
Dynamic Changes of the Gut Microbiota and Its Functional Metagenomic Potential during the Development of Non-Small Cell Lung Cancer
Abstract
:1. Introduction
2. Results
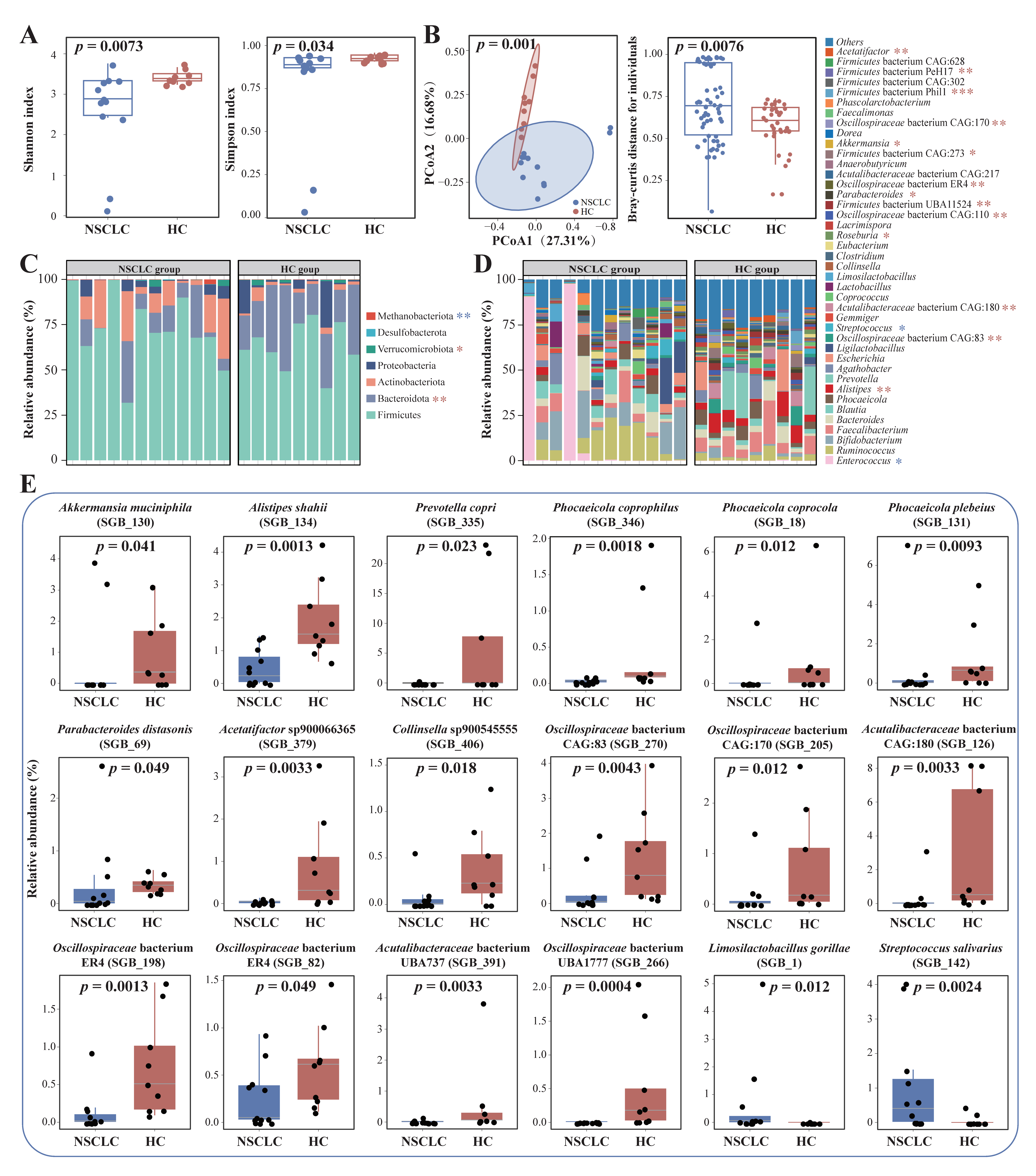
2.1. Fecal Microbiota Characteristics of Patients with NSCLC and Healthy Individuals
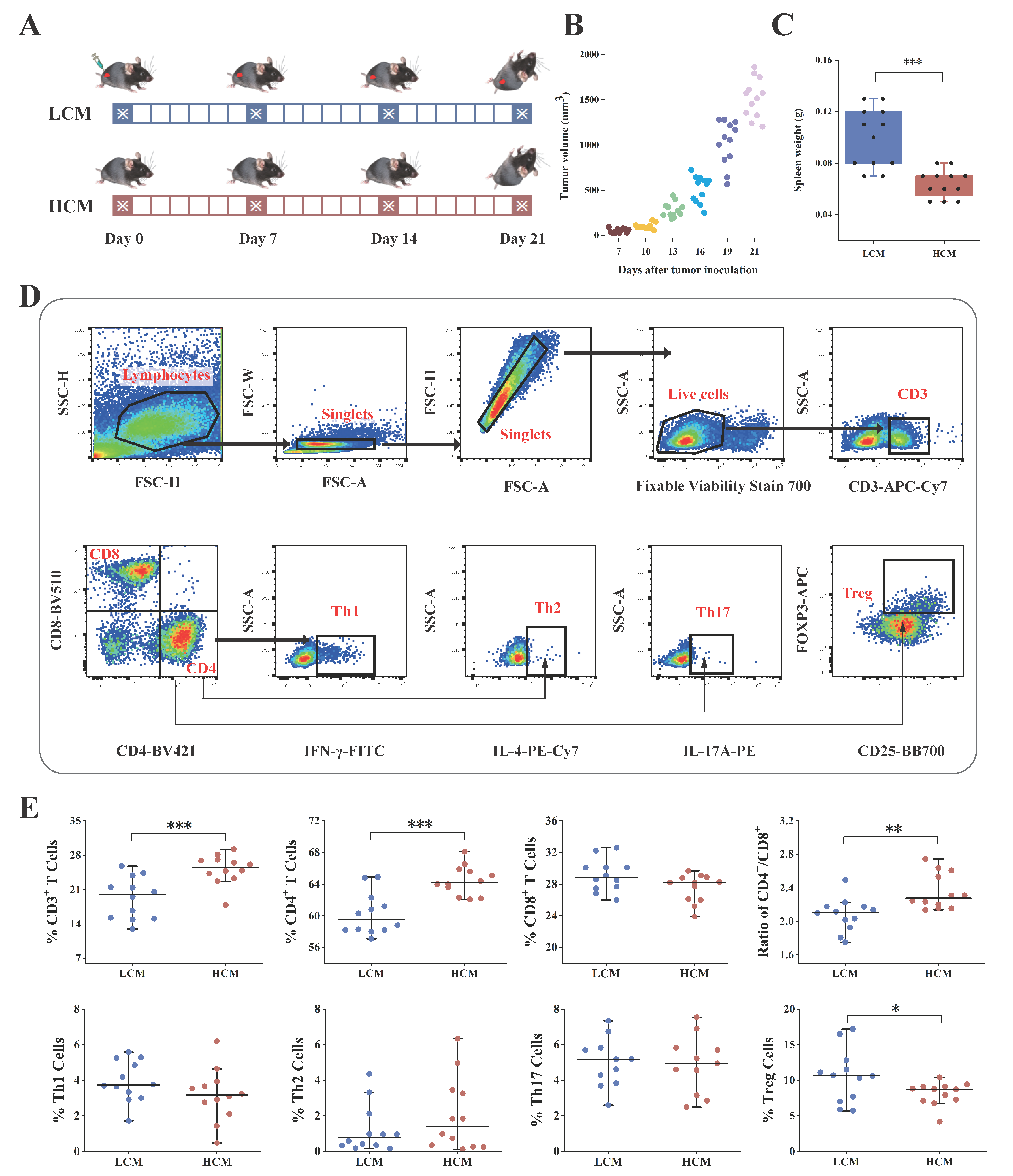
2.2. Lung Tumor Transplantation
2.3. T Cell Immunology in Lung Cancer and Healthy Mice
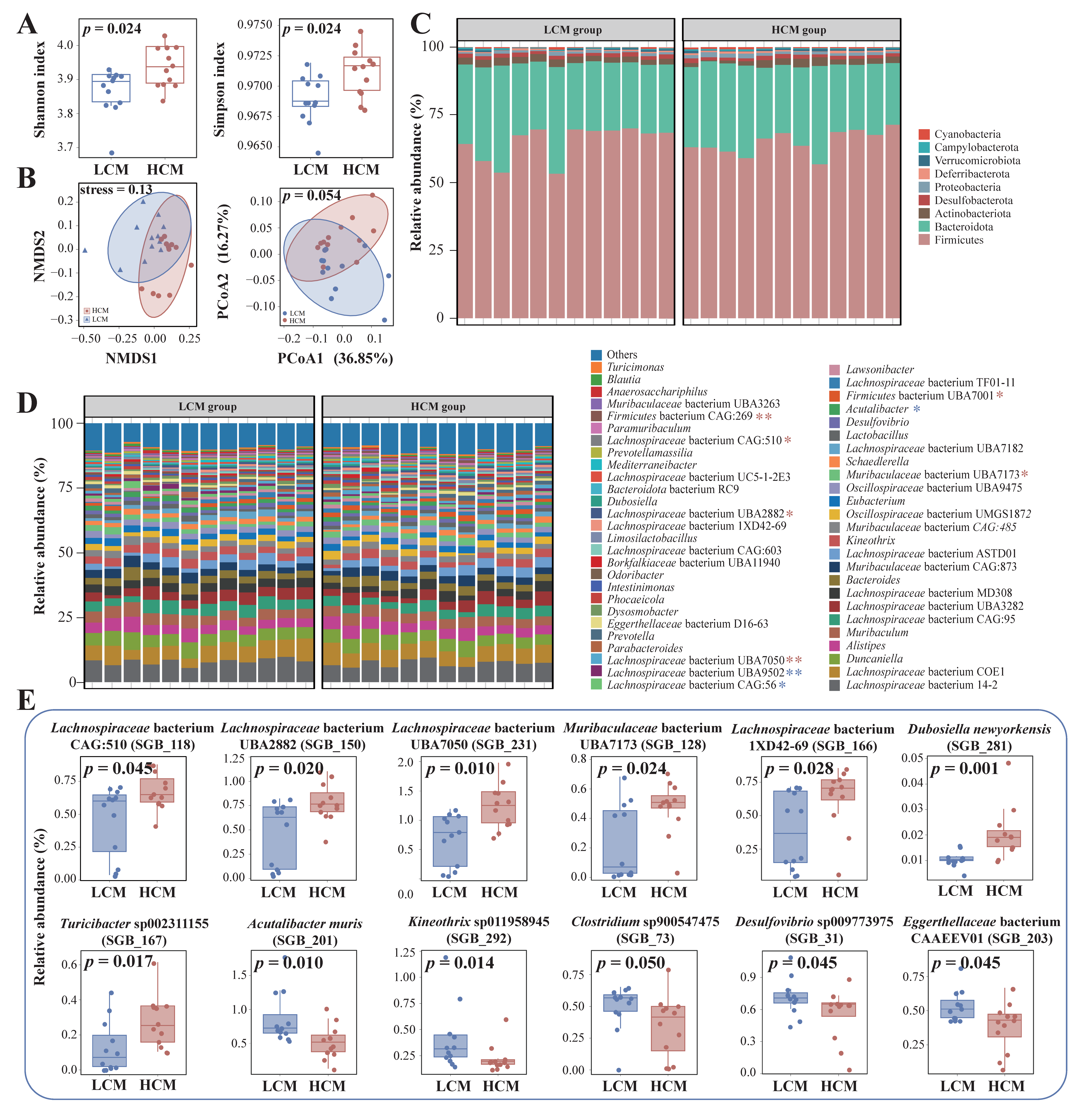
2.4. Fecal Microbiota in Mice with and without Lung Cancer
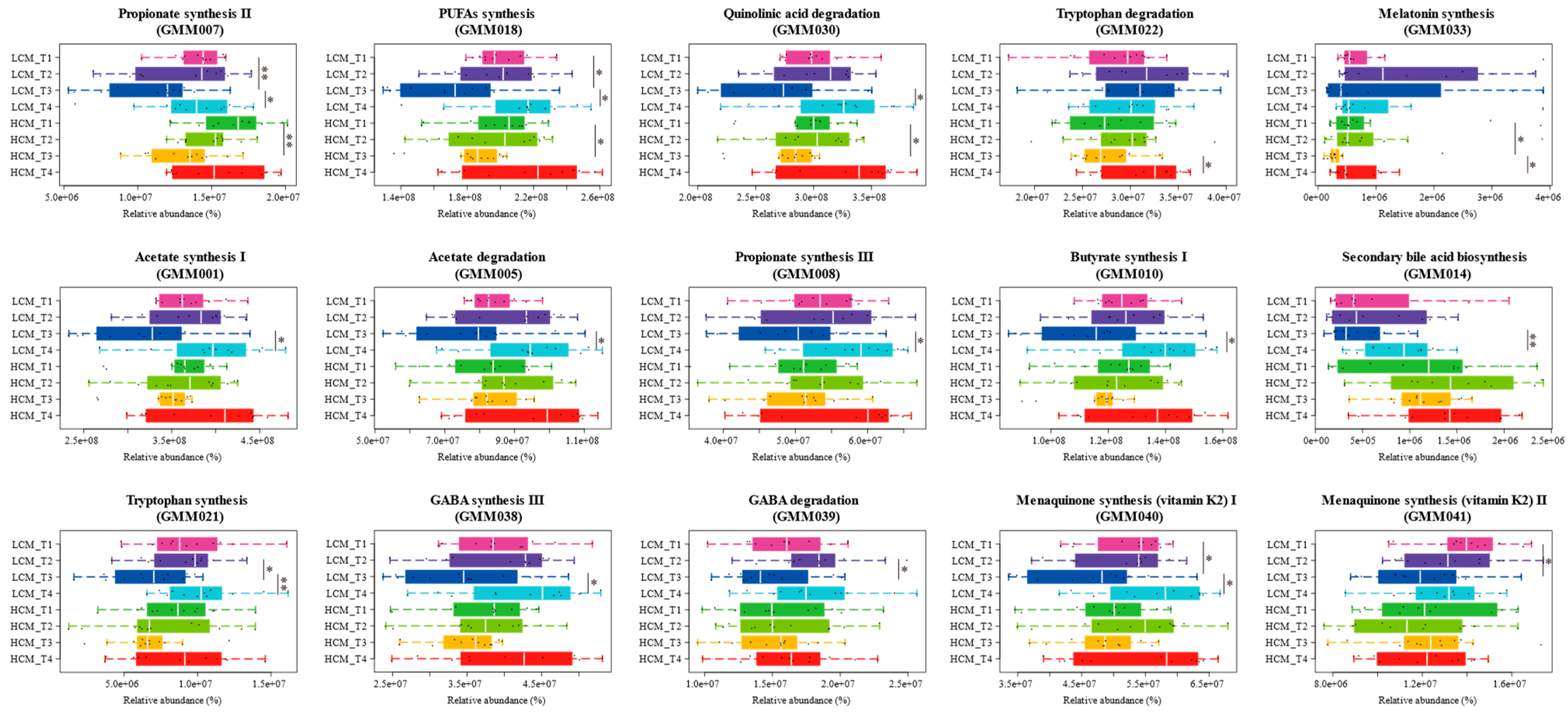
2.5. Fecal Microbiota at Different Tumor Progression Stages
2.6. SGB-Level Disease-Driven Gut Microbiota Responses
2.7. Changes in the Fecal Microbiota-Encoded Metabolic Modules in Lung Cancer Mice
3. Discussion
4. Materials and Methods
4.1. Subjects and Clinical Samples Collection
4.2. Cell Line and Culture
4.3. Mouse Model Construction
4.4. Flow Cytometry Analysis
4.5. Shotgun Metagenomics and Bioinformatics Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wang, X.-R.; Jiang, Z.-B.; Xu, C.; Meng, W.-Y.; Liu, P.; Zhang, Y.-Z.; Xie, C.; Xu, J.-Y.; Xie, Y.-J.; Liang, T.-L. Andrographolide suppresses non-small-cell lung cancer progression through induction of autophagy and antitumor immune response. Pharmacol. Res. 2022, 179, 106198. [Google Scholar] [CrossRef] [PubMed]
- Salloum, R.G.; Braithwaite, D. Expansion of guideline-recommended lung cancer screening eligibility: Implications for health equity of joint screening and cessation interventions. J. Thorac. Oncol. 2022, 17, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Popat, S.; Reinmuth, N.; De Ruysscher, D.; Kerr, K.; Peters, S. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii27–iii39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Joubert, P.; Ansari-Pour, N.; Zhao, W.; Hoang, P.H.; Lokanga, R.; Moye, A.L.; Rosenbaum, J.; Gonzalez-Perez, A.; Martinez-Jimenez, F. Genomic and evolutionary classification of lung cancer in never smokers. Nat. Genet. 2021, 53, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The human gut microbiome–a potential controller of wellness and disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, G.Y. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [Google Scholar] [CrossRef]
- Zhou, A.; Lei, Y.; Tang, L.; Hu, S.; Yang, M.; Wu, L.; Yang, S.; Tang, B. Gut microbiota: The emerging link to lung homeostasis and disease. J. Bacteriol. 2021, 203, e00454-20. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.; Gourgue, F.; Cani, P. Adipose Tissue metabolism and Cancer progression: Novel insights from gut microbiota? Curr. Pathobiol. Rep. 2017, 5, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Chen, G.; Ren, Y.; Zheng, T.; Shen, C.; Li, M.; Chen, X.; Zhai, H.; Li, Z.; Xu, J. Investigating efficacy of “microbiota modulation of the gut-lung Axis” combined with chemotherapy in patients with advanced NSCLC: Study protocol for a multicenter, prospective, double blind, placebo controlled, randomized trial. BMC Cancer 2021, 21, 721. [Google Scholar] [CrossRef]
- Fernandes, M.R.; Aggarwal, P.; Costa, R.G.; Cole, A.M.; Trinchieri, G. Targeting the gut microbiota for cancer therapy. Nat. Rev. Cancer 2022, 22, 703–722. [Google Scholar] [CrossRef]
- Zhao, M.; Jiang, G.; Zhou, H.; Li, J.; Xiang, W.; Li, S.; Wang, H.; Zhou, J. Gut microbiota: A potential target for improved cancer therapy. J. Cancer Res. Clin. Oncol. 2023, 149, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Mao, Q.; Xia, W.; Dong, G.; Yu, C.; Jiang, F. Gut Microbiota Shapes the Efficiency of Cancer Therapy. Front. Microbiol. 2019, 10, 428613. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fang, Z.; Xue, Y.; Zhang, J.; Zhu, J.; Gao, R.; Yao, S.; Ye, Y.; Wang, S.; Lin, C. Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microbes 2020, 11, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Sexton, R.; Alhasan, R.; Rahman, S.; Azmi, A.S.; Sukari, A. Gut microbiome and response to checkpoint inhibitors in non-small cell lung cancer—A review. Crit. Rev. Oncol./Hematol. 2020, 145, 102841. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Hong, S.; Hwang, K.H.; Lim, E.J.; Han, J.Y.; Nam, Y.D. Diagnostic and prognostic potential of the oral and gut microbiome for lung adenocarcinoma. Clin. Transl. Med. 2021, 11, e508. [Google Scholar] [CrossRef] [PubMed]
- Olsson, L.M.; Boulund, F.; Nilsson, S.; Khan, M.T.; Gummesson, A.; Fagerberg, L.; Engstrand, L.; Perkins, R.; Uhlen, M.; Bergström, G. Dynamics of the normal gut microbiota: A longitudinal one-year population study in Sweden. Cell Host Microbe 2022, 30, 726–739.e3. [Google Scholar] [CrossRef] [PubMed]
- El Tekle, G.; Garrett, W.S. Bacteria in cancer initiation, promotion and progression. Nat. Rev. Cancer 2023, 23, 600–618. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Tang, H.; Zhai, Q.; Liu, L.; Xia, L.; Wu, W.; Xu, Y.; Wang, J. Gut Microbiota Dysbiosis in the Development and Progression of Gastric Cancer. J. Oncol. 2022, 2022, 9971619. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, J.; Guan, Y.; Lou, Y.; Cai, X. Dysbiosis of the Gut Microbiome is associated with Tumor Biomarkers in Lung Cancer. Int. J. Biol. Sci. 2019, 15, 2381–2392. [Google Scholar] [CrossRef]
- Mancini, I.; Bruera, E. Constipation in advanced cancer patients. Support. Care Cancer 1998, 6, 356–364. [Google Scholar] [CrossRef]
- Hoegenauer, C.; Hammer, H.F.; Mahnert, A.; Moissl-Eichinger, C. Methanogenic archaea in the human gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Rywotycki, R. Meat nitrosamine contamination level depending on animal breeding factors. Meat Sci. 2003, 65, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Guo, R.; Ni, W.; Liu, K.; Liu, Z.; Dai, J.; Xu, Y.; Abduriyim, S.; Wu, Z. Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse. Microbiome 2023, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Jiang, Z.; Zhang, Z.; Xing, J.; Wang, D.; Tang, D. Akkermansia muciniphila: A potential booster to improve the effectiveness of cancer immunotherapy. J. Cancer Res. Clin. Oncol. 2023, 149, 13477–13494. [Google Scholar] [CrossRef] [PubMed]
- Uppakarn, K.; Bangpanwimon, K.; Hongpattarakere, T.; Wanitsuwan, W. Comparison of the human gut microbiota between normal control subjects and patients with colonic polyps and colorectal cancer. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, N.; Zheng, X.; Liu, Y.; Lu, H.; Yin, X.; Hao, H.; Tan, Y.; Wang, D.; Hu, H. Intratumor Microbiome Analysis Identifies Positive Association Between. Front. Immunol. 2022, 13, 785422. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Shi, F.; Zhang, X.; Zhang, Y.; Bi, K.; Chen, X.; Li, L.; Diao, H. Phospholipid metabolites of the gut microbiota promote hypoxia-induced intestinal injury via CD1d-dependent γδ T cells. Gut Microbes 2022, 14, 2096994. [Google Scholar] [CrossRef]
- Gong, Z.; Ba, L.; Tang, J.; Yang, Y.; Li, Z.; Liu, M.; Yang, C.; Ding, F.; Zhang, M. Gut microbiota links with cognitive impairment in amyotrophic lateral sclerosis: A multi-omics study. J. Biomed. Res. 2023, 37, 125. [Google Scholar] [CrossRef]
- Clemmensen, C.; Müller, T.D.; Woods, S.C.; Berthoud, H.-R.; Seeley, R.J.; Tschöp, M.H. Gut-brain cross-talk in metabolic control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Benakis, C.; Martin-Gallausiaux, C.; Trezzi, J.-P.; Melton, P.; Liesz, A.; Wilmes, P. The microbiome-gut-brain axis in acute and chronic brain diseases. Curr. Opin. Neurobiol. 2020, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Willyard, C. How gut microbes could drive brain disorders. Nature 2021, 590, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kameyama, K.; Miyauchi, E.; Nakanishi, Y.; Kanaya, T.; Fujii, T.; Kato, T.; Sasaki, T.; Tachibana, N.; Negishi, H. Fatty acid overproduction by gut commensal microbiota exacerbates obesity. Cell Metab. 2023, 35, 361–375.e9. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell. Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Bogatyrev, S.R.; Rolando, J.C.; Ismagilov, R.F. Self-reinoculation with fecal flora changes microbiota density and composition leading to an altered bile-acid profile in the mouse small intestine. Microbiome 2020, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Angelis, M.D. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Panis, C.; Victorino, V.; Herrera, A.; Freitas, L.; De Rossi, T.; Campos, F.; Simão, A.C.; Barbosa, D.; Pinge-Filho, P.; Cecchini, R. Differential oxidative status and immune characterization of the early and advanced stages of human breast cancer. Breast Cancer Res. Treat. 2012, 133, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Cassilly, C.D.; Liu, X.; Park, S.-M.; Tusi, B.K.; Chen, X.; Kwon, J.; Filipčík, P.; Bolze, A.S.; Liu, Z. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 2022, 608, 168–173. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; Thompson, J.W.; Yin, T.; Alexander, P.B.; Qin, D.; Mudgal, P.; Wu, H.; Liang, Y.; Tan, L. Cancer-cell-derived GABA promotes β-catenin-mediated tumour growth and immunosuppression. Nat. Cell Biol. 2022, 24, 230–241. [Google Scholar] [CrossRef]
- Tsilimigras, M.C.; Fodor, A.; Jobin, C. Carcinogenesis and therapeutics: The microbiota perspective. Nat. Microbiol. 2017, 2, 17008. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Ho Szeto, C.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Shoji, F.; Yamaguchi, M.; Okamoto, M.; Takamori, S.; Yamazaki, K.; Okamoto, T.; Maehara, Y. Gut microbiota diversity and specific composition during immunotherapy in responders with non-small cell lung cancer. Front. Mol. Biosci. 2022, 9, 1040424. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Gazzaniga, F.S.; Wu, M.; Luthens, A.K.; Gillis, J.; Zheng, W.; LaFleur, M.W.; Johnson, S.B.; Morad, G.; Park, E.M. Targeting PD-L2–RGMb overcomes microbiome-related immunotherapy resistance. Nature 2023, 617, 377–385. [Google Scholar] [CrossRef]
- Stankevicius, V.; Kuodyte, K.; Schveigert, D.; Bulotiene, D.; Paulauskas, T.; Daniunaite, K.; Suziedelis, K. Gene and miRNA expression profiles of mouse Lewis lung carcinoma LLC1 cells following single or fractionated dose irradiation. Oncol. Lett. 2017, 13, 4190–4200. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Dinghua, L.; Chi-Man, L.; Ruibang, L.; Kunihiko, S.; Tak-Wah, L. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Hazen, S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2019, 48, D445–D453. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | mean_NSCLC (Arbitrary Units) | mean_HC (Arbitrary Units) | sd_NSCLC (Arbitrary Units) | sd_HC (Arbitrary Units) | Benjamini-Hochberg Corrected P, Wilcoxon Test | |
|---|---|---|---|---|---|---|
| GMM010 | Butyrate synthesis I | 53,993,846 | 113,000,000 | 31,444,828 | 9,977,743 | 0.000316 |
| GMM033 | Melatonin synthesis | 24,724,187 | 57,855,027 | 15,305,095 | 5,420,989 | 0.000316 |
| GMM013 | Isovaleric acid synthesis II (KADC pathway) | 50,297,742 | 120,000,000 | 29,493,292 | 12,180,467 | 0.000316 |
| GMM038 | GABA synthesis III | 22,555,066 | 55,021,164 | 12,773,931 | 6,999,335 | 0.000316 |
| GMM039 | GABA degradation | 35,005,566 | 73,710,252 | 19,381,261 | 4,117,453 | 0.000316 |
| GMM030 | Quinolinic acid degradation | 182,000,000 | 394,000,000 | 101,000,000 | 29,381,499 | 0.000316 |
| GMM018 | PUFAs synthesis (AA, EPA, DHA) | 99,607,290 | 229,000,000 | 56,642,838 | 20,936,594 | 0.000316 |
| GMM021 | Tryptophan synthesis | 8,385,742 | 18,034,299 | 4,697,625 | 2,370,340 | 0.000316 |
| GMM001 | Acetate synthesis I | 220,000,000 | 475,000,000 | 123,000,000 | 36,742,125 | 0.000316 |
| GMM005 | Acetate degradation | 58,489,189 | 131,000,000 | 32,551,505 | 14,959,711 | 0.000316 |
| GMM008 | Propionate synthesis III | 15,614,304 | 35,211,328 | 9,076,876 | 4,091,735 | 0.000316 |
| GMM014 | Secondary bile acid biosynthesis | 3,438,786 | 9,772,027 | 2,274,418 | 1,859,919 | 0.000357 |
| GMM007 | Propionate synthesis II | 19,529,539 | 42,233,217 | 11,149,791 | 5,828,594 | 0.000357 |
| GMM022 | Tryptophan degradation | 14,821,305 | 31,203,584 | 9,182,576 | 5,132,620 | 0.00071 |
| GMM003 | Acetate synthesis III | 6,374,973 | 15,170,133 | 4,088,844 | 3,289,936 | 0.00071 |
| GMM012 | Isovaleric acid synthesis I (KADH pathway) | 1,526,150 | 5,838,374 | 1,354,434 | 3,053,645 | 0.000819 |
| GMM040 | Menaquinone synthesis (vitamin K2) I | 29,799,019 | 57,445,013 | 18,151,028 | 6,611,267 | 0.000819 |
| GMM041 | Menaquinone synthesis (vitamin K2) II | 3,088,360 | 7,473,864 | 2,634,995 | 2,105,237 | 0.001303 |
| GMM002 | Acetate synthesis II | 185,558.3 | 665,099.1 | 628,913 | 951,112.1 | 0.002039 |
| GMM011 | Butyrate synthesis II | 246,549.1 | 1,263,851 | 578,002.3 | 1,082,580 | 0.002473 |
| GMM015 | Secondary bile acid biosynthesis | 3,571,747 | 6,600,120 | 2,455,394 | 1,036,932 | 0.003611 |
| GMM019 | Histamine synthesis | 7,938,246 | 12,843,787 | 4,291,302 | 1,504,558 | 0.003611 |
| GMM020 | Histamine degradation | 923,024 | 2,241,686 | 1,186,521 | 1,610,406 | 0.040182 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, C.; Li, N.; Gao, G.; He, Q.; Kwok, L.-Y.; Zhang, H. Dynamic Changes of the Gut Microbiota and Its Functional Metagenomic Potential during the Development of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 3768. https://doi.org/10.3390/ijms25073768
Feng C, Li N, Gao G, He Q, Kwok L-Y, Zhang H. Dynamic Changes of the Gut Microbiota and Its Functional Metagenomic Potential during the Development of Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2024; 25(7):3768. https://doi.org/10.3390/ijms25073768
Chicago/Turabian StyleFeng, Cuijiao, Na Li, Guangqi Gao, Qiuwen He, Lai-Yu Kwok, and Heping Zhang. 2024. "Dynamic Changes of the Gut Microbiota and Its Functional Metagenomic Potential during the Development of Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 25, no. 7: 3768. https://doi.org/10.3390/ijms25073768