
Comparative Analysis of Single-Cell RNA Sequencing Methods with and without Sample Multiplexing

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Study Design and Metrics
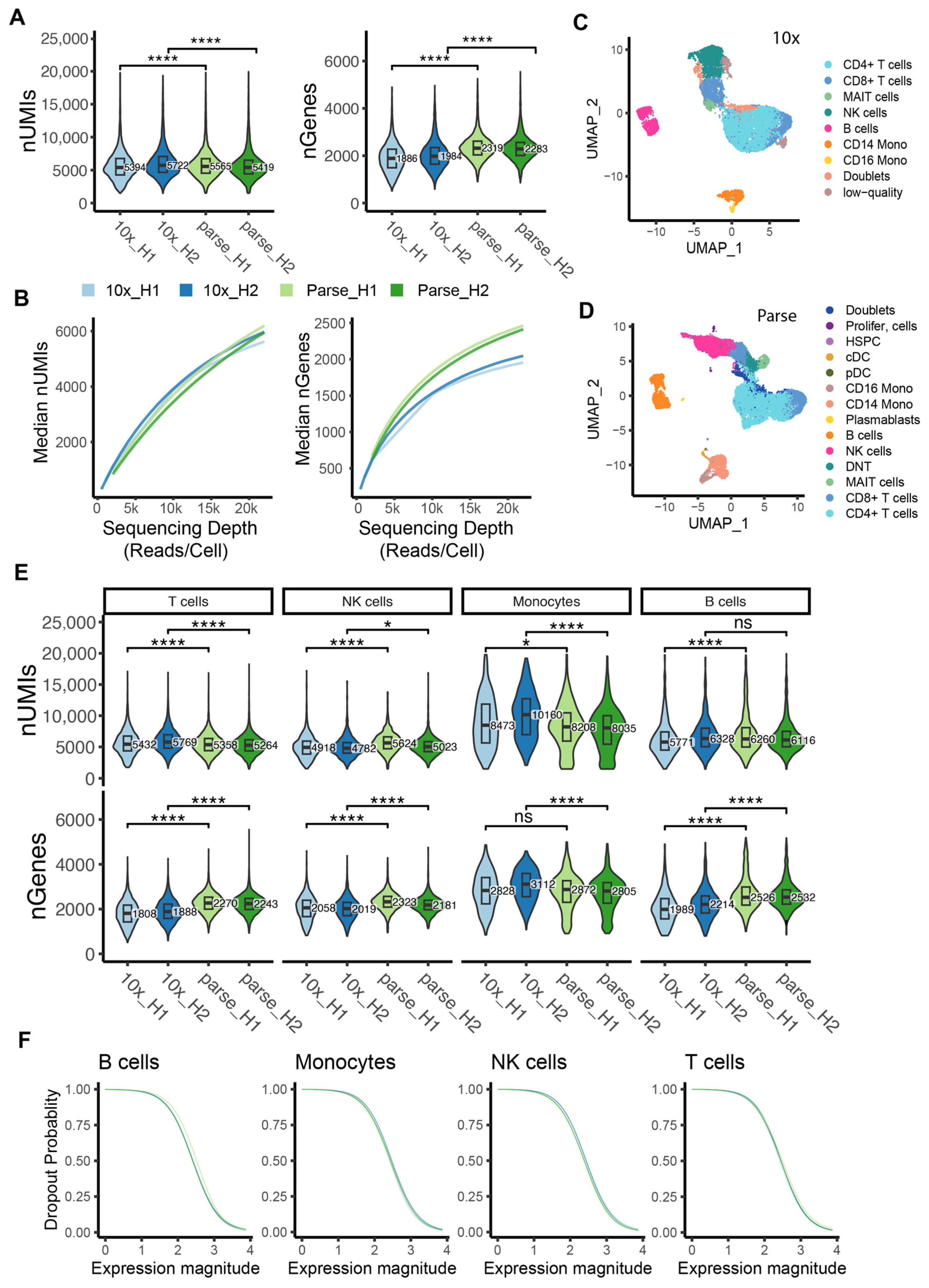
2.2. Comparison of Library Efficiency
2.3. Sensitivity in Gene Detection
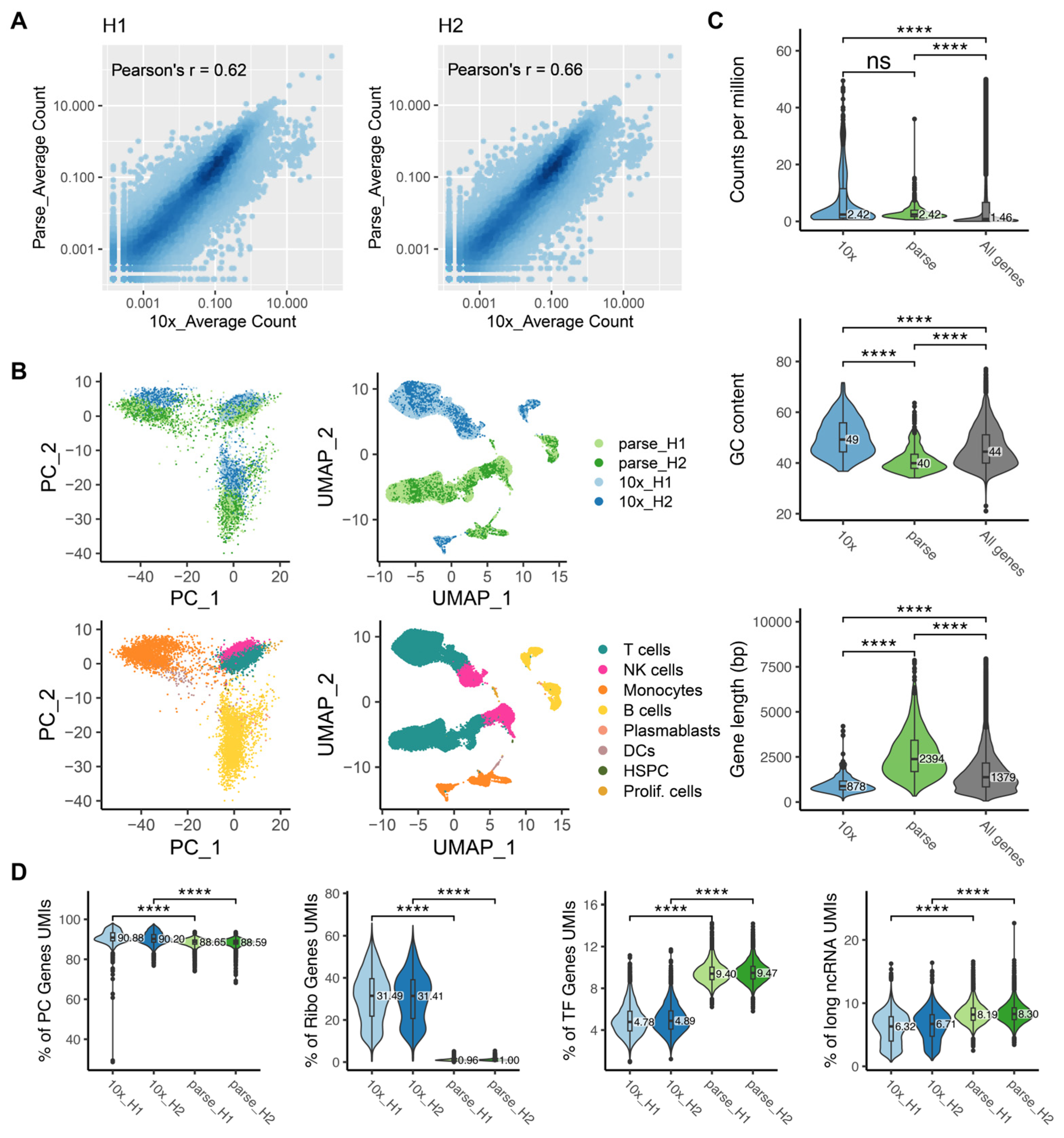
2.4. Bias in Gene Quantification
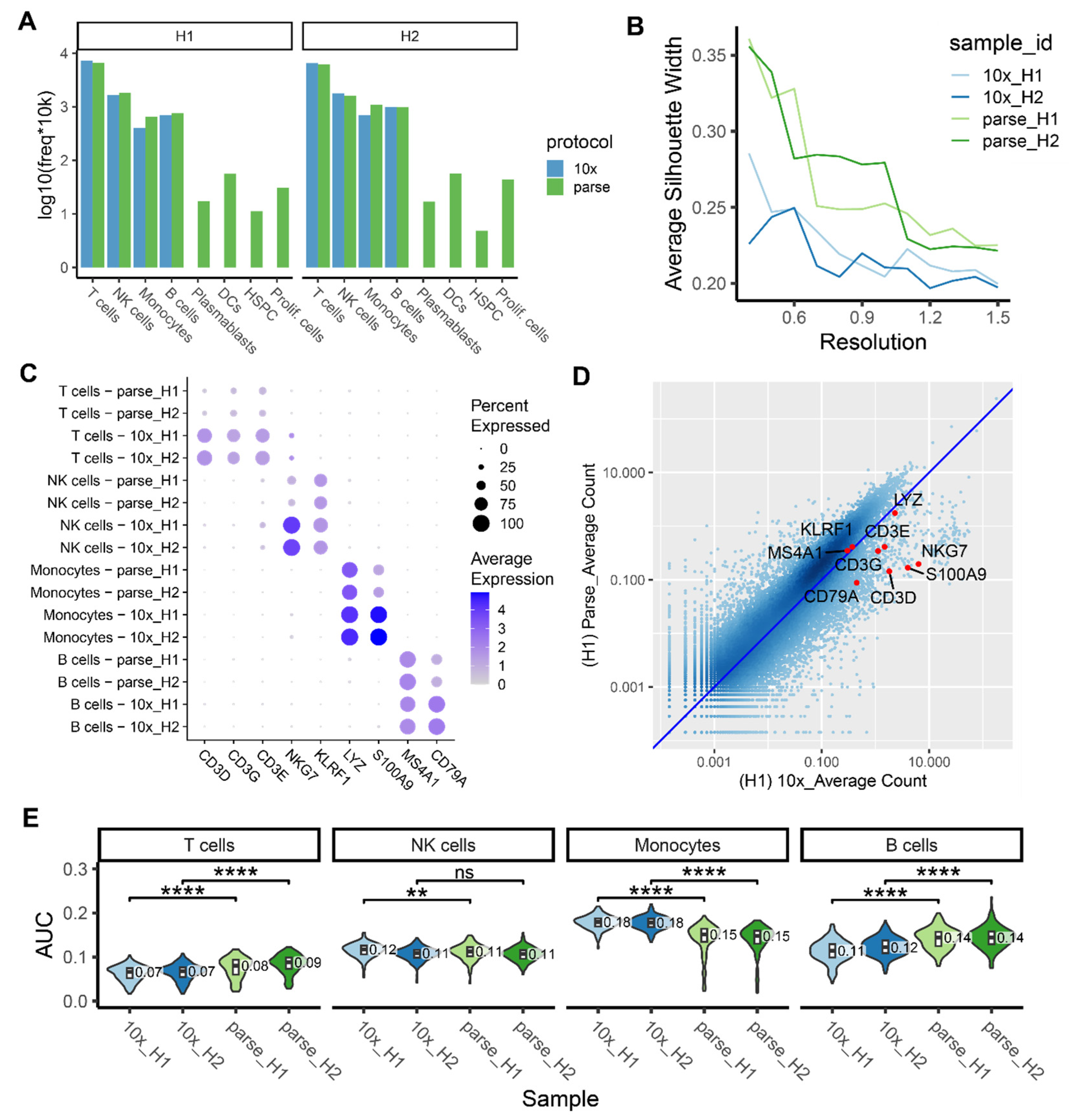
2.5. Ability to Capture and Characterize Cell Type Diversity
3. Discussion
4. Materials and Methods
4.1. Peripheral Blood Mononuclear Cell (PBMC) Collection
4.2. Single-Cell Suspension Preparation
4.3. Parse and 10x Data Downsampling
4.4. Parse and 10x Data Preprocessing and Quality Control
4.5. Cell Type Annotation
4.6. Library Efficiency
4.7. Dropout Probability Estimation
4.8. Correlation Analysis
4.9. Platform-Specific Marker Genes
4.10. Ribosomal Protein-Coding Genes
4.11. Silhouette Score
4.12. Cell-Type Signature Score
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Flores, R.O.R.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.S.; Sikandar, S.S.; Wesche, D.J.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.C.; Townes, F.W.; Teng, M.; Irizarry, R.A. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2018, 19, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.D.; Espeland, M.A.; Langefeld, C.D. A practical solution to pseudoreplication bias in single-cell studies. Nat. Commun. 2021, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liao, J.; Shao, X.; Lu, X.; Fan, X. Multiplexing Methods for Simultaneous Large-Scale Transcriptomic Profiling of Samples at Single-Cell Resolution. Adv. Sci. 2021, 8, e2101229. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-cell sequencing guide for immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed]
- Minervina, A.A.; Pogorelyy, M.V.; Kirk, A.M.; Crawford, J.C.; Allen, E.K.; Chou, C.-H.; Mettelman, R.C.; Allison, K.J.; Lin, C.-Y.; Brice, D.C.; et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8+ T cells. Nat. Immunol. 2022, 23, 781–790. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK+ CD8+ T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e12. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, T.M.; Lu, D.R.; Ellwanger, D.C.; Bhatt, D.; Manzanillo, P.; Arias, V.; Zhou, H.; Yoon, O.K.; Homann, O.; Wang, S.; et al. Systematic comparison of high-throughput single-cell RNA-seq methods for immune cell profiling. BMC Genom. 2021, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Yazar, S.; Alquicira-Hernandez, J.; Wing, K.; Senabouth, A.; Gordon, M.G.; Andersen, S.; Lu, Q.; Rowson, A.; Taylor, T.R.P.; Clarke, L.; et al. Single-cell eQTL mapping identifies cell type–specific genetic control of autoimmune disease. Science 2022, 376, eabf3041. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.; Kliesmete, Z.; Vieth, B.; Adiconis, X.; Simmons, S.; Marshall, J.; McCabe, C.; Heyn, H.; Levin, J.Z.; Enard, W.; et al. The effect of background noise and its removal on the analysis of single-cell expression data. Genome Biol. 2023, 24, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef] [PubMed]
- Rebboah, E.; Reese, F.; Williams, K.; Balderrama-Gutierrez, G.; McGill, C.; Trout, D.; Rodriguez, I.; Liang, H.; Wold, B.J. Mapping and modeling the genomic basis of differential RNA isoform expression at single-cell res-olution with LR-Split-seq. Genome Biol. 2021, 22, 286. [Google Scholar] [CrossRef]
- Mereu, E.; Lafzi, A.; Moutinho, C.; Ziegenhain, C.; McCarthy, D.J.; Álvarez-Varela, A.; Batlle, E.; Sagar; Grün, D.; Lau, J.K.; et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020, 38, 747–755. [Google Scholar] [CrossRef]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chung, L.M.; Zhao, H. Bias detection and correction in RNA-Sequencing data. BMC Bioinform. 2011, 12, 290. [Google Scholar] [CrossRef] [PubMed]
- Phipson, B.; Zappia, L.; Oshlack, A. Gene length and detection bias in single cell RNA sequencing protocols. F1000Research 2017, 6, 595. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, X.; Le, W.; Xie, L.; Li, H.; Wen, W.; Wang, S.; Ma, S.; Huang, Z.; Ye, J.; et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell 2020, 11, 740–770. [Google Scholar] [CrossRef]
- Azuaje, F. A cluster validity framework for genome expression data. Bioinformatics 2002, 18, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018, 175, 1701–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Y.; Zhang, Q.; Ren, X.; Zhang, Z. Direct comparative analyses of 10x genomics chromium and smart-seq2. Genom. Proteom. Bioinform. 2021, 19, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carré, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef]
- Chen, J.; Xu, H.; Tao, W.; Chen, Z.; Zhao, Y.; Han, J.-D.J. Transformer for one stop interpretable cell type annotation. Nat. Commun. 2023, 14, 223. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Naghipourfar, M.; Luecken, M.D.; Khajavi, M.; Büttner, M.; Wagenstetter, M.; Avsec, Ž.; Gayoso, A.; Yosef, N.; Interlandi, M.; et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2022, 40, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, G.; Arias, J.E.R.; Schäfer, P.; Busskamp, V. Automated methods for cell type annotation on scRNA-seq data. Comput. Struct. Biotechnol. J. 2021, 19, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., III; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.; Cambridge Institute of Therapeutic Immunology and Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID-19 BioResource Collaboration; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021, 27, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Wang, X.-M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.-W.; Fan, X.; Xia, P.; Fu, J.-L.; Wang, S.-Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.; Fan, J.; Biederstedt, E. Scde: Single Cell Differential Expression. R Package Version 2.28.2. 2023. Available online: https://pklab.med.harvard.edu/scde (accessed on 15 October 2023).
- Lun, A.T.; McCarthy, D.J.; Marioni, J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLATFORM | 10x | Parse | |
|---|---|---|---|
| STRENGTHS |
|
| |
| WEAKNESSES |
|
| |
| ASSESSED FEATURE | METRICS | 10x | Parse |
| LIBRARY EFFICIENCY | Cell recovery (average %) | 53% | 27% |
| Valid reads (average %) | 98% | 85% | |
| SENSITIVITY IN GENE DETECTION | Number of genes (median [range]) | 1938 [515–4967] | 2302 [860–5548] |
| POTENTIAL BIASES IN GENE QUANTIFICATION | Gene length (median [range]) | 881 bp [255–18,662] | 2394 bp [331–36,798] |
| GC content (median [range]) | 49.4% [36.8–71.6%] | 39.9% [34.1–63.6%] | |
| Ribosomal protein- coding gene abundance (median [range]) | 30.7% [0.4–66.0%] | 1.0% [0–5.3%] | |
| ABILITY TO IDENTIFY CELL TYPES ACCURATELY | Manual annotation1 (average expression2, % of cells expressed3) | B cells: 8.0, 93.9% Monocytes: 102.0, 99.5% NK cells: 26.8, 91.1% T cells: 3.4, 80.3% | B cells: 4.2, 78.0% Monocytes: 14.8, 74.9% NK cells: 2.5, 58.7% T cells: 0.8, 30.4% |
| Reference-based scoring4 (median AUC [range]) | B cells: 0.12 [0.04–0.18] Monocytes: 0.18 [0.12–0.22] NK cells: 0.11 [0.04–0.16] T cells: 0.07 [0.02–0.11] | B cells: 0.14 [0.08–0.24] Monocytes: 0.15 [0.02–0.19] NK cells: 0.11 [0.04–0.16] T cells: 0.08 [0.02–0.12] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Chen, H.; Chellamuthu, V.R.; Lajam, A.b.M.; Albani, S.; Low, A.H.L.; Petretto, E.; Behmoaras, J. Comparative Analysis of Single-Cell RNA Sequencing Methods with and without Sample Multiplexing. Int. J. Mol. Sci. 2024, 25, 3828. https://doi.org/10.3390/ijms25073828
Xie Y, Chen H, Chellamuthu VR, Lajam AbM, Albani S, Low AHL, Petretto E, Behmoaras J. Comparative Analysis of Single-Cell RNA Sequencing Methods with and without Sample Multiplexing. International Journal of Molecular Sciences. 2024; 25(7):3828. https://doi.org/10.3390/ijms25073828
Chicago/Turabian StyleXie, Yi, Huimei Chen, Vasuki Ranjani Chellamuthu, Ahmad bin Mohamed Lajam, Salvatore Albani, Andrea Hsiu Ling Low, Enrico Petretto, and Jacques Behmoaras. 2024. "Comparative Analysis of Single-Cell RNA Sequencing Methods with and without Sample Multiplexing" International Journal of Molecular Sciences 25, no. 7: 3828. https://doi.org/10.3390/ijms25073828
APA StyleXie, Y., Chen, H., Chellamuthu, V. R., Lajam, A. b. M., Albani, S., Low, A. H. L., Petretto, E., & Behmoaras, J. (2024). Comparative Analysis of Single-Cell RNA Sequencing Methods with and without Sample Multiplexing. International Journal of Molecular Sciences, 25(7), 3828. https://doi.org/10.3390/ijms25073828