
More than Just Bread and Wine: Using Yeast to Understand Inherited Cytochrome Oxidase Deficiencies in Humans
 , , ,
, , ,
Abstract
:1. Introduction
Yeast as a Model System for Human Cell Biology
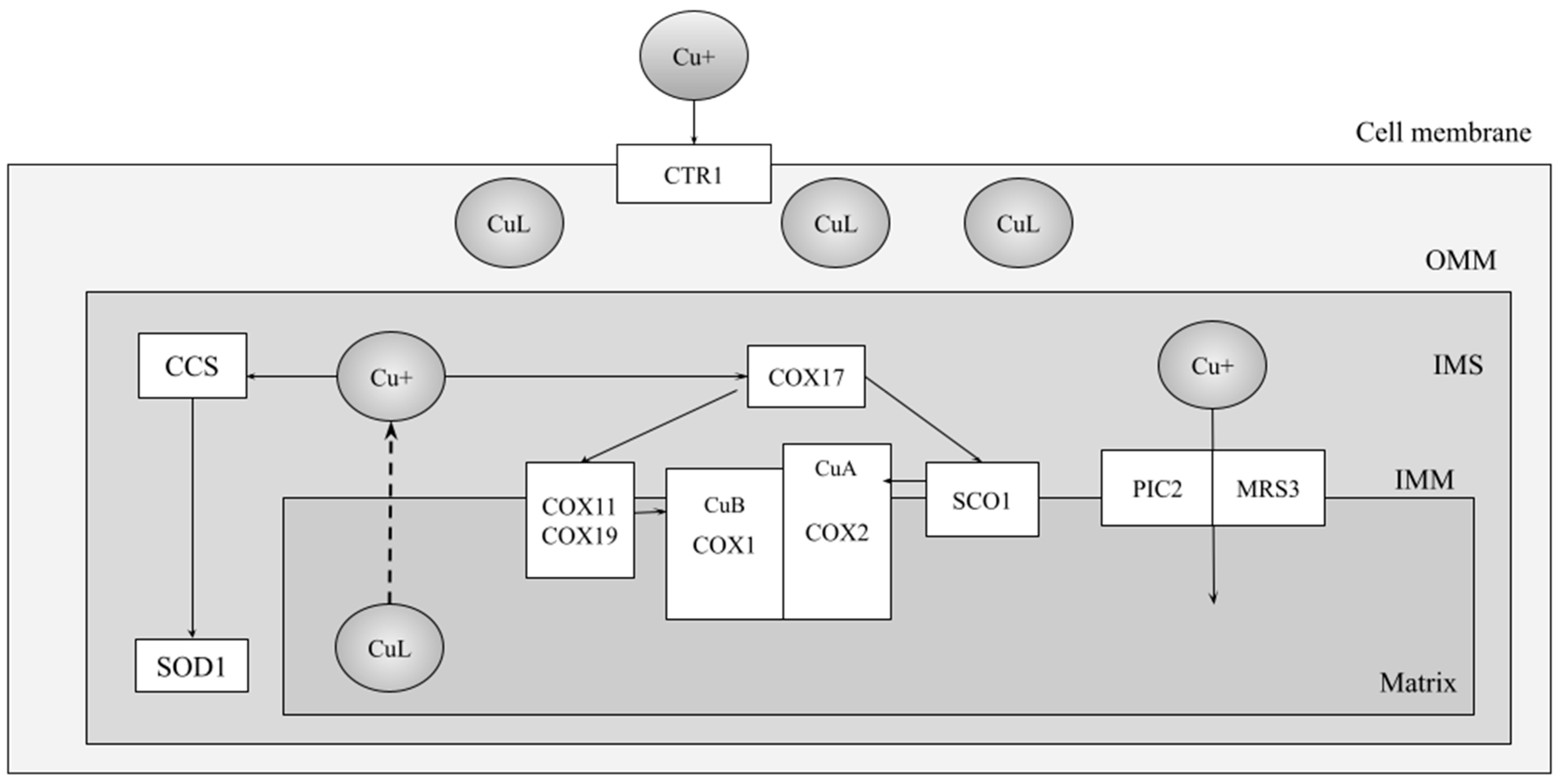
2. The COX Assembly Pathways in Humans and Yeast
3. Defects Affecting Synthesis and Assembly of COX1
3.1. Defects Associated with COX1 Expression
3.1.1. LRPPRC/PET309
3.1.2. TACO1/DPC29
3.1.3. C12ORF62/COX14
3.1.4. MITRAC12/COA3
3.2. Defects Associated with Heme A Biosynthesis and Insertion
3.2.1. COX10
3.2.2. COX15
3.2.3. PET117
3.2.4. SURF1/SHY1
3.3. Defects of Copper Acquisition at the CuB Site
COX11
4. Defects Affecting Synthesis and Assembly of COX2
4.1. Defects Associated with COX2 Expression
4.1.1. OXA1L/OXA1
4.1.2. COX16
4.1.3. COX18
4.1.4. COX20
4.1.5. PET100
4.2. Defects in Copper Provision to the CuA Site
4.2.1. hSCO1
4.2.2. hSCO2
4.2.3. COA6
5. ‘Other’ COX Assembly Factors
6. A Future for Yeast in Studying Human COX Defects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Yeast Systems Biology: Model Organism and Cell Factory. Biotechnol. J. 2019, 14, e1800421. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Vandeloo, M.; Greco, B.M.; Abdullah, M. Humanized yeast to model human biology, disease and evolution. Dis. Model. Mech. 2022, 15, dmm049309. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Fink, G.R. Yeast: An Experimental Organism for 21st Century Biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [PubMed]
- McEwen, J.E.; Ko, C.; Kloeckner-Gruissem, B.; Poyton, R.O. Nuclear functions required for cytochrome c oxidase biogenesis in Saccharomyces cerevisiae. Characterization of mutants in 34 complementation groups. J. Biol. Chem. 1986, 261, 11872–11879. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Dieckmann, C.L. PET genes of Saccaromyces cerevisiae. Microbiol. Rev. 1990, 54, 15. [Google Scholar] [CrossRef] [PubMed]
- Zee, J.M.; Glerum, D.M. Defects in cytochrome oxidase assembly in humans: Lessons from yeast. Biochem. Cell Biol. Biochim. Biol. Cell. 2006, 84, 859–869. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.E.; Gospodaryov, D.V. Alternative NAD(P)H dehydrogenase and alternative oxidase: Proposed physiological roles in animals. Mitochondrion 2018, 45, 7–17. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.E. Unique opportunities for future research on the alternative oxidase of plants. Plant Physiol. 2023, 191, 2084–2092. [Google Scholar] [CrossRef]
- Barrientos, A.; Gouget, K.; Horn, D.; Soto, I.C.; Fontanesi, F. Suppression mechanisms of COX assembly defects in yeast and human: Insights into the COX assembly process. Biochim. Biophys. Acta 2009, 1793, 97–107. [Google Scholar] [CrossRef]
- Dennerlein, S.; Rehling, P.; Richter-Dennerlein, R. Cytochrome c oxidase biogenesis—From translation to early assembly of the core subunit COX1. FEBS Lett. 2023, 597, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Wielburski, A.; Nelson, B.D. Evidence for the sequential assembly of cytochrome oxidase subunits in rat liver mitochondria. Biochem. J. 1983, 212, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.J.; Taanman, J.-W.; Muijsers, A.O.; Speijer, D.; Van Den Bogert, C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998, 254, 389–394. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Su, C.H.; Tzagoloff, A. Modular assembly of yeast cytochrome oxidase. Mol. Biol. Cell 2013, 24, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [PubMed]
- Fernández-Vizarra, E.; Tiranti, V.; Zeviani, M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.A.; McStay, G.P. Functions of Cytochrome c oxidase Assembly Factors. Int. J. Mol. Sci. 2020, 21, 7254. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.V.R.; Su, C.H.; Tzagoloff, A. Modular assembly of yeast mitochondrial ATP synthase and cytochrome oxidase. Biol. Chem. 2020, 401, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.M.; Yanamura, W.; Capaldi, R.A.; Robinson, B.H. Characterization of cytochrome-c oxidase mutants in human fibroblasts. FEBS Lett. 1988, 236, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lepage, P.; Miller, K.; Bunkenborg, J.; Reich, M.; Hjerrild, M.; Delmonte, T.; Villeneuve, A.; Sladek, R.; Xu, F.; et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA 2003, 100, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Manthey, G.M.; McEwen, J.E. The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J. 1995, 14, 4031–4043. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Ochoa, A.; Camacho-Villasana, Y.; García-Guerrero, A.E.; Pérez-Martínez, X. The Pet309 pentatricopeptide repeat motifs mediate efficient binding to the mitochondrial COX1 transcript in yeast. RNA Biol. 2014, 11, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Weraarpachai, W.; Antonicka, H.; Sasarman, F.; Seeger, J.; Schrank, B.; Kolesar, J.E.; Lochmüller, H.; Chevrette, M.; Kaufman, B.A.; Horvath, R.; et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009, 41, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Wanschers, B.F.; Cuypers, T.D.; Esseling, J.J.; Riemersma, M. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012, 13, R12. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.M.; Koerner, T.J.; Tzagoloff, A. Cloning and characterization of COX14, whose product is required for assembly of yeast cytochrome oxidase. J. Biol. Chem. 1995, 270, 15585–15590. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Su, C.H.; Tzagoloff, A. Stabilization of Cox1p intermediates by the Cox14p-Coa3p complex. FEBS Lett. 2013, 587, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A.; Zambrano, A.; Tzagoloff, A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004, 23, 3472–3482. [Google Scholar] [CrossRef]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 2018, 10, e9060. [Google Scholar] [CrossRef] [PubMed]
- Weraarpachai, W.; Sasarman, F.; Nishimura, T.; Antonicka, H.; Auré, K.; Rötig, A.; Lombès, A.; Shoubridge, E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012, 90, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Merante, F.; Petrova-Benedict, R.; MacKay, N.; Mitchell, G.; Lambert, M.; Morin, C.; De Braekeleer, M.; Laframboise, R.; Gagné, R.; Robinson, B.H. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am. J. Hum. Genet. 1993, 53, 481–487. [Google Scholar] [PubMed]
- Mili, S.; Piñol-Roma, S. LRP130, a pentatricopeptide motif protein with a noncanonical RNA-binding domain, is bound in vivo to mitochondrial and nuclear RNAs. Mol. Cell. Biol. 2003, 23, 4972–4982. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Morin, C.; Mitchell, G.; Ackerley, C.; Robinson, B.H. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: Mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004, 382, 331–336. [Google Scholar] [CrossRef]
- Oláhová, M.; Hardy, S.A.; Hall, J.; Yarham, J.W.; Haack, T.B.; Wilson, W.C.; Alston, C.L.; He, L.; Aznauryan, E.; Brown, R.M.; et al. LRPPRC mutations cause early-onset multisystem mitochondrial disease outside of the French-Canadian population. Brain J. Neurol. 2015, 138, 3503–3519. [Google Scholar] [CrossRef] [PubMed]
- Manthey, G.M.; Przybyla-Zawislak, B.D.; McEwen, J.E. The Saccharomyces cerevisiae Pet309 protein is embedded in the mitochondrial inner membrane. Eur. J. Biochem. 1998, 255, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Herbert, C.J.; Golik, P.; Bonnefoy, N. Yeast PPR proteins, watchdogs of mitochondrial gene expression. RNA Biol. 2013, 10, 1477–1494. [Google Scholar] [CrossRef] [PubMed]
- Seeger, J.; Schrank, B.; Pyle, A.; Stucka, R.; Lörcher, U.; Müller-Ziermann, S.; Abicht, A.; Czermin, B.; Holinski-Feder, E.; Lochmüller, H.; et al. Clinical and neuropathological findings in patients with TACO1 mutations. Neuromuscul. Disord. NMD 2010, 20, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Hubble, K.A.; Henry, M.F. DPC29 promotes post-initiation mitochondrial translation in Saccharomyces cerevisiae. Nucleic Acids Res. 2023, 51, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, F.; Clemente, P.; Barrientos, A. Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Vukotic, M.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Deckers, M.; Rehling, P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 2010, 191, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Weraarpachai, W.; Ravn, K.; Born, A.P.; Jønson, L.; Duno, M.; Wibrand, F.; Shoubridge, E.A.; Vissing, J. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 2015, 52, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Merz, S.; Westermann, B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009, 10, R95. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Nobrega, M.; Gorman, N.; Sinclair, P. On the functions of the yeast COX10 and COX11 gene products. Biochem. Mol. Biol. Int. 1993, 31, 593–598. [Google Scholar] [PubMed]
- Glerum, D.M.; Tzagoloff, A. Isolation of a human cDNA for heme A:farnesyltransferase by functional complementation of a yeast cox10 mutant. Proc. Natl. Acad. Sci. USA 1994, 91, 8452–8456. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, M.P.; Nobrega, F.G.; Tzagoloff, A. COX10 codes for a protein homologous to the ORF1 product of Paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase. J. Biol. Chem. 1990, 265, 14220–14226. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.M.; Muroff, I.; Jin, C.; Tzagoloff, A. COX15 Codes for a Mitochondrial Protein Essential for the Assembly of Yeast Cytochrome Oxidase. J. Biol. Chem. 1997, 272, 19088–19094. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; Carlson, C.G.; Glerum, D.M.; Tzagoloff, A. Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 2001, 492, 133–138. [Google Scholar] [CrossRef] [PubMed]
- McEwen, J.E.; Hong, K.H.; Park, S.; Preciado, G.T. Sequence and chromosomal localization of two PET genes required for cytochrome c oxidase assembly in Saccharomyces cerevisiae. Curr. Genet. 1993, 23, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.G.; Swenson, S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The assembly factor Pet117 couples heme a synthase activity to cytochrome oxidase assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Mashkevich, G.; Repetto, B.; Glerum, D.M.; Jin, C.; Tzagoloff, A. SHY1, the yeast homolog of the mammalian SURF-1 gene, encodes a mitochondrial protein required for respiration. J. Biol. Chem. 1997, 272, 14356–14364. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Gray, J.; Mitchell, L.; Antholine, W.E.; Hosler, J.P. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J. Biol. Chem. 2005, 280, 17652–17656. [Google Scholar] [CrossRef] [PubMed]
- Nývltová, E.; Dietz, J.V.; Seravalli, J.; Khalimonchuk, O.; Barrientos, A. Coordination of metal center biogenesis in human cytochrome c oxidase. Nat. Commun. 2022, 13, 3615. [Google Scholar] [CrossRef] [PubMed]
- Valnot, I.; von Kleist-Retzow, J.-C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.-W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rötig, A. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Pankratz, N.; Nichols, W.C.; Uniacke, S.K.; Halter, C.; Murrell, J.; Rudolph, A.; Shults, C.W.; Conneally, P.M.; Foroud, T. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Coenen, M.J.H.; van der Heuvel, L.P.; Ugalde, C.; Brinke, M.T.; Nijtmans, L.G.J.; Trijbels, F.J.M.; Beblo, S.; Maier, E.M.; Muntau, A.C.; Smeitink, J.A.M. Cytochrome c oxidase biogenesis in a patient with a mutation in COX10 gene. Ann. Neurol. 2004, 56, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Pierrel, F.; Khalimonchuk, O.; Cobine, P.A.; Bestwick, M.; Winge, D.R. Coa2 is an assembly factor for yeast cytochrome c oxidase biogenesis that facilitates the maturation of Cox1. Mol. Cell. Biol. 2008, 28, 4927–4939. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.; Khalimonchuk, O.; Pierrel, F.; Winge, D.R. The role of Coa2 in hemylation of yeast Cox1 revealed by its genetic interaction with Cox10. Mol. Cell. Biol. 2010, 30, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Oquendo, C.E.; Antonicka, H.; Shoubridge, E.A.; Reardon, W.; Brown, G.K. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, M.; Tiranti, V.; Farina, L.; Uziel, G.; Zeviani, M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005, 42, e28. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Lillquist, Y.P.; Waters, P.J.; Sinclair, G.; Struys, E.; McFadden, D.; Hendson, H.; Hyams, L.; Shoffner, J.; Vallance, H.D. Infantile cardioencephalopathy due to a COX15 gene defect: Report and review. Am. J. Med. Genet. A. 2011, 155A, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Rivett, E.D.; Heo, L.; Feig, M.; Hegg, E.L. Biosynthesis and trafficking of heme o and heme a: New structural insights and their implications for reaction mechanisms and prenylated heme transfer. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 640–668. [Google Scholar] [CrossRef] [PubMed]
- Saiki, K.; Mogi, T.; Ogura, K.; Anraku, Y. In vitro heme O synthesis by the cyoE gene product from Escherichia coli. J. Biol. Chem. 1993, 268, 26041–26044. [Google Scholar] [CrossRef] [PubMed]
- Hederstedt, L. Diversity of Cytochrome c Oxidase Assembly Proteins in Bacteria. Microorganisms 2022, 10, 926. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Wang, Z.; Brown, K.R.; Cricco, J.A.; Hegg, E.L. Heme O synthase and heme A synthase from Bacillus subtilis and Rhodobacter sphaeroides interact in Escherichia coli. Biochemistry 2004, 43, 13541–13548. [Google Scholar] [CrossRef] [PubMed]
- Khalimonchuk, O.; Kim, H.; Watts, T.; Perez-Martinez, X.; Winge, D.R. Oligomerization of heme o synthase in cytochrome oxidase biogenesis is mediated by cytochrome oxidase assembly factor Coa2. J. Biol. Chem. 2012, 287, 26715–26726. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.R.; Brown, B.M.; Hoagland, E.; Mayne, C.L.; Hegg, E.L. Heme A synthase does not incorporate molecular oxygen into the formyl group of heme A. Biochemistry 2004, 43, 8616–8624. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Visser, G.; Baertling, F.; Wintjes, L.T.; Wolters, V.M.; van Montfrans, J.; de Kort, G.A.P.; Nikkels, P.G.J.; van Hasselt, P.M.; van der Crabben, S.N.; et al. Mutated PET117 causes complex IV deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 2017, 136, 759–769. [Google Scholar] [CrossRef]
- Sun, Q.; Shi, L.; Li, S.; Li, J.; Zhang, R.; Huang, X.; Shao, Y.; Feng, Z.; Peng, Y.; Yang, Z.; et al. PET117 assembly factor stabilizes translation activator TACO1 thereby upregulates mitochondria-encoded cytochrome C oxidase 1 synthesis. Free Radic. Biol. Med. 2023, 205, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Church, C.; Chapon, C.; Poyton, R.O. Cloning and Characterization of PET100, a Gene Required for the Assembly of Yeast Cytochrome c Oxidase. J. Biol. Chem. 1996, 271, 18499–18507. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; et al. MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; De Bie, I.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.-C.; Chevrette, M.; et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Jaksch, M.; Hofmann, S.; Galimberti, C.; Hoertnagel, K.; Lulli, L.; Freisinger, P.; Bindoff, L.; Gerbitz, K.D.; Comi, G.-P.; et al. Loss-of-function mutations of SURF-1 are specifically associated with Leigh syndrome with cytochrome c oxidase deficiency. Ann. Neurol. 1999, 46, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, M.; Yokoyama, Y.; Ninomiya, S.; Inoue, C.; Yamashita, S.; Seino, Y. Two novel mutations of SURF1 in Leigh syndrome with cytochrome c oxidase deficiency. Hum. Genet. 1999, 105, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Poyau, A.; Buchet, K.; Fouad Bouzidi, M.; Zabot, M.T.; Echenne, B.; Yao, J.; Shoubridge, E.A.; Godinot, C. Missense mutations in SURF1 associated with deficient cytochrome c oxidase assembly in Leigh syndrome patients. Hum. Genet. 2000, 106, 194–205. [Google Scholar] [PubMed]
- Rahman, S.; Brown, R.M.; Chong, W.K.; Wilson, C.J.; Brown, G.K. A SURF1 gene mutation presenting as isolated leukodystrophy. Ann. Neurol. 2001, 49, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Freehauf, C.; Sacconi, S.; DiMauro, S.; Thoma, J.; Tsai, A.C. Novel SURF1 mutation in a child with subacute encephalopathy and without the radiological features of Leigh Syndrome. Am. J. Med. Genet. A 2004, 128A, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Ghezzi, D.; Chassagne, M.; Mayençon, M.; Padet, S.; Melchionda, L.; Rouvet, I.; Lannes, B.; Bozon, D.; Latour, P.; et al. SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease. Neurology 2013, 81, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Shoubridge, E.A. Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet. 1999, 8, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Valnot, I.; Rustin, P.; Taanman, J.-W. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J. Biol. Chem. 2004, 279, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A. Shy1p is necessary for full expression of mitochondrial COX1 in the yeast model of Leigh’s syndrome. EMBO J. 2002, 21, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Khalimonchuk, O.; Bestwick, M.; Meunier, B.; Watts, T.C.; Winge, D.R. Formation of the redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase. Mol. Cell. Biol. 2010, 30, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, R.; Bareth, B.; Balleininger, M.; Wissel, M.; Rehling, P.; Mick, D.U. Mimicking a SURF1 allele reveals uncoupling of cytochrome c oxidase assembly from translational regulation in yeast. Hum. Mol. Genet. 2011, 20, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Bundschuh, F.A.; Hannappel, A.; Anderka, O.; Ludwig, B. Surf1, associated with Leigh syndrome in humans, is a heme-binding protein in bacterial oxidase biogenesis. J. Biol. Chem. 2009, 284, 25735–25741. [Google Scholar] [CrossRef] [PubMed]
- Vest, K.E.; Leary, S.C.; Winge, D.R.; Cobine, P.A. Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 2013, 288, 23884–23892. [Google Scholar] [CrossRef] [PubMed]
- Vest, K.E.; Wang, J.; Gammon, M.G.; Maynard, M.K.; White, O.L.; Cobine, J.A.; Mahone, W.K.; Cobine, P.A. Overlap of copper and iron uptake systems in mitochondria in Saccharomyces cerevisiae. Open Biol. 2016, 6, 150223. [Google Scholar] [CrossRef] [PubMed]
- Cobine, P.A.; Ojeda, L.D.; Rigby, K.M.; Winge, D.R. Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem. 2004, 279, 14447–14455. [Google Scholar] [CrossRef] [PubMed]
- Cobine, P.A.; Pierrel, F.; Bestwick, M.L.; Winge, D.R. Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J. Biol. Chem. 2006, 281, 36552–36559. [Google Scholar] [CrossRef]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar] [CrossRef] [PubMed]
- Horng, Y.-C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 2004, 279, 35334–35340. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Capitanio, N.; Nobrega, M.P.; Gatti, D. Cytochrome oxidase assembly in yeast requires the product of COX11, a homolog of the P. denitrificans protein encoded by ORF3. EMBO J. 1990, 9, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Rödel, G. Accumulation of the cytochrome c oxidase subunits I and II in yeast requires a mitochondrial membrane-associated protein, encoded by the nuclear SCO1 gene. Mol. Gen. Genet. MGG 1989, 216, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 20531–20535. [Google Scholar] [CrossRef] [PubMed]
- Hiser, L.; Di Valentin, M.; Hamer, A.G.; Hosler, J.P. Cox11p Is required for stable formation of the CuB and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Banting, G.S.; Glerum, D.M. Mutational analysis of the Saccharomyces cerevisiae cytochrome c oxidase assembly protein Cox11p. Eukaryot. Cell 2006, 5, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, E.K.; Adams, D.L.; Schon, E.A.; Glerum, D.M. A human SCO2 mutation helps define the role of Sco1p in the cytochrome oxidase assembly pathway. J. Biol. Chem. 2000, 275, 26780–26785. [Google Scholar] [CrossRef]
- Nobrega, M.P.; Bandeira, S.C.B.; Beers, J.; Tzagoloff, A. Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J. Biol. Chem. 2002, 277, 40206–40211. [Google Scholar] [CrossRef] [PubMed]
- Rigby, K.; Zhang, L.; Cobine, P.A.; George, G.N.; Winge, D.R. Characterization of the cytochrome c oxidase assembly factor Cox19 of Saccharomyces cerevisiae. J. Biol. Chem. 2007, 282, 10233–10242. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; Laan, M.V.D.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 2015, 26, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Rius, R.; Bennett, N.K.; Bhattacharya, K.; Riley, L.G.; Yüksel, Z.; Formosa, L.E.; Compton, A.G.; Dale, R.C.; Cowley, M.J.; Gayevskiy, V.; et al. Biallelic pathogenic variants in COX11 are associated with an infantile-onset mitochondrial encephalopathy. Hum. Mutat. 2022, 43, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Caron-Godon, C.A.; Della Vecchia, S.; Romano, A.; Doccini, S.; Canto, F.D.; Pasquariello, R.; Rubegni, A.; Battini, R.; Santorelli, F.M.; Glerum, D.M.; et al. Novel COX11 Mutations Associated with Mitochondrial Disorder: Functional Characterization in Patient Fibroblasts and Saccharomyces cerevisiae. Int. J. Mol. Sci. 2023, 24, 16636. [Google Scholar] [CrossRef] [PubMed]
- Glerum, D.M.; Tzagoloff, A. Submitochondrial distributions and stabilities of subunits 4, 5, and 6 of yeast cytochrome oxidase in assembly defective mutants. FEBS Lett. 1997, 412, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Carr, H.S.; George, G.N.; Winge, D.R. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 2002, 277, 31237–31242. [Google Scholar] [CrossRef] [PubMed]
- Veniamin, S.; Sawatzky, L.G.; Banting, G.S.; Glerum, D.M. Characterization of the peroxide sensitivity of COX-deficient yeast strains reveals unexpected relationships between COX assembly proteins. Free Radic. Biol. Med. 2011, 51, 1589–1600. [Google Scholar] [CrossRef]
- Bode, M.; Longen, S.; Morgan, B.; Peleh, V.; Dick, T.P.; Bihlmaier, K.; Herrmann, J.M. Inaccurately assembled cytochrome c oxidase can lead to oxidative stress-induced growth arrest. Antioxid. Redox Signal. 2013, 18, 1597–1612. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fox, T.D. Membrane translocation of mitochondrially coded Cox2p: Distinct requirements for export of N and C termini and dependence on the conserved protein Oxa1p. Mol. Biol. Cell 1997, 8, 1449–1460. [Google Scholar] [CrossRef]
- Meyer, W.; Bauer, M.; Pratje, E. A mutation in cytochrome oxidase subunit 2 restores respiration of the mutant pet ts1402. Curr. Genet. 1997, 31, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Herrmann, J.M.; Pratje, E.; Neupert, W.; Stuart, R.A. Oxa1p, an essential component of the N-tail protein export machinery in mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Fiumera, H.L.; Dujardin, G.; Fox, T.D. Roles of Oxa1-related inner-membrane translocases in assembly of respiratory chain complexes. Biochim. Biophys. Acta 2009, 1793, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.G.; Barrientos, A.; Tzagoloff, A.; Glerum, D.M. COX16 encodes a novel protein required for the assembly of cytochrome oxidase in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 3770–3775. [Google Scholar] [CrossRef]
- Aich, A.; Wang, C.; Chowdhury, A.; Ronsör, C.; Pacheu-Grau, D.; Richter-Dennerlein, R.; Dennerlein, S.; Rehling, P. COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis. eLife 2018, 7, e32572. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.L.; Green-Willms, N.S.; Fox, T.D.; Tzagoloff, A.; Nobrega, F.G. Cloning and characterization of COX18, a Saccharomyces cerevisiae PET gene required for the assembly of cytochrome oxidase. J. Biol. Chem. 2000, 275, 14898–14902. [Google Scholar] [CrossRef] [PubMed]
- Saracco, S.A.; Fox, T.D. Cox18p is required for export of the mitochondrially encoded Saccharomyces cerevisiae Cox2p C-tail and interacts with Pnt1p and Mss2p in the inner membrane. Mol. Biol. Cell 2002, 13, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Barrientos, A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem. 2017, 292, 7774–7783. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Tzagoloff, A.; Neupert, W.; Stuart, R.A. Identification of Cox20p, a novel protein involved in the maturation and assembly of cytochrome oxidase subunit 2. J. Biol. Chem. 2000, 275, 4571–4578. [Google Scholar] [CrossRef] [PubMed]
- Elliott, L.E.; Saracco, S.A.; Fox, T.D. Multiple Roles of the Cox20 Chaperone in Assembly of Saccharomyces cerevisiae Cytochrome c Oxidase. Genetics 2012, 190, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Boulet, A.; Leary, S.C.; Barrientos, A. Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 2014, 23, 2901–2913. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.L.; Gohil, V.M. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.A.; Maher, M.J.; Lindau, C.; Vögtle, F.N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Singh, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Wasilewski, M.; Oeljeklaus, S.; Gibhardt, C.S.; Aich, A.; Chudenkova, M.; Dennerlein, S.; Deckers, M.; Bogeski, I.; Warscheid, B.; et al. COA6 facilitates cytochrome c oxidase biogenesis as thiol-reductase for copper metallochaperones in mitochondria. J. Mol. Biol. 2020, 432, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, I.; Oeljeklaus, S.; Aich, A.; Ronsör, C.; Callegari, S.; Dudek, J.; Warscheid, B.; Dennerlein, S.; Rehling, P. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 323–333. [Google Scholar] [CrossRef]
- Stiburek, L.; Fornuskova, D.; Wenchich, L.; Pejznochova, M.; Hansikova, H.; Zeman, J. Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J. Mol. Biol. 2007, 374, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Chalvet, F.; Hamel, P.; Slonimski, P.P.; Dujardin, G. OXA1, a Saccharomyces cerevisiae nuclear gene whose sequence is conserved from prokaryotes to eukaryotes controls cytochrome oxidase biogenesis. J. Mol. Biol. 1994, 239, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Herrmann, J.; Pratje, E.; Neupert, W.; Stuart, R.A. Oxa1p mediates the export of the N- and C-termini of pCoxII from the mitochondrial matrix to the intermembrane space. FEBS Lett. 1997, 418, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Hildenbeutel, M.; Theis, M.; Geier, M.; Haferkamp, I.; Neuhaus, H.E.; Herrmann, J.M.; Ott, M. The membrane insertase Oxa1 is required for efficient import of carrier proteins into mitochondria. J. Mol. Biol. 2012, 423, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.; Kauff, F.; van der Sluis, E.O.; Ott, M.; Herrmann, J.M. Evolution of YidC/Oxa1/Alb3 insertases: Three independent gene duplications followed by functional specialization in bacteria, mitochondria and chloroplasts. Biol. Chem. 2011, 392, 13–19. [Google Scholar] [CrossRef]
- Anghel, S.A.; McGilvray, P.T.; Hegde, R.S.; Keenan, R.J. Identification of Oxa1 Homologs Operating in the Eukaryotic Endoplasmic Reticulum. Cell Rep. 2017, 21, 3708–3716. [Google Scholar] [CrossRef] [PubMed]
- Homberg, B.; Rehling, P.; Cruz-Zaragoza, L.D. The multifaceted mitochondrial OXA insertase. Trends Cell Biol. 2023, 33, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Poerschke, S.; Oeljeklaus, S.; Cruz-Zaragoza, L.D.; Schenzielorz, A.; Dahal, D.; Hillen, H.S.; Das, H.; Kremer, L.S.; Valpadashi, A.; Breuer, M.; et al. Identification of TMEM126A as OXA1L-interacting protein reveals cotranslational quality control in mitochondria. Mol. Cell 2024, 84, 345–358.e5. [Google Scholar] [CrossRef] [PubMed]
- Wintjes, L.T.M.; Kava, M.; van den Brandt, F.A.; van den Brand, M.A.; Lapina, O.; Bliksrud, Y.T.; Kulseth, M.A.; Amundsen, S.S.; Selberg, T.R.; Ybema-Antoine, M.; et al. A novel variant in COX16 causes cytochrome c oxidase deficiency, severe fatal neonatal lactic acidosis, encephalopathy, cardiomyopathy, and liver dysfunction. Hum. Mutat. 2021, 42, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.K.H.; Oeljeklaus, S.; Cruz-Zaragoza, L.D.; Schenzielorz, A.; Dahal, D.; Hillen, H.S.; Das, H.; Kremer, L.S.; Valpadashi, A.; Breuer, M. Studies of COX16, COX19, and PET191 in human cytochrome-c oxidase deficiency. Arch. Neurol. 2004, 61, 1935–1937. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; Tzagoloff, A. Cox16 protein is physically associated with Cox1p assembly intermediates and with cytochrome oxidase. J. Biol. Chem. 2017, 292, 16277–16283. [Google Scholar] [CrossRef]
- Cerqua, C.; Morbidoni, V.; Desbats, M.A.; Doimo, M.; Frasson, C.; Sacconi, S.; Baldoin, M.C.; Sartori, G.; Basso, G.; Salviati, L.; et al. COX16 is required for assembly of cytochrome c oxidase in human cells and is involved in copper delivery to COX2. Biochim. Biophys. Acta BBA-Bioenerg. 2018, 1859, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Garbellini, M.; Magri, F.; Menni, F.; Meneri, M.; Bedeschi, M.F.; Dilena, R.; Cecchetti, V.; Picciolli, I.; Furlan, F.; et al. A biallelic variant in COX18 cause isolated Complex IV deficiency associated with neonatal encephalo-cardio-myopathy and axonal sensory neuropathy. Eur. J. Hum. Genet. EJHG 2023, 31, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Fiumera, H.L.; Broadley, S.A.; Fox, T.D. Translocation of mitochondrially synthesized Cox2 domains from the matrix to the intermembrane space. Mol. Cell. Biol. 2007, 27, 4664–4673. [Google Scholar] [CrossRef] [PubMed]
- Fiumera, H.L.; Dunham, M.J.; Saracco, S.A.; Butler, C.A.; Kelly, J.A.; Fox, T.D. Translocation and assembly of mitochondrially coded Saccharomyces cerevisiae cytochrome c oxidase subunit Cox2 by Oxa1 and Yme1 in the absence of Cox18. Genetics 2009, 182, 519–528. [Google Scholar] [CrossRef] [PubMed]
- van Bloois, E.; Koningstein, G.; Bauerschmitt, H.; Herrmann, J.M.; Luirink, J. Saccharomyces cerevisiae Cox18 complements the essential Sec-independent function of Escherichia coli YidC. FEBS J. 2007, 274, 5704–5713. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Trevisson, E.; Pistollato, F.; Baldoin, M.C.; Rezzonico, R.; Bourget, I.; Desnuelle, C.; Tenconi, R.; Basso, G.; DiMauro, S.; et al. hCOX18 and hCOX19: Two human genes involved in cytochrome c oxidase assembly. Biochem. Biophys. Res. Commun. 2005, 337, 832–839. [Google Scholar] [CrossRef]
- Ban, R.; Kopajtich, R.; Lv, J.; Stenton, S.L.; Shimura, M.; Wang, Z.; Yuan, Y.; Wang, J.; Han, X.; Liu, Z.; et al. The phenotypic spectrum of COX20 -associated mitochondrial disorder. Brain 2022, 145, e125–e127. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Wanschers, B.F.J.; Nijtmans, L.G.; Rodenburg, R.J.; Zschocke, J.; Dikow, N.; van den Brand, M.A.M.; Hendriks-Franssen, M.G.M.; Gilissen, C.; Veltman, J.A.; et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 2013, 22, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.G.; Tiongson, E.; Diaz, F.; Haude, K.; Panzer, K.; Collier, A.; Kim, J.; Adams, D.; Tifft, C.J.; Cui, H.; et al. Novel pathogenic COX20 variants causing dysarthria, ataxia, and sensory neuropathy. Ann. Clin. Transl. Neurol. 2019, 6, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ozcanyuz, D.G.; Incecik, F.; Herguner, O.M.; Mungan, N.O.; Bozdogan, S.T. Dysarthria, Ataxia, and Dystonia Associated with COX20 (FAM36A) Gene Mutation: A Case Report of a Turkish Child. Ann. Indian Acad. Neurol. 2020, 23, 399–401. [Google Scholar] [PubMed]
- Kumar, V.; Hart, A.J.; Keerthiraju, E.R.; Waldron, P.R.; Tucker, G.A.; Greetham, D. Expression of Mitochondrial Cytochrome C Oxidase Chaperone Gene (COX20) Improves Tolerance to Weak Acid and Oxidative Stress during Yeast Fermentation. PLoS ONE 2015, 10, e0139129. [Google Scholar] [CrossRef] [PubMed]
- Keerthiraju, E.; Du, C.; Tucker, G.; Greetham, D. A Role for COX20 in Tolerance to Oxidative Stress and Programmed Cell Death in Saccharomyces cerevisiae. Microorganisms 2019, 7, 575. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Smith, K.R.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Oláhová, M.; Haack, T.B.; Alston, C.L.; Houghton, J.A.; He, L.; Morris, A.A.; Brown, G.K.; McFarland, R.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N.; et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur. J. Hum. Genet. EJHG 2015, 23, 935–939. [Google Scholar] [CrossRef]
- Church, C.; Goehring, B.; Forsha, D.; Wazny, P.; Poyton, R.O. A Role for Pet100p in the Assembly of Yeast Cytochrome c Oxidase. J. Biol. Chem. 2005, 280, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Nittis, T.; George, G.N.; Winge, D.R. Yeast Sco1, a protein essential for cytochrome c oxidase function is a Cu(I)-binding protein. J. Biol. Chem. 2001, 276, 42520–42526. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Glerum, D.M.; Tzagoloff, A. Purification and characterization of yeast Sco1p, a mitochondrial copper protein. J. Biol. Chem. 2002, 277, 22185–22190. [Google Scholar] [CrossRef] [PubMed]
- Lode, A.; Kuschel, M.; Paret, C.; Rödel, G. Mitochondrial copper metabolism in yeast: Interaction between Sco1p and Cox2p. FEBS Lett. 2000, 485, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.-H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef]
- Abriata, L.A.; Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Gkazonis, P.; Spyroulias, G.A.; Vila, A.J.; Wang, S. Mechanism of Cu(A) assembly. Nat. Chem. Biol. 2008, 4, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Canonica, F.; Klose, D.; Ledermann, R.; Sauer, M.M.; Abicht, H.K.; Quade, N.; Gossert, A.D.; Chesnov, S.; Fischer, H.-M.; Jeschke, G.; et al. Structural basis and mechanism for metallochaperone-assisted assembly of the Cu(A) center in cytochrome oxidase. Sci. Adv. 2019, 5, eaaw8478. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Sasarman, F.; Nishimura, T.; Shoubridge, E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009, 18, 2230–2240. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Burkhart, J.M.; Rao, S.; Gerbeth, C.; Hinrichs, J.; Martinou, J.-C.; Chacinska, A.; Sickmann, A.; Zahedi, R.P.; Meisinger, C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteomics MCP 2012, 11, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Lochmüller, H.; Stucka, R.; Yao, J.; Shoubridge, E.A.; Kim, S.H.; Gerbitz, K.-D.; Jaksch, M. Characterization of human SCO1 and COX17 genes in mitochondrial cytochrome-c-oxidase deficiency. Biochem. Biophys. Res. Commun. 2000, 276, 530–533. [Google Scholar] [CrossRef]
- Valnot, I.; Osmond, S.; Gigarel, N.; Mehaye, B.; Amiel, J.; Cormier-Daire, V.; Munnich, A.; Bonnefont, J.-P.; Rustin, P.; Rötig, A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000, 67, 1104–1109. [Google Scholar] [CrossRef]
- Stiburek, L.; Vesela, K.; Hansikova, H.; Hulkova, H.; Zeman, J. Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol. 2009, 296, C1218–C1226. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Antonicka, H.; Sasarman, F.; Weraarpachai, W.; Cobine, P.A.; Pan, M.; Brown, G.K.; Brown, R.; Majewski, J.; Ha, K.C.H.; et al. Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat. 2013, 34, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Rödel, G. SCO1, a yeast nuclear gene essential for accumulation of mitochondrial cytochrome c oxidase subunit II. Mol. Gen. Genet. MGG 1988, 211, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, A.; Krummeck-Weiß, G.; Hofer, A.; Bartuschka, A.; Ostermann, K.; Rödel, G. Mitochondrial copper metabolism in yeast: Mutational analysis of Sco1p involved in the biogenesis of cytochrome c oxidase. Curr. Genet. 1999, 35, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Abajian, C.; Rosenzweig, A.C. Crystal structure of yeast Sco1. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 2006, 11, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Sue, C.; Banting, G.S.; Yang, H.; Glerum, D.M.; Hendrickson, W.A.; Schon, E.A. Crystal structure of human SCO1: Implications for redox signaling by a mitochondrial cytochrome c oxidase ‘assembly’ protein. J. Biol. Chem. 2005, 280, 15202–15211. [Google Scholar] [CrossRef]
- Khalimonchuk, O.; Bird, A.; Winge, D.R. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J. Biol. Chem. 2007, 282, 17442–17449. [Google Scholar] [CrossRef] [PubMed]
- Cobine, P.A.; Pierrel, F.; Leary, S.C.; Sasarman, F.; Horng, Y.-C.; Shoubridge, E.A.; Winge, D.R. The P174L mutation in human Sco1 severely compromises Cox17-dependent metallation but does not impair copper binding. J. Biol. Chem. 2006, 281, 12270–12276. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Sacconi, S.; Rasalan, M.M.; Kronn, D.F.; Braun, A.; Canoll, P.; Davidson, M.; Shanske, S.; Bonilla, E.; Hays, A.P.; et al. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch. Neurol. 2002, 59, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A.; Bourgeois, J.; Fu, M.; Kataeva, G.; Shah, J.; Simon, D.; Mahoney, D.; Johns, D.; MacKay, N.; Robinson, B. Novel SCO2 mutation (G1521A) presenting as a spinal muscular atrophy type I phenotype. Am. J. Med. Genet. A 2004, 125A, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Horvath, R.; Horn, N.; Auer, D.P.; Macmillan, C.; Peters, J.; Gerbitz, K.; Kraegeloh–Mann, I.; Muntau, A.; Karcagi, V.; et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 2001, 57, 1440–1446. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Cavallaro, G.; Ciofi-Baffoni, S. Seeking the determinants of the elusive functions of Sco proteins. FEBS J. 2011, 278, 2244–2262. [Google Scholar] [CrossRef] [PubMed]
- Morgada, M.N.; Abriata, L.A.; Cefaro, C.; Gajda, K.; Banci, L.; Vila, A.J. Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 11771–11776. [Google Scholar] [CrossRef]
- Lode, A.; Paret, C.; Rödel, G. Molecular characterization of Saccharomyces cerevisiae Sco2p reveals a high degree of redundancy with Sco1p. Yeast Chichester Engl. 2002, 19, 909–922. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Pratt, A.T.; Soma, S.; Theriault, S.G.; Griffin, A.T.; Trivedi, P.P.; Gohil, V.M. Mitochondrial disease genes COA6, COX6B and SCO2 have overlapping roles in COX2 biogenesis. Hum. Mol. Genet. 2016, 25, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Huigsloot, M.; Nijtmans, L.G.; Szklarczyk, R.; Baars, M.J.; van den Brand, M.A.; HendriksFranssen, M.G.; van den Heuvel, L.P.; Smeitink, J.A.; Huynen, M.; Rodenburg, R.J. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Khalimonchuk, O.; Rigby, K.; Bestwick, M.; Pierrel, F.; Cobine, P.A.; Winge, D.R. Pet191 Is a Cytochrome c Oxidase Assembly Factor in Saccharomyces cerevisiae. Eukaryot. Cell 2018, 7, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Longen, S.; Bien, M.; Bihlmaier, K.; Kloeppel, C.; Kauff, F.; Hammermeister, M.; Westermann, B.; Herrmann, J.M.; Riemer, J. Systematic analysis of the twin CX(9)cCprotein family. J. Mol. Biol. 2009, 393, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.; Jeong, M.-Y.; Khalimonchuk, O.; Kim, H.; Winge, D.R. Analysis of Leigh syndrome mutations in the yeast SURF1 homolog reveals a new member of the cytochrome oxidase assembly factor family. Mol. Cell. Biol. 2010, 30, 4480–4491. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, A.B.; Soma, S.; Vicary, A.C.; Zulkifli, M.; Kaur, H.; Gohil, V.M. A yeast suppressor screen links Coa4 to the mitochondrial copper delivery pathway for cytochrome c oxidase. Genetics 2022, 221, iyac090. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.M.; Ardissone, A.; Reyes, A.; Robinson, A.J.; Moroni, I.; Ghezzi, D.; Fernandez-Vizarra, E.; Zeviani, M. COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome c oxidase deficiency. J. Med. Genet. 2016, 53, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, L.; Haack, T.B.; Hardy, S.; Abbink, T.E.; Fernandez-Vizarra, E.; Lamantea, E.; Marchet, S.; Morandi, L.; Moggio, M.; Carrozzo, R.; et al. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2014, 95, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Hedberg-Oldfors, C.; Darin, N.; Thomsen, C.; Lindberg, C.; Oldfors, A. COX deficiency and leukoencephalopathy due to a novel homozygous APOPT1/COA8 mutation. Neurol. Genet. 2020, 6, e464. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bhalla, N. Moonlighting Proteins. Annu. Rev. Genet. 2020, 54, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jeffery, C.J. Moonlighting Proteins in the Fuzzy Logic of Cellular Metabolism. Molecules 2020, 25, 3440. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Wang, M.; Wei, R.; Li, G.; Bi, H.-L.; Jia, Z.; Zhang, M.; Pang, M.; Li, X.; Ma, L.; Tang, Y. SUMOylation of SYNJ2BP-COX16 promotes breast cancer progression through DRP1-mediated mitochondrial fission. Cancer Lett. 2022, 547, 215871. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, D.; Giordo, R.; Kleinbrink, E.L.; Banisad, E.; Grossman, L.I.; Arshad, R.; Stark, A.; Maroun, M.-C.; Lipovich, L.; Fernandez-Madrid, F. Non-coding regions of nuclear-DNA-encoded mitochondrial genes and intergenic sequences are targeted by autoantibodies in breast cancer. Front. Genet. 2022, 13, 970619. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Sähr, H.; Zhao, Z.; Yin, L.; Omlor, G.; Lehner, B.; Fellenberg, J. MiR-127 and miR-376a act as tumor suppressors by in vivo targeting of COA1 and PDIA6 in giant cell tumor of bone. Cancer Lett. 2017, 409, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Beyfuss, K.; Hood, D.A. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. Commun. Free Radic. Res. 2018, 23, 100–117. [Google Scholar] [CrossRef]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Jeyaraju, D.V.; Voisin, V.; Hurren, R.; Xu, C.; Hawley, J.R.; Barghout, S.H.; Khan, D.H.; Gronda, M.; Wang, X.; et al. Disrupting Mitochondrial Copper Distribution Inhibits Leukemic Stem Cell Self-Renewal. Cell Stem Cell 2020, 26, 926–937.e10. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H. Yeast and cancer. Biosci. Rep. 2004, 24, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Vanderwaeren, L.; Dok, R.; Voordeckers, K.; Nuyts, S.; Verstrepen, K.J. Saccharomyces cerevisiae as a Model System for Eukaryotic Cell Biology, from Cell Cycle Control to DNA Damage Response. Int. J. Mol. Sci. 2002, 23, 11665. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.T.; Craven, L.; Russell, O.M.; Turnbull, D.M.; Vincent, A.E. Diagnosis and Treatment of Mitochondrial Myopathies. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Human Protein | Yeast Homologue | Role(s) |
|---|---|---|
| COX1 Module-Associated | ||
| LRPPRC | PET309 | COX1 mRNA stabilization, activation of transcription |
| TACO1 | DPC29 | Translational activator for COX1, other mtDNA transcripts |
| C12ORF62 | COX14 | Regulates COX1 expression, part of MITRAC |
| MITRAC12 | COA3 | Regulates translation of COX1; modulates binding to COX2 module via COX16 |
| COX10 | COX10 | Farnesyl transferase (heme O synthase)—converts heme B to heme O |
| COX15 | COX15 | Heme A synthase—converts heme O intermediate to heme A |
| PET117 | PET117 | Required for oligomerization of COX15, hemylation of COX1 |
| SURF1 | SHY1 | Involved in the final hemylation of COX1 |
| COX11 | COX11 | Delivers copper to COX1 |
| COX2 Module-Associated | ||
| OXA1L | OXA1 | Insertion of mitochondrially encoded subunits into IMM |
| COX16 | COX16 | Chaperone for COX2, recruits SCO proteins; helps COX2 module associate with S2; brings COX1 and COX2 modules together |
| COX18 | COX18 | Insertion of the C-terminus of COX2 in the IMM |
| COX20 | COX20 | Binds to COX2 before and after cleavage; stabilizes complex with SCO proteins |
| PET100 | PET100 | Interacts with MR-1S, PET117 in late stages of biogenesis; essential to assembly in humans; stabilizes S3 intermediate |
| hSCO1 | SCO1 | Insertion of copper into CuA site; hSCO1 associates with PET191 prior to copper delivery by COX17, passes one Cu to COX2. |
| hSCO2 | SCO1 | hSCO2 undergoes disulfide exchange with COX2 and delivers Cu; yeast SCO2 function unknown |
| COA6 | COA6 | Thiol reductase activity, CuA site assembly; perhaps overlapping role with hSCO2 |
| Unspecified Role | ||
| COA5 | PET191 | Essential to human assembly; associates with SCO1 until Cu is delivered |
| Assembly Factor | Phenotype | Citations |
|---|---|---|
| Factors Associated with COX1 Module | ||
| LRPPRC | French-Canadian Leigh syndrome | [23,37] |
| TACO1 | Leigh syndrome, ocular and cognitive impairments | [26,40] |
| COX14 | Fatal neonatal lactic acidosis | [33] |
| COA3 | Obesity, exercise intolerance, short stature, neuropathy | [45] |
| COX10 | Tubulopathy and leukodystrophy, Leigh syndrome and fatal infantile hypertrophic cardiomyopathy, sensorineural hearing loss | [57,58,59] |
| COX15 | Fatal infantile hypertrophic cardiomyopathy, Leigh syndrome | [62,63,64,65] |
| PET117 | Neurodevelopmental regression, medulla oblongata lesions | [72] |
| SURF1 | Leigh syndrome, leukodystrophy, mild encephalopathy, Charcot–Marie–Tooth disease | [76,77,82,83,84] |
| COX11 | Infantile-onset mitochondrial encephalopathy, Leigh-like features | [106,107] |
| Factors Associated with COX2 Module | ||
| OXA1L | Mitochondrial encephalopathy and combined oxidative phosphorylation defect | [32] |
| COX16 | Hypertrophic cardiomyopathy, encephalopathy and severe fatal lactic acidosis, liver dysfunction | [136] |
| COX18 | Neonatal mitochondrial cardioencephalomyopathy and axonal sensory neuropathy | [140] |
| COX20 | Early-onset hypotonia, ataxia, areflexia, dystonia, dysarthria, and sensory neuropathy | [145,146] |
| PET100 | Leigh syndrome, Infantile lactic acidosis | [151,152] |
| SCO1 | Neonatal-onset hepatic failure and encephalopathy, hypertrophic cardiomyopathy | [164,165,166] |
| SCO2 | Fatal infantile cardioencephalomyopathy, hypertrophic cardiomyopathy, spinal muscular atrophy | [154,173,174,175,176] |
| COA6 | Neonatal hypertrophic cardiomyopathy | [180] |
| Unspecified Role | ||
| COA5 | Fatal infantile cardioencephalomyopathy | [182] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caron-Godon, C.A.; Collington, E.; Wolf, J.L.; Coletta, G.; Glerum, D.M. More than Just Bread and Wine: Using Yeast to Understand Inherited Cytochrome Oxidase Deficiencies in Humans. Int. J. Mol. Sci. 2024, 25, 3814. https://doi.org/10.3390/ijms25073814
Caron-Godon CA, Collington E, Wolf JL, Coletta G, Glerum DM. More than Just Bread and Wine: Using Yeast to Understand Inherited Cytochrome Oxidase Deficiencies in Humans. International Journal of Molecular Sciences. 2024; 25(7):3814. https://doi.org/10.3390/ijms25073814
Chicago/Turabian StyleCaron-Godon, Chenelle A., Emma Collington, Jessica L. Wolf, Genna Coletta, and D. Moira Glerum. 2024. "More than Just Bread and Wine: Using Yeast to Understand Inherited Cytochrome Oxidase Deficiencies in Humans" International Journal of Molecular Sciences 25, no. 7: 3814. https://doi.org/10.3390/ijms25073814
APA StyleCaron-Godon, C. A., Collington, E., Wolf, J. L., Coletta, G., & Glerum, D. M. (2024). More than Just Bread and Wine: Using Yeast to Understand Inherited Cytochrome Oxidase Deficiencies in Humans. International Journal of Molecular Sciences, 25(7), 3814. https://doi.org/10.3390/ijms25073814