.; Russell, S.; et al. Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2116730119. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef] [PubMed]
- Prebensen, C.; Lefol, Y.; Myhre, P.L.; Luders, T.; Jonassen, C.; Blomfeldt, A.; Omland, T.; Nilsen, H.; Berdal, J.E. Longitudinal whole blood transcriptomic analysis characterizes neutrophil activation and interferon signaling in moderate and severe COVID-19. Sci. Rep. 2023, 13, 10368. [Google Scholar] [CrossRef] [PubMed]
- Loftus, T.J.; Ungaro, R.; Dirain, M.; Efron, P.A.; Mazer, M.B.; Remy, K.E.; Hotchkiss, R.S.; Zhong, L.; Bacher, R.; Starostik, P.; et al. Overlapping but Disparate Inflammatory and Immunosuppressive Responses to SARS-CoV-2 and Bacterial Sepsis: An Immunological Time Course Analysis. Front. Immunol. 2021, 12, 792448. [Google Scholar] [CrossRef]
- Tarbiah, N.; Todd, I.; Tighe, P.J.; Fairclough, L.C. Cigarette smoking differentially affects immunoglobulin class levels in serum and saliva: An investigation and review. Basic Clin. Pharmacol. Toxicol. 2019, 125, 474–483. [Google Scholar] [CrossRef]
- Gonzalez-Quintela, A.; Alende, R.; Gude, F.; Campos, J.; Rey, J.; Meijide, L.M.; Fernandez-Merino, C.; Vidal, C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin. Exp. Immunol. 2007, 151, 42–50. [Google Scholar] [CrossRef]
- Khan, S.R.; van der Burgh, A.C.; Peeters, R.P.; van Hagen, P.M.; Dalm, V.A.S.H.; Chaker, L. Determinants of Serum Immunoglobulin Levels: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 1103. [Google Scholar] [CrossRef]
- Roseman, C.; Truedsson, L.; Kapetanovic, M.C. The effect of smoking and alcohol consumption on markers of systemic inflammation, immunoglobulin levels and immune response following pneumococcal vaccination in patients with arthritis. Arthritis Res. Ther. 2012, 14, R170. [Google Scholar] [CrossRef]
- Gunsolley, J.C.; Pandey, J.P.; Quinn, S.M.; Tew, J.; Schenkein, H.A. The effect of race, smoking and immunoglobulin allotypes on IgG subclass concentrations. J. Periodontal Res. 1997, 32, 381–387. [Google Scholar] [CrossRef]
- Tollerud, D.J.; Weiss, S.T.; Brown, L.M.; Blattner, W.A.; Maloney, E.M.; Kurman, C.C.; Nelson, D.L.; Hoover, R.N. Racial differences in serum immunoglobulin levels: Relationship to cigarette smoking, t-cell subsets, and soluble interleukin-2 receptors. J. Clin. Lab. Anal. 1995, 9, 37–41. [Google Scholar] [CrossRef]
- Rao, D.R.; Maple, K.L.; Dettori, A.; Afolabi, F.; Francis, J.K.R.; Artunduaga, M.; Lieu, T.J.; Aldy, K.; Cao, D.J.; Hsu, S.; et al. Clinical Features of E-cigarette, or Vaping, Product Use-Associated Lung Injury in Teenagers. Pediatrics 2020, 146, e20194104. [Google Scholar] [CrossRef]
- Steigerwald, S.; Wong, P.O.; Cohen, B.E.; Ishida, J.H.; Vali, M.; Madden, E.; Keyhani, S. Smoking, Vaping, and Use of Edibles and Other Forms of Marijuana Among U.S. Adults. Ann. Intern. Med. 2018, 169, 890–892. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef]
- Bastos, P.G.; Knaus, W.A. APACHE III study: A summary. Intensive Care World 1991, 8, 35–38. [Google Scholar]
- Cleves, M.A.; Sanchez, N.; Draheim, M. Evaluation of two competing methods for calculating Charlson’s comorbidity index when analyzing short-term mortality using administrative data. J. Clin. Epidemiol. 1997, 50, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Laudanski, K.; Okeke, T.; Siddiq, K.; Hajj, J.; Restrepo, M.; Gullipalli, D.; Song, W.-C. A disturbed balance between blood complement protective factors (FH, ApoE) and common pathway effectors (C5a, TCC) in acute COVID-19 and during convalesce. Sci. Rep. 2022, 12, 13658. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Ashton, N.J.; Dobson, R.J.B.; Andersson, L.M.; Yilmaz, A.; Blennow, K.; Gisslen, M.; Zetterberg, H. Proteomic blood profiling in mild, severe and critical COVID-19 patients. Sci. Rep. 2021, 11, 6357. [Google Scholar] [CrossRef]
- Olink Proteomics Data Science Team. OlinkRPackage, GitHub, 2021. Available online: https://github.com/Olink-Proteomics/OlinkRPackage(accessed on 15 September 2023).
- Halpern, A. Critical Care Statistics. Available online: https://www.sccm.org/Communications/Critical-Care-Statistics (accessed on 15 September 2023).
- Nakagawa, S.; Cuthill, I.C. Effect size, confidence interval and statistical significance: A practical guide for biologists. Biol. Rev. 2007, 82, 591–605. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients (n = 80) | ||
|---|---|---|
| Demographics |
Cotininepositive vs. Cotininenegative | |
| Age * (years) [X ± SD] * | 60.4 ± 18.2 | ns |
| Gender (female) [%] * | 39.2% | ns |
| Race [%] * | 20.3% (Caucasian) 67.1% (African American) | ns |
| BMI [X ± SD] * | 32.1 ± 8.69 | ns |
| Comorbidities | ||
| CCI [X ± SD] | 6.1 ± 2.16 | ns |
| CVA [%] | 12.5% (n = 10) | ns |
| CHF [%] | 20.0% (n = 16) | ns |
| PVD [%] | 10.0% (n = 8) | ns |
| COPD [%] | 18.75% (n = 15) | ns |
| DM [%] | 36.25% (n = 29) | ns |
| CKD [%] | 25.0% (n = 20) | ns |
| ESRD [%] | 2.5% (n = 2) | ns |
| Hospital Trajectory | ||
| Length of Stay (days) [X + SD] | 12.9 ± 15.68 | ns |
| Admitted to the ICU (%) Duration of ICU stay (days) [X ± SD] | 50.6%; 8.49 ± 18.47 | ns |
| Mechanical ventilation (%) Duration of ICU stay (days) [X ± SD] | 27.8% 5.05 ± 14.09 | ns |
| ECMO (%) Duration of ECMO (days) [X ± SD] | 3.8% 1.9 ± 12.88 | ns |
| Treatment | ||
| Remdesivir [%] | 53.2% | ns |
| Steroids [%] | 63.3% | ns |
| Plasma [%] | 8.9% | ns |
| Outcome | ||
| Alive at 6 months [%] | 73.8% | ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudanski, K.; Mahmoud, M.A.; Ahmed, A.S.; Susztak, K.; Mathew, A.; Chen, J. Immunological Signatures in Blood and Urine in 80 Individuals Hospitalized during the Initial Phase of COVID-19 Pandemic with Quantified Nicotine Exposure. Int. J. Mol. Sci. 2024, 25, 3714. https://doi.org/10.3390/ijms25073714
Laudanski K, Mahmoud MA, Ahmed AS, Susztak K, Mathew A, Chen J. Immunological Signatures in Blood and Urine in 80 Individuals Hospitalized during the Initial Phase of COVID-19 Pandemic with Quantified Nicotine Exposure. International Journal of Molecular Sciences. 2024; 25(7):3714. https://doi.org/10.3390/ijms25073714
Chicago/Turabian StyleLaudanski, Krzysztof, Mohamed A. Mahmoud, Ahmed Sayed Ahmed, Kaitlin Susztak, Amal Mathew, and James Chen. 2024. "Immunological Signatures in Blood and Urine in 80 Individuals Hospitalized during the Initial Phase of COVID-19 Pandemic with Quantified Nicotine Exposure" International Journal of Molecular Sciences 25, no. 7: 3714. https://doi.org/10.3390/ijms25073714
APA StyleLaudanski, K., Mahmoud, M. A., Ahmed, A. S., Susztak, K., Mathew, A., & Chen, J. (2024). Immunological Signatures in Blood and Urine in 80 Individuals Hospitalized during the Initial Phase of COVID-19 Pandemic with Quantified Nicotine Exposure. International Journal of Molecular Sciences, 25(7), 3714. https://doi.org/10.3390/ijms25073714