
Endoplasmic Reticulum Stress Induces Vasodilation in Liver Vessels That Is Not Mediated by Unfolded Protein Response
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
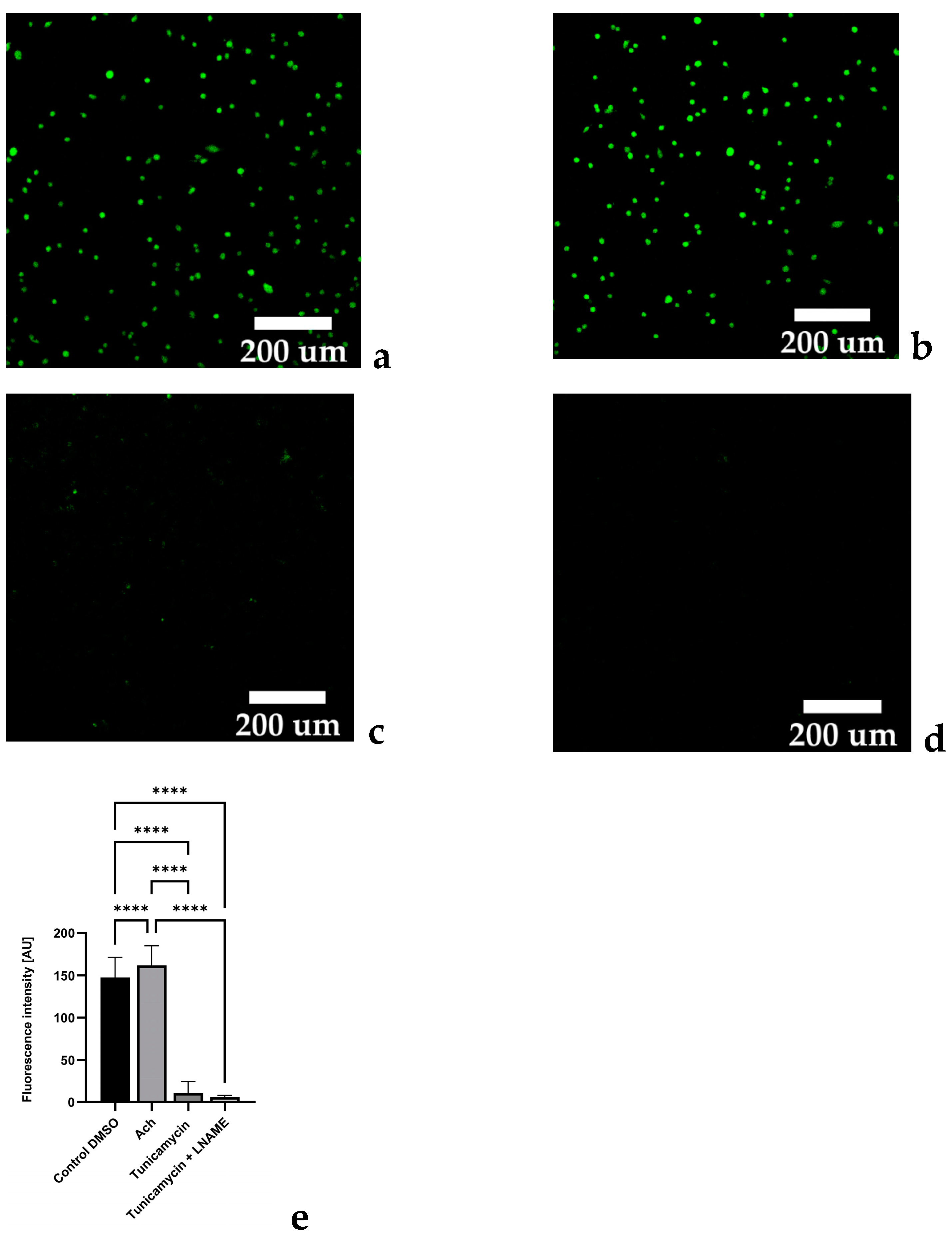
2.1. Tunicamycin Induces Protein Aggregation Characteristics for ER Stress
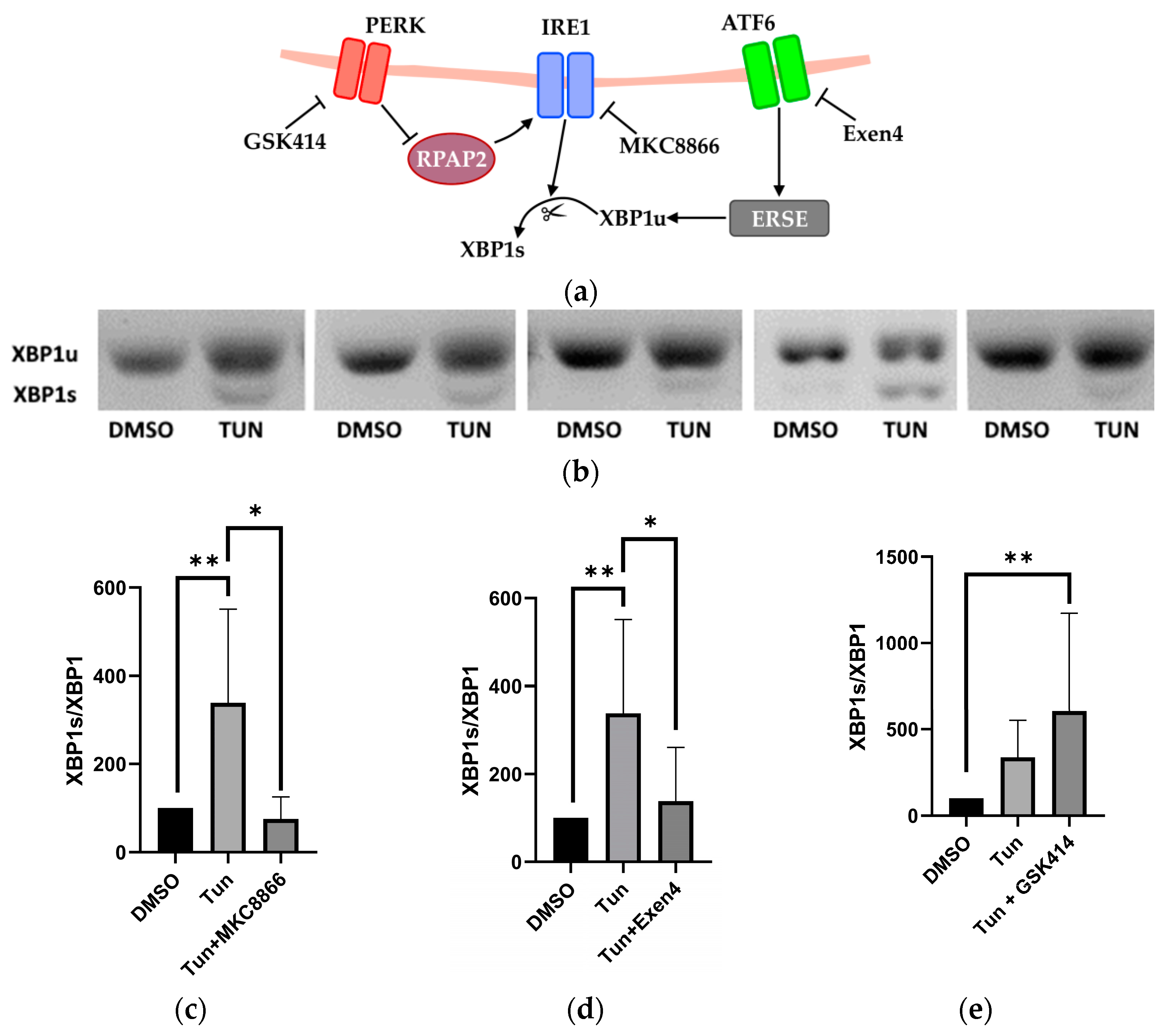
2.2. Activation and Inhibition of UPR Pathways
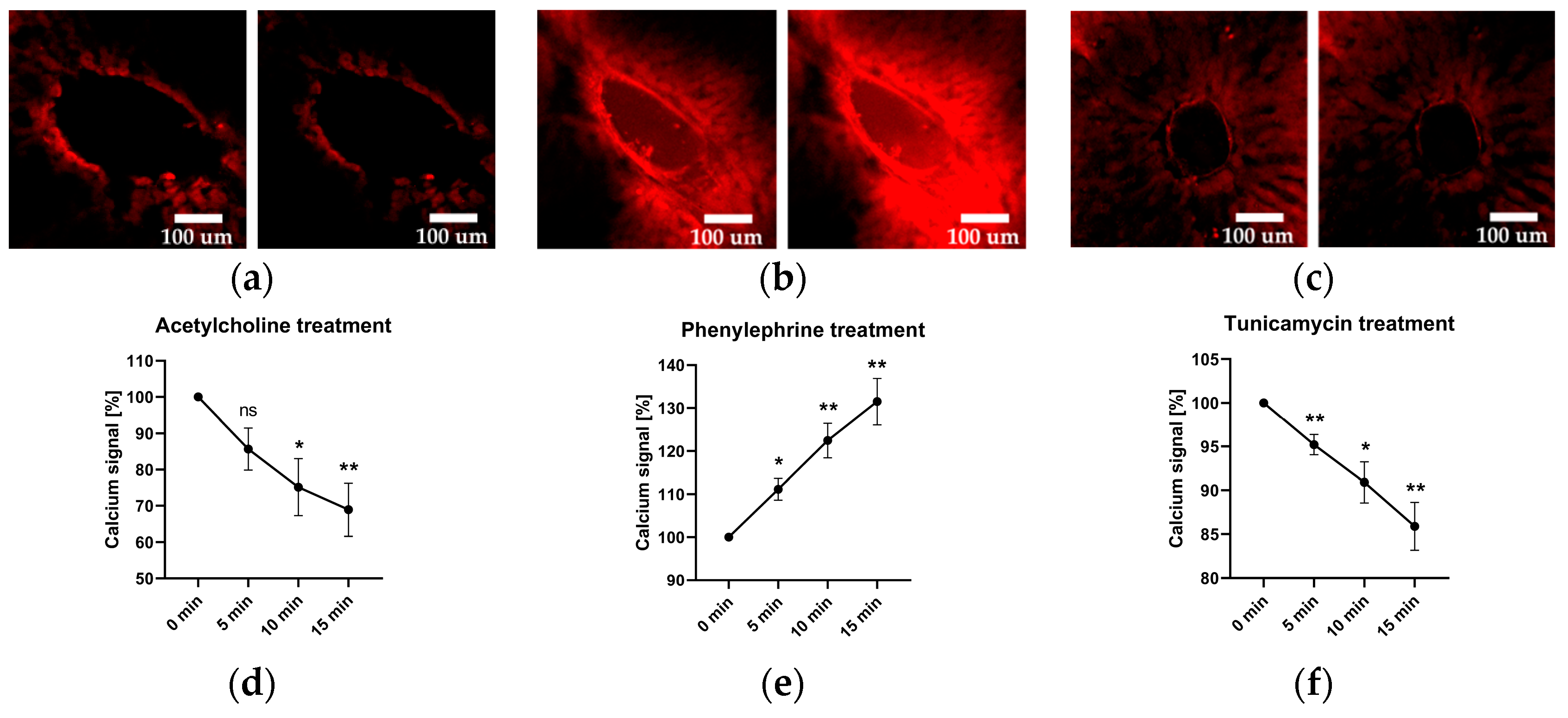
2.3. Vascular Calcium Levels
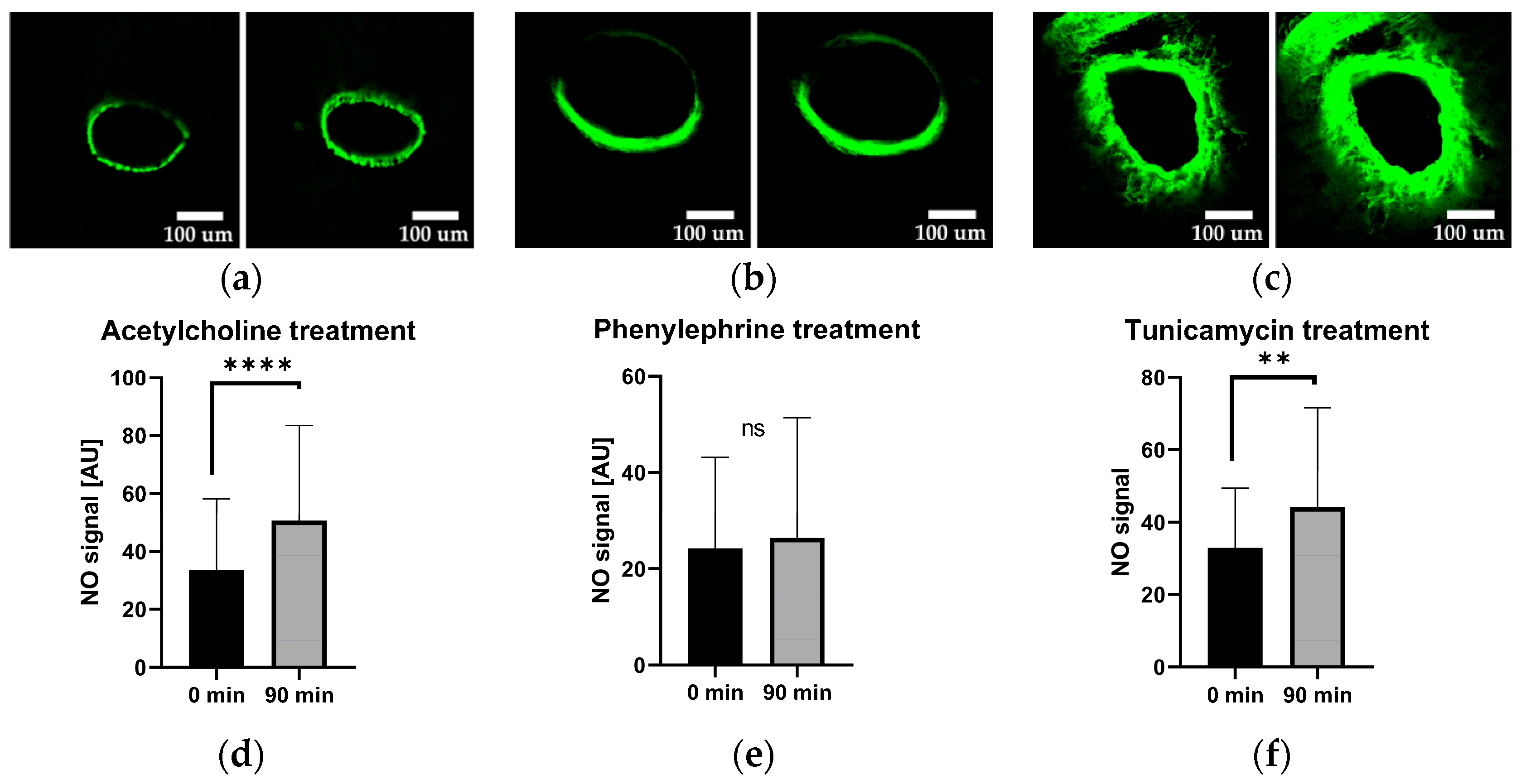
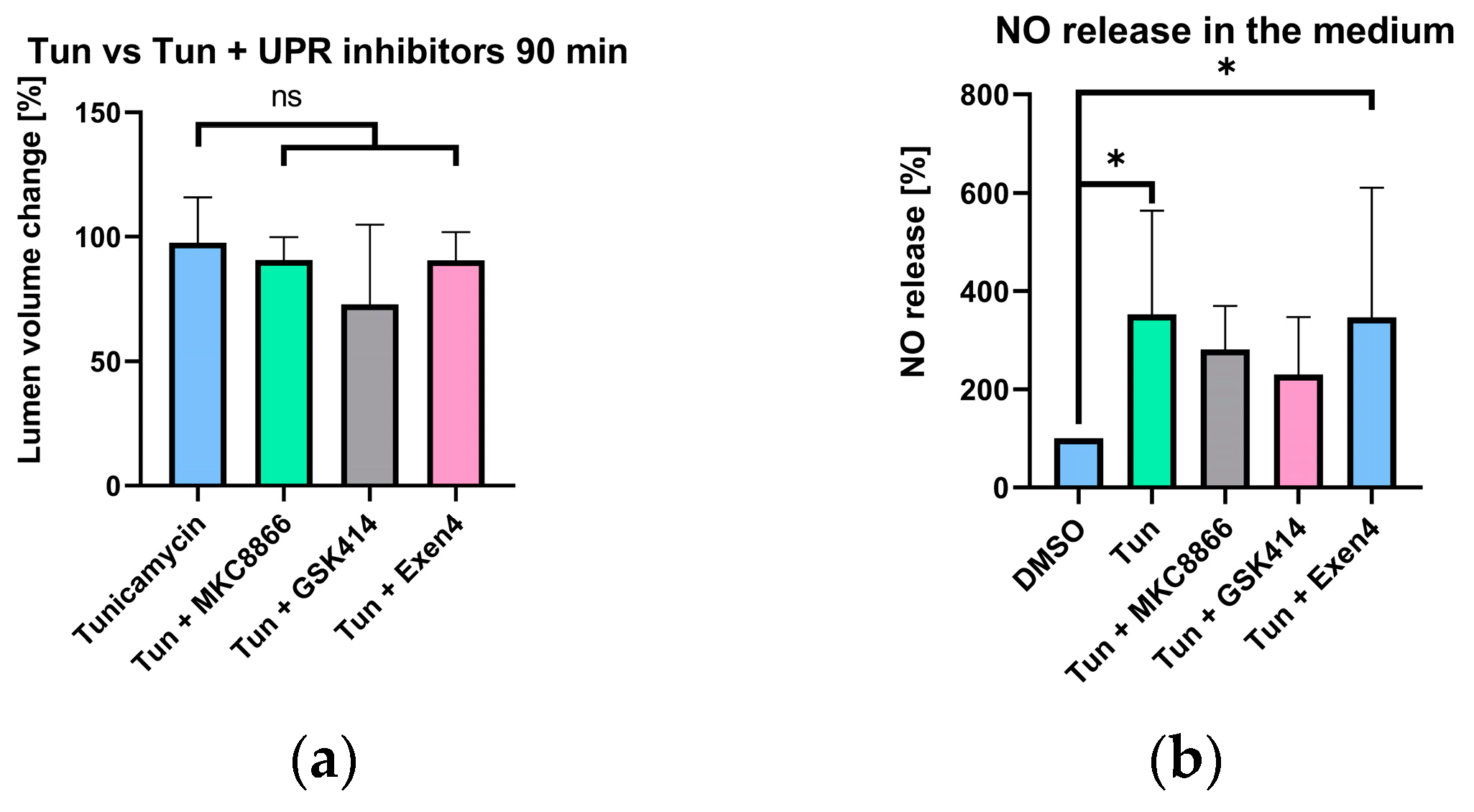
2.4. Vascular Nitric Oxide Levels
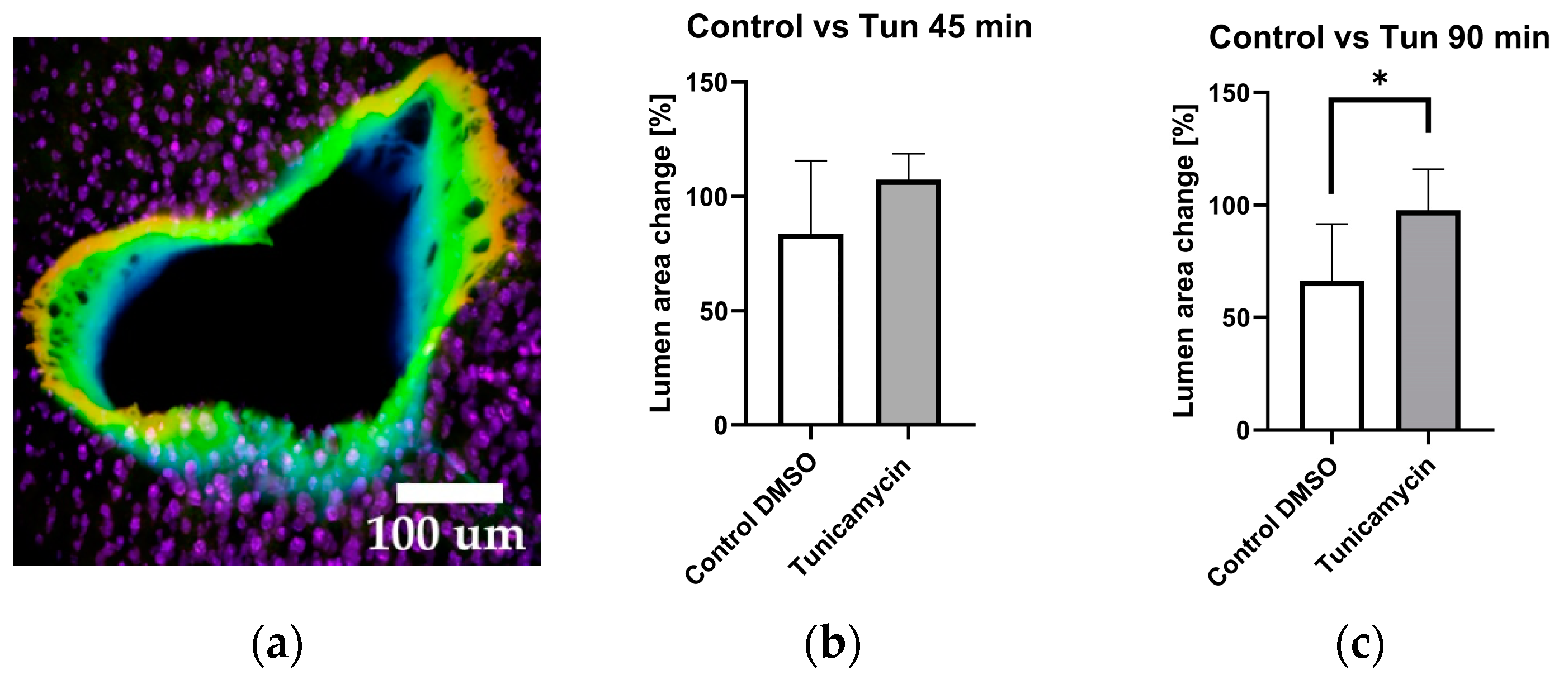
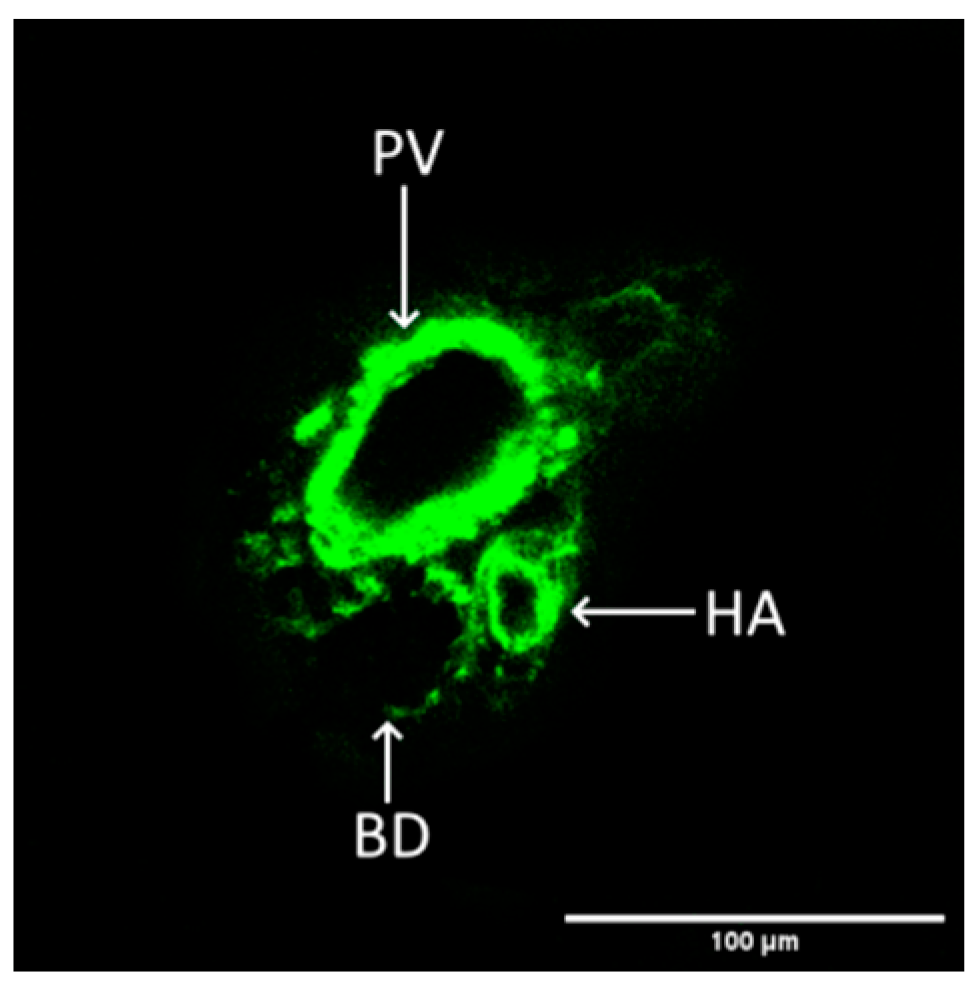
2.5. Vessel Volume Changes in Response to Tunicamycin
2.6. Selective UPR Pathway Inhibition
2.7. Experiments with Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Animals and Respective Materials
4.2. Human Umbilical Vein Endothelial Cell Isolation and Culture
4.3. Reagents
- DAF-FM Diacetate (Cat. Nr. D23842, Thermo Fischer, Waltham, MA, USA)
- FLUO-4 AM (Cat. Nr. F14201, Thermo Fischer, Waltham, MA, USA).
- Thioflavin-T (Cat. Nr. T3516-5g, f.c. 50 µM, Thermo Fischer, Waltham, MA, USA).
- Acetylcholine (Cat. Nr. AA6625-10MG, f.c 300 µM, Merck/Sigma-Aldrich, Darmstadt, Germany).
- Phenylephrine (Cat. Nr. P1240000, f.c 300 µM, Merck/Sigma-Aldrich, Darmstadt, Germany).
- Tunicamycin (Cat. Nr. Cay11445-10, f.c. 200 µg/mL, Cayman Chemical, Ann Arbor, MA, USA).
4.4. Preparation Methods
4.5. Determination of XBP1 Splicing
4.6. Determination of NO Levels
4.7. Microscopic Imaging and Analysis
- run(“Convert to Mask”, “method = Huang background = Dark calculate black”);
- run(“Despeckle”, “stack”);
- close(“ROI Manager”);
- run(“Analyze Particles...”, “size = 1500-Infinity summarize add slice”);
- roiManager(“Select”, 0);
- run(“Measure”);
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, S.-W.; Park, S.E.; Rhee, E.-J.; Park, C.-Y.; Oh, K.-W.; Park, S.-W.; Lee, W.-Y. Exendin-4 attenuates endoplasmic reticulum stress through a SIRT1-dependent mechanism. Cell Stress Chaperones 2014, 19, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanism, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Kozlov, A.V.; Zifko, C.; Postl, A.; Hartl, R.T.; Miller, I.; Gille, L.; Staniek, K.; Moldzio, R.; Gregor, W.; et al. Reperfusion does not induce oxidative stress but sustained endoplasmic reticulum stress in livers of rats subjected to traumatic-hemorrhagic shock. Shock 2010, 33, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Karmaniolou, I.I.; Theodoraki, K.A.; Orfanos, N.F.; Kostopanagiotou, G.G.; Smyrniotis, V.E.; Mylonas, A.I.; Arkadopoulos, N.F. Resuscitation after hemorrhagic shock: The effect on the liver—A review of experimental data. J. Anesth. 2013, 27, 447–460. [Google Scholar] [CrossRef]
- Tian, J.-L.; Zhang, J.-S. Hepatic perfusion disorders: Etiopathogenesis and related diseases. World J. Gastroenterol. 2006, 12, 3265–3270. [Google Scholar] [CrossRef]
- Shreya, S.; Grosset, C.F.; Jain, B.P. Unfolded Protein Response Signaling in Liver Disorders: A 2023 Updated Review. Int. J. Mol. Sci. 2023, 24, 14066. [Google Scholar] [CrossRef]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef]
- Li, R.-L.; Zhuo, C.-L.; Yan, X.; Li, H.; Lin, L.; Li, L.-Y.; Jiang, Q.; Zhang, D.; Wang, X.-M.; Liu, L.-L.; et al. Irisin attenuates vascular remodeling in hypertensive mice induced by Ang II by suppressing Ca2+-dependent endoplasmic reticulum stress in VSMCs. Int. J. Biol. Sci. 2024, 20, 680–700. [Google Scholar] [CrossRef]
- Ando, M.; Matsumoto, T.; Taguchi, K.; Kobayashi, T. Decreased contraction induced by endothelium-derived contracting factor in prolonged treatment of rat renal artery with endoplasmic reticulum stress inducer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Poburko, D.; Sahota, P.; Sandhu, J.; Ruehlmann, D.O.; van Breemen, C. The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J. Physiol. 2001, 534 Pt 3, 641–650. [Google Scholar] [CrossRef]
- Lenna, S.; Han, R.; Trojanowska, M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef]
- Galán, M.; Kassan, M.; Kadowitz, P.J.; Trebak, M.; Belmadani, S.; Matrougui, K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Schroeder, H.; Power, G.G.; Blood, A.B. A physiologically relevant role for NO stored in vascular smooth muscle cells: A novel theory of vascular NO signaling. Redox Biol. 2022, 53, 102327. [Google Scholar] [CrossRef] [PubMed]
- Petzelbauer, P.; Bender, J.R.; Wilson, J.; Pober, J.S. Heterogeneity of dermal microvascular endothelial cell antigen expression and cytokine responsiveness in situ and in cell culture. J. Immunol. 1993, 151, 5062–5072. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Dungel, P.; Perlinger, M.; Singer, K.; Ghebes, C.; Duvigneau, J.C.; Müllebner, A.; Schäfer, U.; Redl, H.; Kozlov, A.V. Experimental data suggesting that inflammation mediated rat liver mitochondrial dysfunction results from secondary hypoxia rather than from direct effects of inflammatory mediators. Front. Physiol. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Müllebner, A.; Herminghaus, A.; Miller, I.; Kames, M.; Luís, A.; Picker, O.; Bauer, I.; Kozlov, A.V.; Duvigneau, J.C. Tissue Damage, Not Infection, Triggers Hepatic Unfolded Protein Response in an Experimental Rat Peritonitis Model. Front. Med. 2022, 9, 785285. [Google Scholar] [CrossRef] [PubMed]
- Paulo, L.L.; Cruz, J.C.; Zhuge, Z.; Carvalho-Galvão, A.; Brandão, M.C.; Diniz, T.F.; Haworth, S.M.; Athayde-Filho, P.F.; Lemos, V.S.; Lundberg, J.O.; et al. The novel organic mononitrate NDHP attenuates hypertension and endothelial dysfunction in hypertensive rats. Redox Biol. 2017, 15, 182–191. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavadskis, S.; Shiganyan, A.; Müllebner, A.; Oesterreicher, J.; Holnthoner, W.; Duvigneau, J.C.; Kozlov, A.V. Endoplasmic Reticulum Stress Induces Vasodilation in Liver Vessels That Is Not Mediated by Unfolded Protein Response. Int. J. Mol. Sci. 2024, 25, 3865. https://doi.org/10.3390/ijms25073865
Zavadskis S, Shiganyan A, Müllebner A, Oesterreicher J, Holnthoner W, Duvigneau JC, Kozlov AV. Endoplasmic Reticulum Stress Induces Vasodilation in Liver Vessels That Is Not Mediated by Unfolded Protein Response. International Journal of Molecular Sciences. 2024; 25(7):3865. https://doi.org/10.3390/ijms25073865
Chicago/Turabian StyleZavadskis, Sergejs, Anna Shiganyan, Andrea Müllebner, Johannes Oesterreicher, Wolfgang Holnthoner, Johanna Catharina Duvigneau, and Andrey V. Kozlov. 2024. "Endoplasmic Reticulum Stress Induces Vasodilation in Liver Vessels That Is Not Mediated by Unfolded Protein Response" International Journal of Molecular Sciences 25, no. 7: 3865. https://doi.org/10.3390/ijms25073865