
What Is the “Hydrogen Bond”? A QFT-QED Perspective


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theoretical Background and Comments on Experimental Data
2.1. Recap of the Main Problems within the Corpuscular QM View (First Quantization)
- 0.05 eV for direct dipole-dipole Keesom interactions (Equation (2))
- 0.03 eV for Debye interactions between permanent and induced dipoles (Equation (3))
- 0.12 eV for dispersive London interactions between two induced dipoles (Equation (4))
“The claim that chemistry has been completely explained in terms of quantum theory is now received wisdom among physicists and chemists. Yet quantum physics is able neither to predict nor explain the strong association of water molecules in liquid or ice. Quantum chemistry algorithms either exclude hydrogen bonded (H-bonded) systems, or treat them by modelling a water molecule as an asymmetric tetrahedron having two positive and two negative electrical charges at its vertices. Recent calculations of the potential energy surface of the simple water dimer {H2O}2 yield 30,000 ab initio energies at the coupled clustering techniques (CCT) level [21]. But free OH-stretches [deviate from] experimental values by 30–40 cm−1 and their dissociation energy 1.1 kJ·mol−1 [are likewise] below benchmark experimental values. To obtain satisfactory agreement with experiment, it is necessary to replace ab initio potentials with spectroscopically accurate measurements. This is hardly a ringing endorsement of the underlying theory”(despite Dirac’s 1929 claims [22]).
2.2. Synthesis of the Theoretical Background in QFT-QED for Liquid Water

- The first case refers to infrared (IR) and near IR (NIR) analysis of water or water solutions spectra (of O-H stretching region, IR, or of its first harmonic, NIR) taken at different temperatures [62,63] whose trends showed the clear existence of an isosbestic points, expressing the existence of two populations of molecules that depend on T in a reciprocal way. This, of course, is not a novelty, but what is worth looking at is the resulting van’t Hoff plots (i.e., the Log (equilibrium constant of the transition from one population to the other) vs. 1/T) are linear, revealing that (i) the energy difference between the two states does not depend on T and (ii) that its slope is in good agreement with the energy gap predicted by QED theory. Furthermore, in [63] it has been shown how the plot of the logarithm of the ratios between the spectral intensity of one population (distinguished from the other one by the isosbestic point) with respect to the total, taken at each temperature, plotted as a function of log T yields a straight line. This accounts for a scale-free behaviour, revealing the coherent dynamics underlying the demonstrated isomorphism existing between self-similar (fractal) topologies and squeezed quantum coherent states [64,65,66].
- The second case deals with fitting of the dielectric permittivity of pure water and electrolyte water solutions in the range 0.2–1.5 THz [61,67]. The fit to the experimental data requires a two-fluid Debye model that mimics the electrical permittivity (both for the real and the imaginary parts). However, in order to be effective over the full spectral range, it requires an additional linear term (ξω, where ξ ≈ 0.47 ps) to the imaginary part of the dielectric function. This fact has a profound physical meaning because it implies the violation of the Kramers–Kronig (KK) relation [68] within the time span ξ. The KK relation expresses the causal relationship between the forcing field and the charge displacement. This tiny violation, on a time scale of the order of the renormalized oscillation period of the coherent field within the CDs (which excite and relax in a few hundred femtoseconds, τr ≈ 1/ωr ~ 300–500 fs) witnesses temporally non-local correlations in the medium (i.e., phase correlations), possible when the system is in an entangled coherent state (a phase eigenstate). As Li has pointed out [69,70], the concept of coherence is closely related to Heisenberg’s uncertainty principle, i.e., coherence space-time being actually equivalent to the uncertainty space-time. This is the region of space and time within which particles lose their classical properties of individuality and countability (the operator becomes undefined). The particles and fields within the coherence space-time region must be considered as an indivisible whole in which the phase is well-defined: thus, what happens to “a part” of a CD, within its coherent space-time range, happens to the whole CD [69]. This is a noteworthy point also for overcoming the prevailing naïve picture of the HBs [40], which are still conceived as forces acting between “particles”. As described, this classic idea originated from the first quantization can be fruitfully replaced by the QFT perspective (second quantization) where the apparent (non-directional) force is the emergent property resulting from an energy gradient that is NOT primarily bound to the binding between molecules, but is established at the ground energy level (vacuum) [37] as a consequence of the “em-field + matter-field” coupling over the whole high-numbered system [39].
- Another crucial issue concerns ions and their solvation in water. Within an electrostatic conception of the dissolution of electrolytes in water, the initial dynamics has no physical consistency, since few layers of water molecules should be able to keep some Na+ and Cl− ions away from their crystal lattice, if the energy barrier to be overcome—in order to break the ionic bond—is in the order of 5 eV. A single water layer could at most produce a dielectric drop in the Coulomb force equal to 13 (εr = 13 and not εr = 80 which applies to bulk water). Again, only by abandoning an ingenuous “sticks-and-balls” interpretation of condensed matter, and by taking into account the quantum electrodynamic nature of objects like ions and their coupling with the vacuum, is it possible to consistently describe the spontaneous solvation process, showing that ions in the incoherent fraction of water establish their own coherence domains with their energy gaps (larger than the ion-bond energy) and dissolve in the liquid phase of the solvent without collisions [17]. This explains (i) why the solvent power of water increases with rising temperatures (although the net value of the bulk dielectric permittivity decreases), (ii) why there is no emission of bremsstrahlung radiation from an electrolyte solution and (iii) why the phenomenon of ion-cyclotron resonance occurs [71].
- There are numerous other cases, which we will only briefly mention here, as they are beyond the scope of this topic and, therefore, will be dealt with in future papers. These include the morphogenic role of water in biological matter [72], interfacial water [73], dispersion properties of biologically bound water upon exposure in the 10 Hz to 100 GHz range [74], floating water bridge [75], burning salt water upon RF-exposure [76], branching chain reaction of water [77], coherent water and cellular information processing [78] as well as stable water mixtures of both hydrophobic/hydrophilic liquids [79].
3. Discussion
- water is necessarily a two-fluid system, like already proposed by Röntgen more than a century ago [81];
- the two phases in liquid water differ from one another for much deeper physical reasons than “different arrangements” (moreover unjustifiable) of the classical “HB-networks”;
- the short-range (electrostatic or perturbatively electrodynamic) forces—such as van der Waals interactions—act mainly in the non-coherent fraction and do not change their typicality depending on the aggregation state (clusters, normal liquid, supercooled liquid, types of ice, etc.) and, together with the long-range forces, determine the maximum achievable close-packing density in coherent fraction;
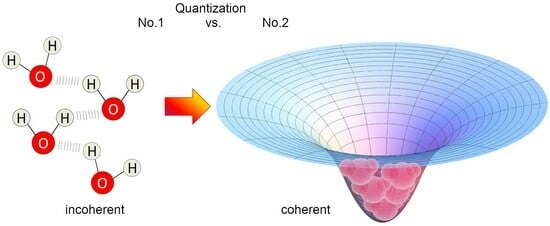
- the main cause of the cohesion of the system cannot be attributed primarily to local, directional, short-range forces among molecules (which, even if attractive, would not be sufficient at room temperature [14,39,40]). Instead, the emergence of a coherent matter field consisting of in-phase oscillating electric charges and photons creates potential wells (as large as the volume of the photons) at the ground level (vacuum) that are experienced by nearby molecules. An analogy can be made with marbles placed on an elastic cloth, which cluster together in the depression created by their own weight (if they are sufficiently close to one another, i.e., dense enough), and not by the existence of a net attractive force between them. Coherence causes water molecules to flip into such a minimum potential energy well, see Figure 7;
- the differences found experimentally in the emergent intermolecular “attraction”, called “hydrogen bonding” in a QM-corpuscular perspective, are due to the dependence on the energy-well profile within the CD. In this way, we can understand why this apparent “intermolecular” force depends on the thermodynamic boundary conditions and on the type of aggregation experienced by the molecules (see Figure 8).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Definition of the Hydrogen Bond within First Quantization
- (E1)
- The forces involved in the formation of a hydrogen bond include those of an electrostatic origin, those arising from charge transfer between the donor and acceptor leading to partial covalent bond formation between H and Y, and those originating from dispersion.
- (E2)
- The atoms X and H are covalently bonded to one another and the X–H bond is polarized, the H•••Y bond strength increasing with the increase in electronegativity of X.
- (E3)
- The X–H•••Y angle is usually linear (180°) and the closer the angle is to 180°, the stronger is the hydrogen bond and the shorter is the H•••Y distance.
- (E4)
- The length of the X–H bond usually increases on hydrogen bond formation leading to a red shift in the infrared X–H stretching frequency and an increase in the infrared absorption cross-section for the X–H stretching vibration. The greater the lengthening of the X–H bond in X–H•••Y, the stronger is the H•••Y bond. Simultaneously, new vibrational modes associated with the formation of the H•••Y bond are generated.
- (E5)
- The X–H•••Y–Z hydrogen bond leads to characteristic NMR signatures that typically include pronounced proton deshielding for H in X–H, through hydrogen bond spin–spin couplings between X and Y, and nuclear Overhauser enhancements.
- (E6)
- The Gibbs energy of formation for the HB should be greater than the thermal energy of the system for the hydrogen bond to be detected experimentally.
- (C1)
- The pKa of X–H and pKb of Y–Z in a given solvent correlate strongly with the energy of the hydrogen bond formed between them.
- (C2)
- Hydrogen bonds are involved in proton-transfer reactions (X–H•••Y → X•••H–Y) and may be considered the partially activated precursors to such reactions.
- (C3)
- Networks of hydrogen bonds can show the phenomenon of co-operativity, leading to deviations from pair-wise additivity in hydrogen bond properties.
- (C4)
- Hydrogen bonds show directional preferences and influence packing modes in crystal structures.
- (C5)
- Estimates of charge transfer in hydrogen bonds show that the interaction energy correlates well with the extent of charge transfer between the donor and the acceptor.
- (C6)
- Analysis of the electron density topology of hydrogen-bonded systems usually shows a bond path connecting H and Y and a (3, −1) bond critical point between H and Y.
References
- Simons, J. Hydrogen Fluoride and its Solutions. Chem. Rev. 1931, 8, 213–235. [Google Scholar] [CrossRef]
- Nernst, W. Verteilung eines Stoffes zwischen zwei Lösungsmitteln und zwischen Lösungsmittel und Dampfraum. Zeitschr Phys. Chem. 1891, 8, 110–139. [Google Scholar] [CrossRef]
- Moore, T.; Winmill, T. The States of Amines in Aqueous Solutions. J. Chem. Soc. Trans. 1912, 101, 1635. [Google Scholar] [CrossRef]
- Bragg, B.W. The Crystal Structure of Ice. Proc. Phys. Soc. Lond. 1922, 34, 98–103. [Google Scholar] [CrossRef]
- Huggins, M.L. 50 Years of Hydrogen Bond Theory. Angew. Chem. Int. Ed. 1971, 10, 147–152. [Google Scholar] [CrossRef]
- Pauling, L. The Shared-Electron Chemical Bond. Proc. Natl. Acad. Sci. USA 1928, 14, 359–362. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond. Application of Results Obtained from the Quantum Mechanics and from a Theory of Paramagnetic Susceptibility to the Structure of Molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Pauling, L.; Brockway, L. The Structure of the Carboxyl Group: I. The Investigation of Formic Acid by the Diffraction of Electrons. Proc. Natl. Acad. Sci. USA 1934, 20, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.; Fowler, R. A Theory of Water and Ionic Solution, with Particular Reference to Hydrogen and Hydroxyl Ions. J. Chem. Phys. 1933, 1, 515. [Google Scholar] [CrossRef]
- Pauling, L. The Structure and Entropy of Ice and of Other Crystals with Some Randomness of Atomic Arrangement. J. Am. Chem. Soc. 1935, 57, 2680–2684. [Google Scholar] [CrossRef]
- Zachariasen, W.H. The Liquid “Structure” of Methyl Alcohol. J. Chem. Phys. 1935, 3, 158–161. [Google Scholar] [CrossRef]
- Elangannan, A.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.; Branson, H. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Henry, M. The Hydrogen Bond. Inference: Int. Rev. Sci. 2015, 1, 1–19. Available online: https://www.researchgate.net/publication/339900495_The_hydrogen_bond (accessed on 24 January 2024). [CrossRef]
- Henry, M. The topological and quantum structure of zoemorphic water. In Aqua Incognita: Why Ice Floats on Water and Galileo 400 Years on; Nostro, P.L., Ninham, B.W., Eds.; Connor Court Publisher: Ballarat, VIC, Australia, 2014; pp. 197–239. ISBN 978-1925138214. [Google Scholar]
- Henry, M. L’Eau et la Physique Quantique (Water and Quantum Physics—Progressing towards a Medical Revolution, in French); Dangles Editions: Escalquens, France, 2016; ISBN 978-2703311478. [Google Scholar]
- Del Giudice, E.; Preparata, G. QED coherence and electrolyte solutions. J. Electroanal. Chem. 2000, 482, 110–116. [Google Scholar] [CrossRef]
- Matcha, R.L.; King, S.C. Theory of the chemical bond. 1. Implicit perturbation theory and dipole moment model for diatomic molecules. J. Am. Chem. Soc. 1976, 98, 3415–3420. [Google Scholar] [CrossRef]
- Ghanty, T.K.; Staroverov, V.N.; Koren, P.R.; Davidson, E.R. Is the Hydrogen Bond in Water Dimer and Ice Covalent? J. Am. Chem. Soc. 2000, 122, 1210. [Google Scholar] [CrossRef]
- Isaacs, E.D.; Shukla, A.; Platzman, P.M.; Hamann, D.R.; Barbiellini, B.; Tulk, C. Covalency of the Hydrogen Bond in Ice: A Direct X-Ray Measurement. Phys. Rev. Lett. 1999, 82, 600–603. [Google Scholar] [CrossRef]
- Shank, A.; Wang, Y.M.; Kaledin, A.; Braams, B.J.; Bowman, J.M. Accurate ab initio and “hybrid” potential energy surfaces, intramolecular vibrational energies, and classical ir spectrum of the water dimer. J. Chem. Phys. 2009, 130, 144314. [Google Scholar] [CrossRef]
- Dirac, P. Quantum Mechanics of Many-Electron Systems. Proc. R. Soc. Lond. A 1929, 123, 714–733. [Google Scholar] [CrossRef]
- Flurry, R.L. Symmetry Groups: Theory and Chemical Applications; Prentice-Hall: Hoboken, NJ, USA, 1980; ISBN 0-13-880013-8. [Google Scholar]
- Chaplin, M. Water Structure and Science—Molecular Orbitals for Water (H2O). 2022. Available online: https://water.lsbu.ac.uk/water/h2o_orbitals.html (accessed on 24 January 2024).
- Locke, W. Introduction to Molecular Orbital Theory; ICSTM Department of Chemistry; Imperial College: London, UK, 1996; Available online: https://www.ch.ic.ac.uk/vchemlib/course/mo_theory/ (accessed on 24 January 2024).
- Nilsson, A.; Ogasawara, H.; Cavalleri, M.; Nordlund, D.; Nyberg, M.; Wernet, P.; Pettersson, L.G.M. The hydrogen bond in ice probed by soft X-ray spectroscopy and density functional theory. J. Chem. Phys. 2005, 122, 154505. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Silvestrelli, P.; Parrinello, M. Compton scattering and the character of the hydrogen bond in ice Ih. J. Chem. Phys. 2001, 115, 115. [Google Scholar] [CrossRef]
- Bader, V.R. Atoms in Molecules: A Quantum Theory; Clanderson Press: Oxford, UK, 1990; Volume 22. [Google Scholar] [CrossRef]
- Guo, J.H.; Luo, Y.; Augustsson, A.; Rubensson, J.E.; Såthe, C.; Ågren, H.; Siegbahn, H.; Nordgren, J. X-ray Emission Spectroscopy of Hydrogen Bonding and Electronic Structure of Liquid Water. Phys. Rev. Lett. 2002, 89, 137402. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.; Edgecombe, K. A simple measure of electron localization in atomic and molecular systems. J. Ournal. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Kumar, A.; Gadre, S.R.; Mohan, N.; Suresh, C.H. Lone Pairs: An Electrostatic Viewpoint. J. Phys. Chem. 2014, 118, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Gürtler, P.; Saile, V.; Koch, E. Rydberg series in the absorption spectra of H2O and D2O in the vacuum ultraviolet. Chem. Phys. Lett. 1977, 51, 386–391. [Google Scholar] [CrossRef]
- Price, W.C. The Far Ultraviolet Absorption Spectra and Ionization Potentials of H2O and H2S. J. Chem. Phys. 1936, 4, 147–153. [Google Scholar] [CrossRef]
- Bukowski, R.; Szalewicz, K.; Groenenboom, G.C.; van der Avoird, A. Predictions of the Properties of Water from First Principles. Science 2007, 315, 1249–1252. [Google Scholar] [CrossRef]
- Wernet, P.; Nordlund, D.; Bergmann, U.; Cavalleri, M.; Odelius, M.; Ogasawara, H.; Näslund, L.A.; Hirsch, T.K.; Ojamäe, L.; Glatzel, P.; et al. The Structure of the First Coordination Shell in Liquid Water. Science 2004, 304, 995–999. [Google Scholar] [CrossRef]
- Teixeira, J. Experimental determination of the nature of diffusive motions of water molecules at low temperatures. Phys. Rev. A 1985, 31, 1913–1917. [Google Scholar] [CrossRef]
- Preparata, G. QED Coherence in Matter; World Scientific: Singapore, 1995. [Google Scholar] [CrossRef]
- Arani, R.; Bono, I.; Del Giudice, E.; Preparata, G. QED coherence and the thermodynamics of water. Int. J. Mod. Phys. B 1995, 9, 1813–1841. [Google Scholar] [CrossRef]
- Bono, I.; Del Giudice, E.; Gamberale, L.; Henry, M. Emergence of the Coherent Structure of Liquid Water. Water 2012, 4, 510–532. [Google Scholar] [CrossRef]
- Del Giudice, E.; Galimberti, U.; Gamberale, L.; Preparata, G. Electrodynamic Coherence in water: A possible origin of the tetrahedral coordination. Mod. Phys. Lett. B 1995, 9, 953–961. [Google Scholar] [CrossRef]
- Preparata, G.; Del Giudice, E.; Vitiello, G. Water as a Free Electric Dipole Laser. Phys. Rev. Lett. 1988, 61, 1085–1088. [Google Scholar] [CrossRef]
- Rao, C.R.R. Theory of Hydrogen Bonding in Water. In Water a Comprehensive Treatise in 7 Volumes, 2nd ed.; Franks, F., Ed.; Plenum Press: New York, NY, USA, 1972; Volume 1. [Google Scholar] [CrossRef]
- Preparata, G. An Introduction to a Realistic Quantum Physics; World Scientific: Singapore, 2002. [Google Scholar] [CrossRef]
- von Neumann, J. Mathematical Foundations of Quantum Theory; Princeton University Press: Princeton, NJ, USA, 1955; ISBN 9780691178561. [Google Scholar]
- Blasone, M.; Jizba, J.; Vitiello, G. Quantum Field Theory and Its Macroscopic Manifestations; Imperial College Press: London, UK, 2011. [Google Scholar] [CrossRef]
- Lamb, W.E.; Retherford, R.C. Fine structure of the hydrogen atom by a microwave method. Phys. Rev. Lett. 1947, 72, 241–243. [Google Scholar] [CrossRef]
- Teixeira, J.; Luzar, A. Physics of liquid water: Structure and dynamics. In Hydration Processes in Biology: Theoretical and Experimental Approaches; Bellissent-Funel, M., Ed.; IOS Press: Amsterdam, The Netherlands, 1999; ISBN 9-051-9943-9-7. [Google Scholar]
- Anderson, P. Coherent Excited States in the Theory of Superconductivity: Gauge Invariance and the Meissner Effect. Phys. Rev. 1958, 110, 827–835. [Google Scholar] [CrossRef]
- Anderson, P. Basic Notions of Condensed Matter Physics; Basic Books; CRC Press: Boca Raton, FL, USA, 1984; ISBN 9780429494116. [Google Scholar] [CrossRef]
- Del Giudice, E.; Vitiello, G. The Role of the electromagnetic field in the formation of domains in the process of symmetry-breaking phase transitions. Phys. Rev. A 2006, 74, 022105. [Google Scholar] [CrossRef]
- Tokushima, T.; Harada, Y.; Takahashi, O.; Senba, Y.; Ohashi, H.; Pettersson, L.G.; Nilsson, A.; Shin, S. High resolution X-ray emission spectroscopy of liquid water: The observation of two structural motifs. Chem. Phys. Lett. 2008, 460, 387–400. [Google Scholar] [CrossRef]
- Huang, C.; Wikfeldt, K.; Tokushima, T.; Nordlund, D.; Harada, Y.; Bergmann, U.; Niebuhr, M.; Weiss, T.M.; Horikawa, Y.; Leetmaa, M.; et al. The inhomogeneous structure of water at ambient conditions. Proc. Natl. Acad. Sci. USA 2009, 106, 15214–15218. [Google Scholar] [CrossRef]
- Taschin, A.; Bartolini, P.; Eramo, R.; Righini, R.; Torre, R. Evidence of two distinct local structures of water from ambient to supercooled conditions. Nat. Comm. 2013, 4, 2401. [Google Scholar] [CrossRef]
- Garbelli, A. Proprietà Termodinamiche e Dielettriche Dell’acqua alla luce della Teoria Complessa delle Interazioni Molecolari Rlettrodinamiche ed Elettrostatiche (Thermodynamic and Dielectric Properties of Water in the Light of the Complex Theory of Electrodinamic and Electrostatic Molecular Interactions). Ph.D. Thesis, University of Milan, Milan, Italy, 2000. [Google Scholar]
- Del Giudice, E.; Tedeschi, A. Water and Autocatalysis in Living Matter. Electromagn. Biol. Med. 2009, 28, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, E.; Spinetti, P.R.; Tedeschi, A. Water Dynamics at the Root of Metamorphosis in Living Organisms. Water 2012, 2, 566–586. [Google Scholar] [CrossRef]
- Del Giudice, E.; Voeikov, V.; Tedeschi, A.; Vitiello, G. The origin and the special role of coherent water in living systems. In Fields of the Cell; Fels, M.C.D., Ed.; Research Signpost: Trivandrum, India, 2015; pp. 95–111. [Google Scholar] [CrossRef]
- Renati, P. Electrodynamic Coherence as a Physical Basis for Emergence of Perception, Semantics, and Adaptation in Living Systems. J. Genet. Mol. Cell. Biol. 2020, 7, 2020110686. [Google Scholar] [CrossRef]
- Madl, P.; Renati, P. Quantum Electrodynamics Coherence and Hormesis: Foundations of Quantum Biology. Int. J. Mol. Sci. 2023, 24, 14003. [Google Scholar] [CrossRef] [PubMed]
- Buzzacchi, M.; Del Giudice, E.; Preparata, G. Coherence of the Glassy State. Int J Mod Phys B 2002, 16, 3771–3786. [Google Scholar] [CrossRef]
- De Ninno, A.; Nikollari, E.; Missori, M.; Frezza, F. Dielectric permittivity of aqueous solutions of electrolytes probed by THz time-domain and FTIR spectroscopy. Phys. Lett. A 2020, 384, 126865. [Google Scholar] [CrossRef]
- De Ninno, A.; Del Giudice, E.; Gamberale, L.; Castellano, C. The structure of liquid water emerging from the vibrational spectroscopy interpretation with QED theory. Water 2014, 6, 13–25. [Google Scholar] [CrossRef]
- Renati, P.; Kovacs, Z.; De Ninno, A.; Tsenkova, R. Temperature dependence analysis of the NIR spectra of liquid water confirms the existence of two phases, one of which is in a coherent state. J. Mol. Liq. 2019, 292, 111449. [Google Scholar] [CrossRef]
- Celeghini, E.; De Martino, S.; De Siena, S.; Rasetti, M.; Vitiello, G. Quantum Groups, Coherent States, Squeezing and Lattice Quantum Mechanics. Ann. Phys. 1995, 241, 50–67. [Google Scholar] [CrossRef]
- Celeghini, E.; Rasetti, M.; Vitiello, G. Quantum dissipation. Ann. Phys. 1992, 215, 156–170. [Google Scholar] [CrossRef]
- Vitiello, G. Coherent states, fractals and brain waves. New Math. Nat. Comp. 2009, 5, 245–264. [Google Scholar] [CrossRef]
- Nikollari, E.; De Ninno, A.; Frezza, A. Dielectric response of liquid water and aqueous solutions: Two-fluid behaviour. In Proceedings of the 3rd European Aquaphotomics Conference, Rome, Italy, 2–4 September 2023. [Google Scholar]
- Toll, J.S. Causality and the Dispersion Relation: Logical Foundations. Phys. Rev. J. Arch. 1956, 104, 1760–1770. [Google Scholar] [CrossRef]
- Li, K. Uncertainty Principle, Coherence and Structures. In On Self-Organization; Springer Series in Synergetics; Springer: Berlin/Heidelberg, Germany, 1994; Volume 61, pp. 113–155. [Google Scholar] [CrossRef]
- Li, K. Coherence in physics and biology. In Recent Advances in Biophoton Research and Its Applications; Popp, F., Li, K., Gu, Q., Eds.; World Scientific Publishing: Singapore, 1992. [Google Scholar] [CrossRef]
- Del Giudice, E.; Fleischmann, M.; Preparata, G.; Talpo, G. On the “unreasonable” effects of ELF magnetic fields upon a system of ions. Bioelectromagnetics 2002, 23, 522–530. [Google Scholar] [CrossRef]
- Henry, M. L’eau Morphogenique—Sante, Information et Champs de Coscience, Escalquens; Éditions Dangles: Labege France, 2020; ISBN 978-2-7033-1269-7. [Google Scholar]
- Pollack, G.H. The Fourth Phase of Water—Beyond Solid, Liquid, and Vapor; Ebner & Sons: Seattle, WA, USA, 2013; ISBN 978-0962-6895-4-3. [Google Scholar]
- Schwan, H.P. Field interaction with biological matter. Ann. N. Y. Acad. Sci. 1977, 303, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.C.; Woisetschläger, J.; Gatterer, K.; Maier, E.; Pecnik, R.; Holler, G.; Eisenkölbl, H. The floating water bridge. J. Phys. D Appl. Phys 2007, 40, 6112–6114. [Google Scholar] [CrossRef]
- Roy, R.; Rao, M.L.; Kanzius, J. Observations of polarised RF radiation catalysis of dissociation of H2O–NaCl solutions. Mater. Res. Innov. 2008, 12, 3–6. [Google Scholar] [CrossRef]
- Voeikov, V. Reactive oxygen species, water, photons and life. Riv. Biol. 2010, 103, 321–342. [Google Scholar] [PubMed]
- Henry, M. De l’information a l’exformation-une Historie de vide, d’eau ou d’AND? (From Information to Exformation—A Story of Vacuum, Water or DNA?); NAQ: Strasbourg, France, 2015; ISBN 10-95620-03-7. [Google Scholar]
- Schauberger, W. Verfahren und Vorrichtung zur Herstellung von Gemischen, Lösungen, Emulsionen, Suspensionen u. dgl. sowie zur biologischen Reinigung von freien Gewässern. (Process and Apparatus for the Preparation of Mixtures, Solutions, Emulsions, Suspensions and the Like and for the Biological Purification of Open Waters). Patent 265991, 3 June 1966. Available online: https://depatisnet.dpma.de/DepatisNet/depatisnet?action=bibdat&docid=AT000000265991B (accessed on 24 January 2024).
- Robinson, G.W.; Cho, C.H.; Urquidi, J. Isosbestic points in liquid water: Further strong evidence for the two-state mixture model. J. Chem. Phys. 1999, 111, 698. [Google Scholar] [CrossRef]
- Röntgen, W. Über die Constitution des flüssigen Wassers. Ann. Phys. 1892, 281, 91–97. [Google Scholar] [CrossRef]
- Buzzacchi, M.; Del Giudice, E.; Preparata, G. Glasses: A new view from QED coherence. arXiv 1999, arXiv:cond-mat/9906395. [Google Scholar] [CrossRef]
- Chaplin, M. Water Absorption Spectrum. Retrieved from Water Structure and Science. Available online: https://water.lsbu.ac.uk/water/water_anomalies.html (accessed on 24 January 2024).
- Nilsson, A.; Petterson, L. The structural origin of anomalous properties of liquid water. Nat. Commun. 2015, 6, 8998. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renati, P.; Madl, P. What Is the “Hydrogen Bond”? A QFT-QED Perspective. Int. J. Mol. Sci. 2024, 25, 3846. https://doi.org/10.3390/ijms25073846
Renati P, Madl P. What Is the “Hydrogen Bond”? A QFT-QED Perspective. International Journal of Molecular Sciences. 2024; 25(7):3846. https://doi.org/10.3390/ijms25073846
Chicago/Turabian StyleRenati, Paolo, and Pierre Madl. 2024. "What Is the “Hydrogen Bond”? A QFT-QED Perspective" International Journal of Molecular Sciences 25, no. 7: 3846. https://doi.org/10.3390/ijms25073846