
Metabolic Profiling as an Approach to Differentiate T-Cell Acute Lymphoblastic Leukemia Cell Lines Belonging to the Same Genetic Subgroup
 ,
,  ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
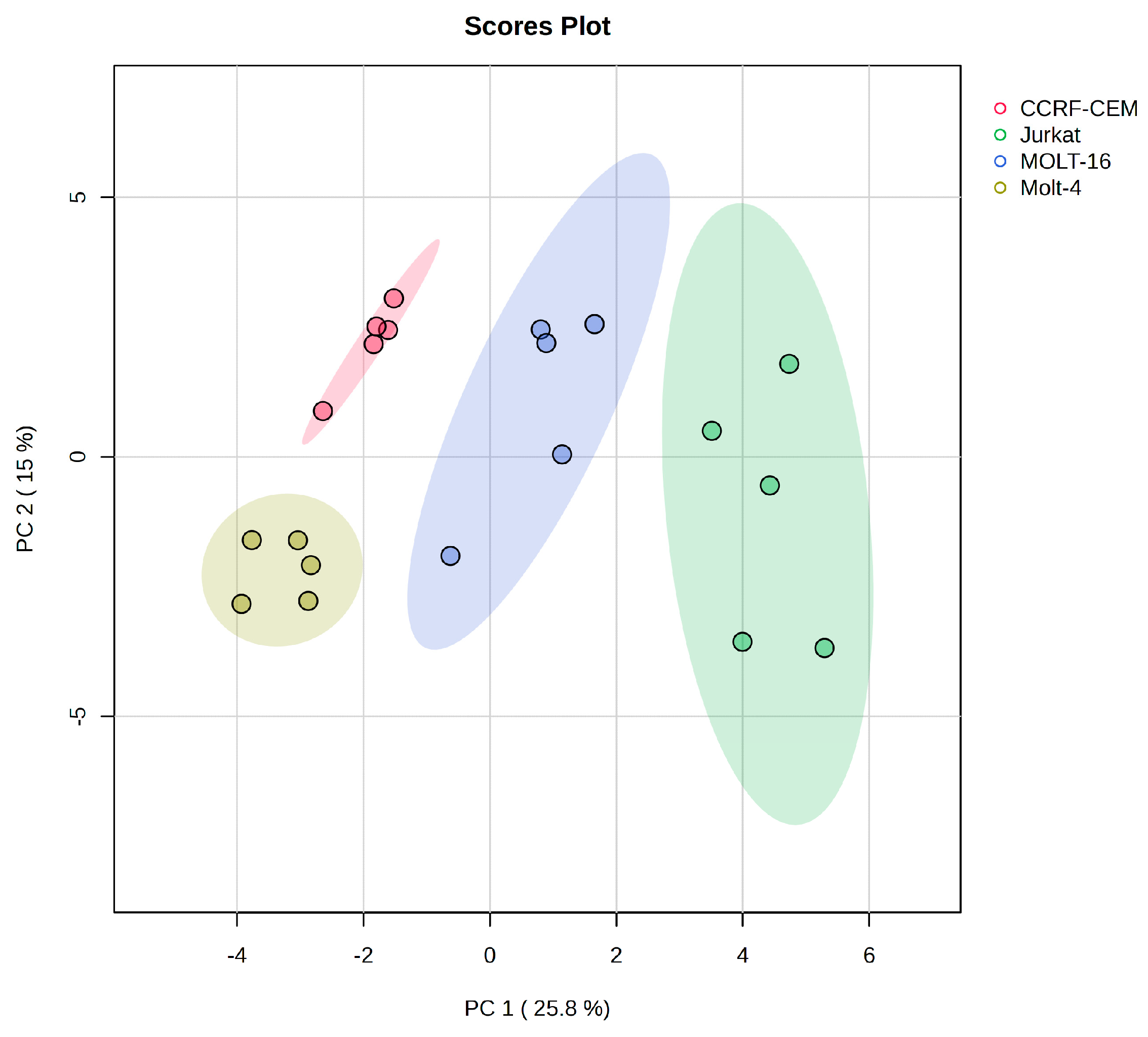
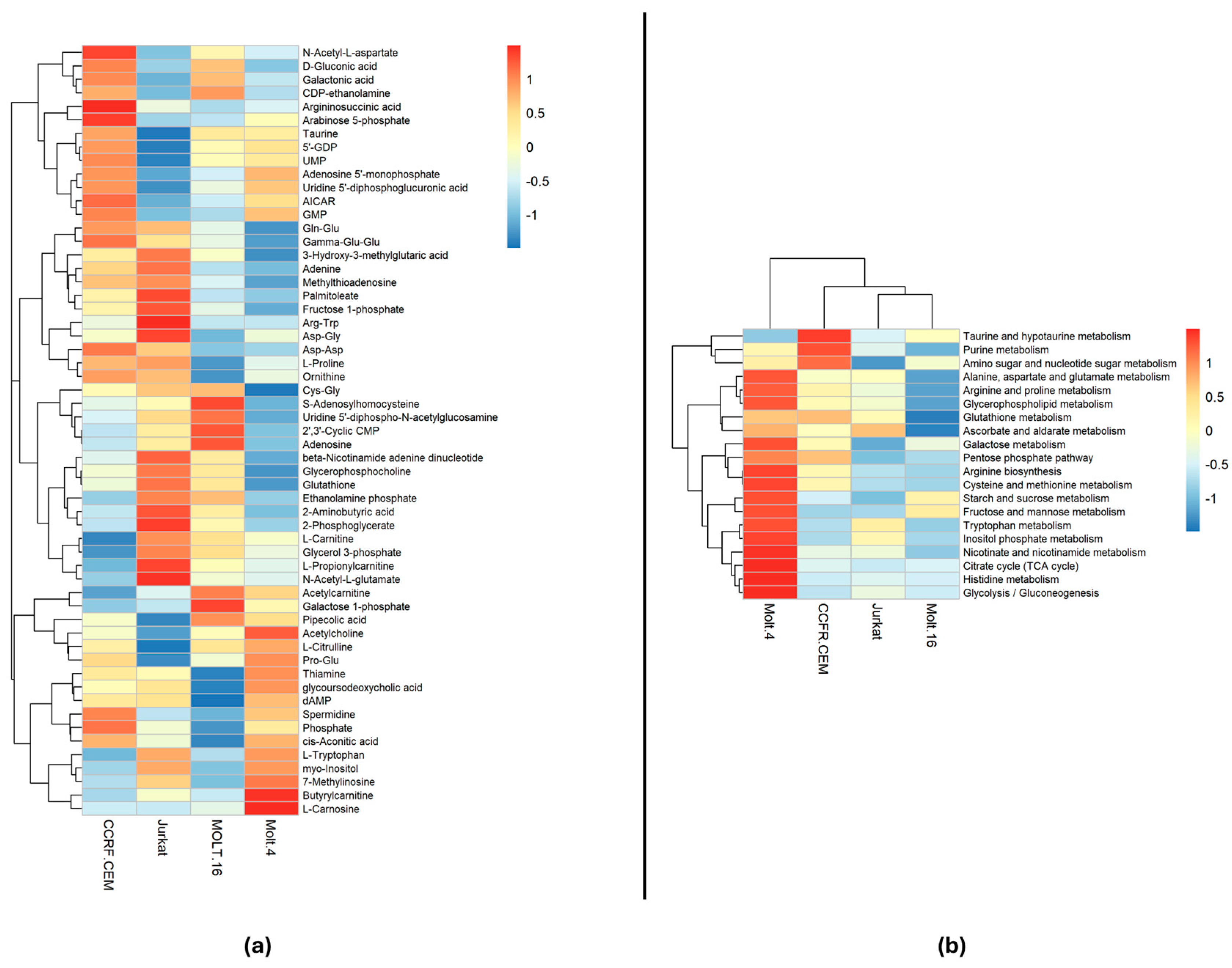
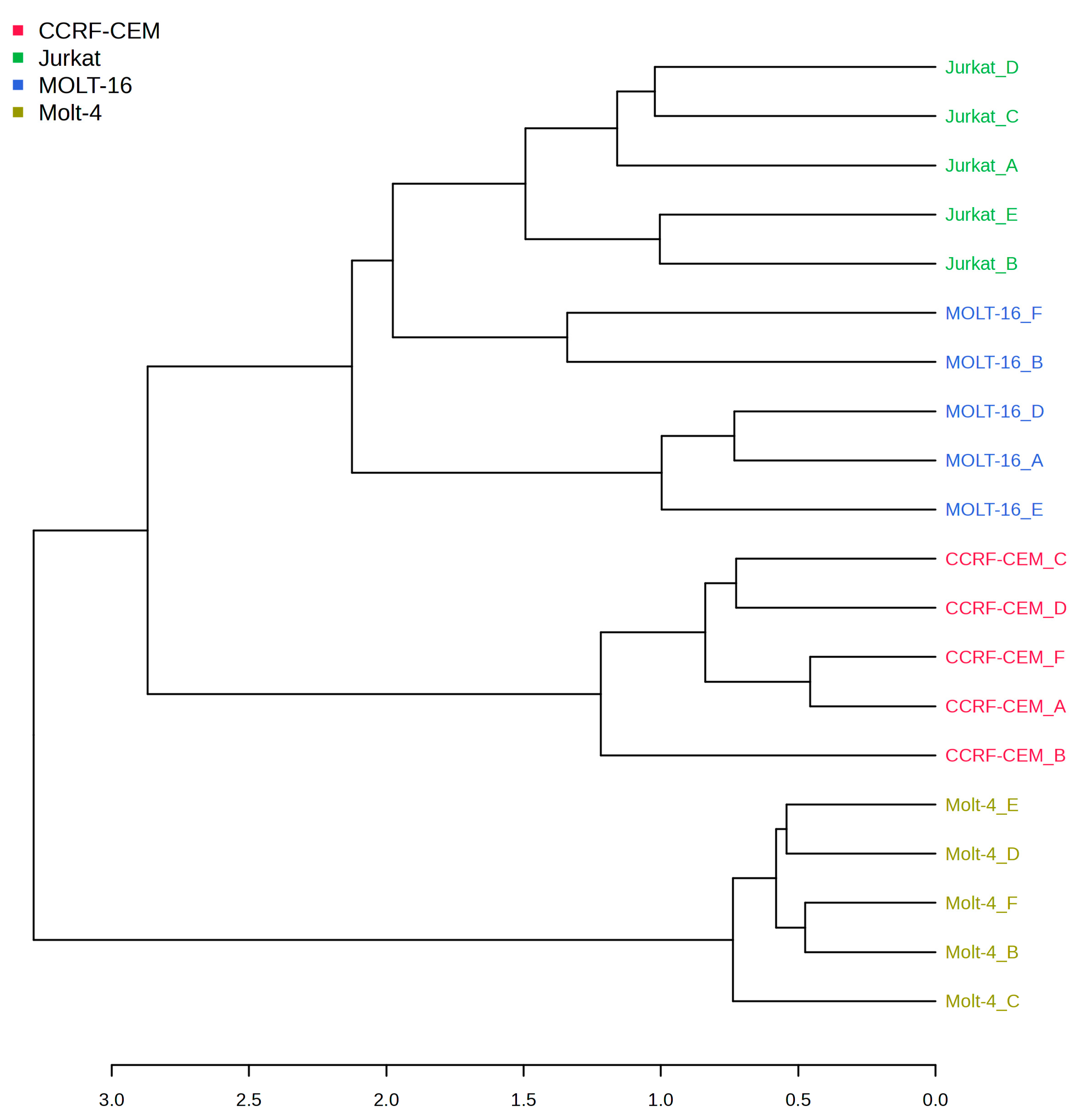
2.1. Polar Metabolite Analysis
2.2. Lipid Profile Analysis
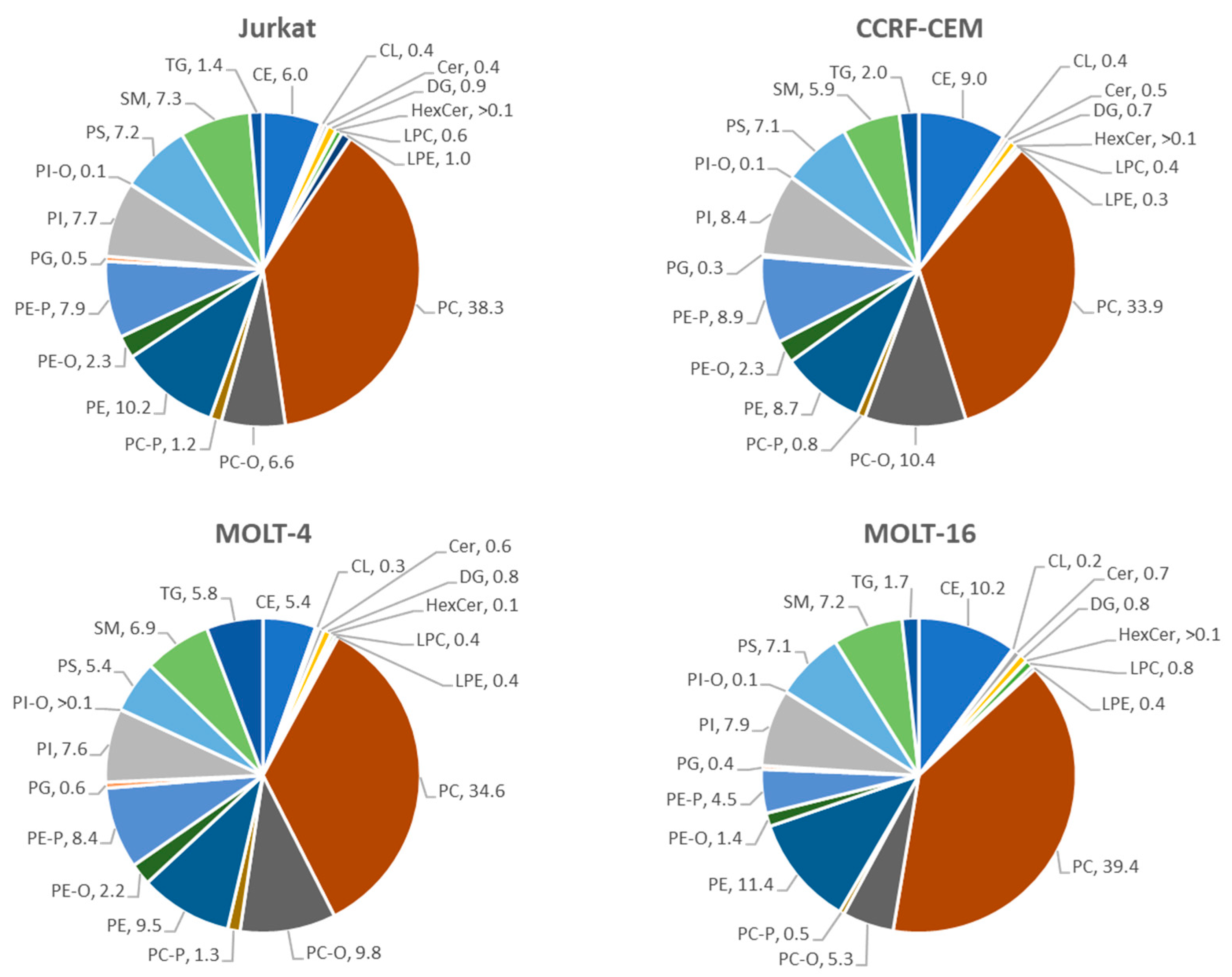
2.2.1. Analysis at the Level of Lipid Classes
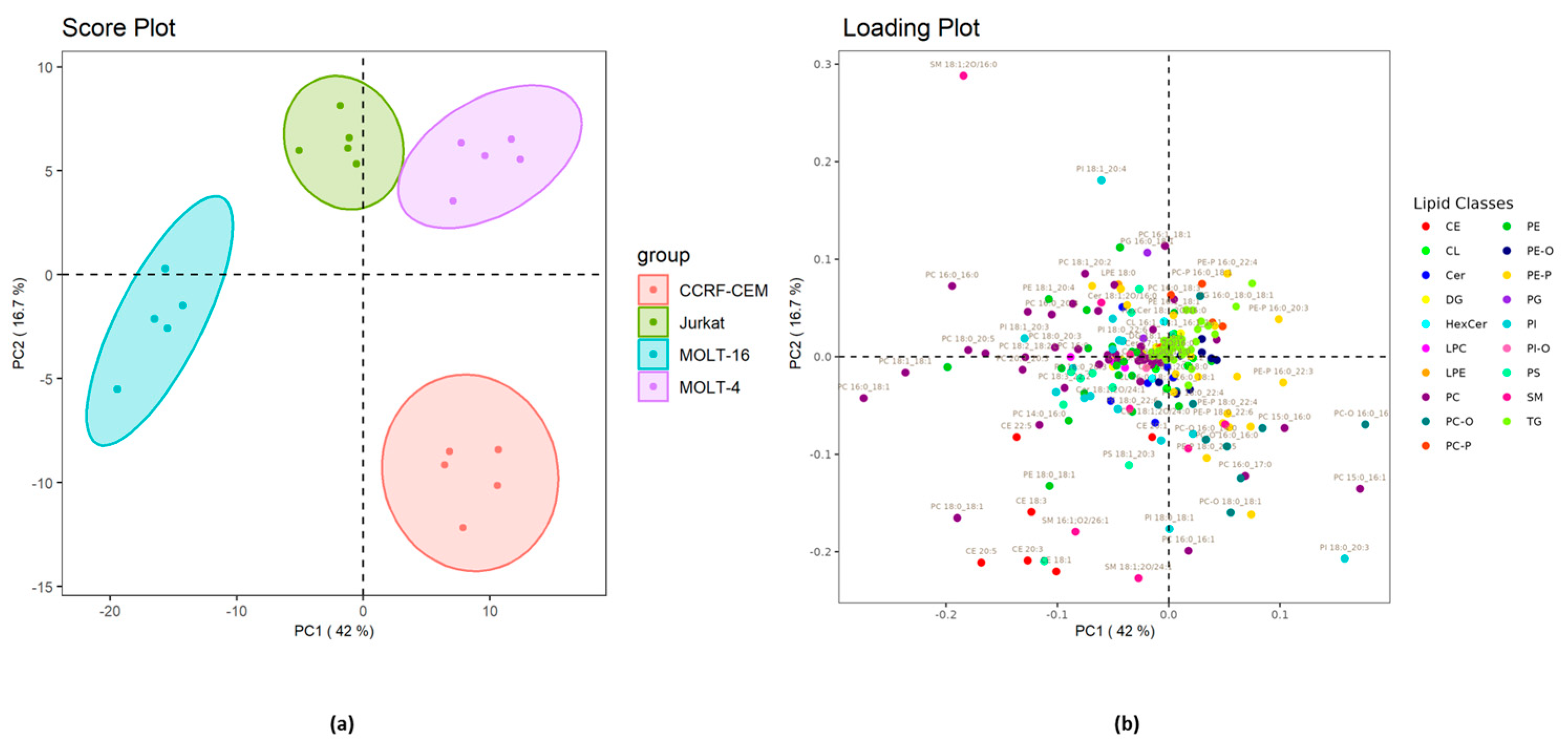
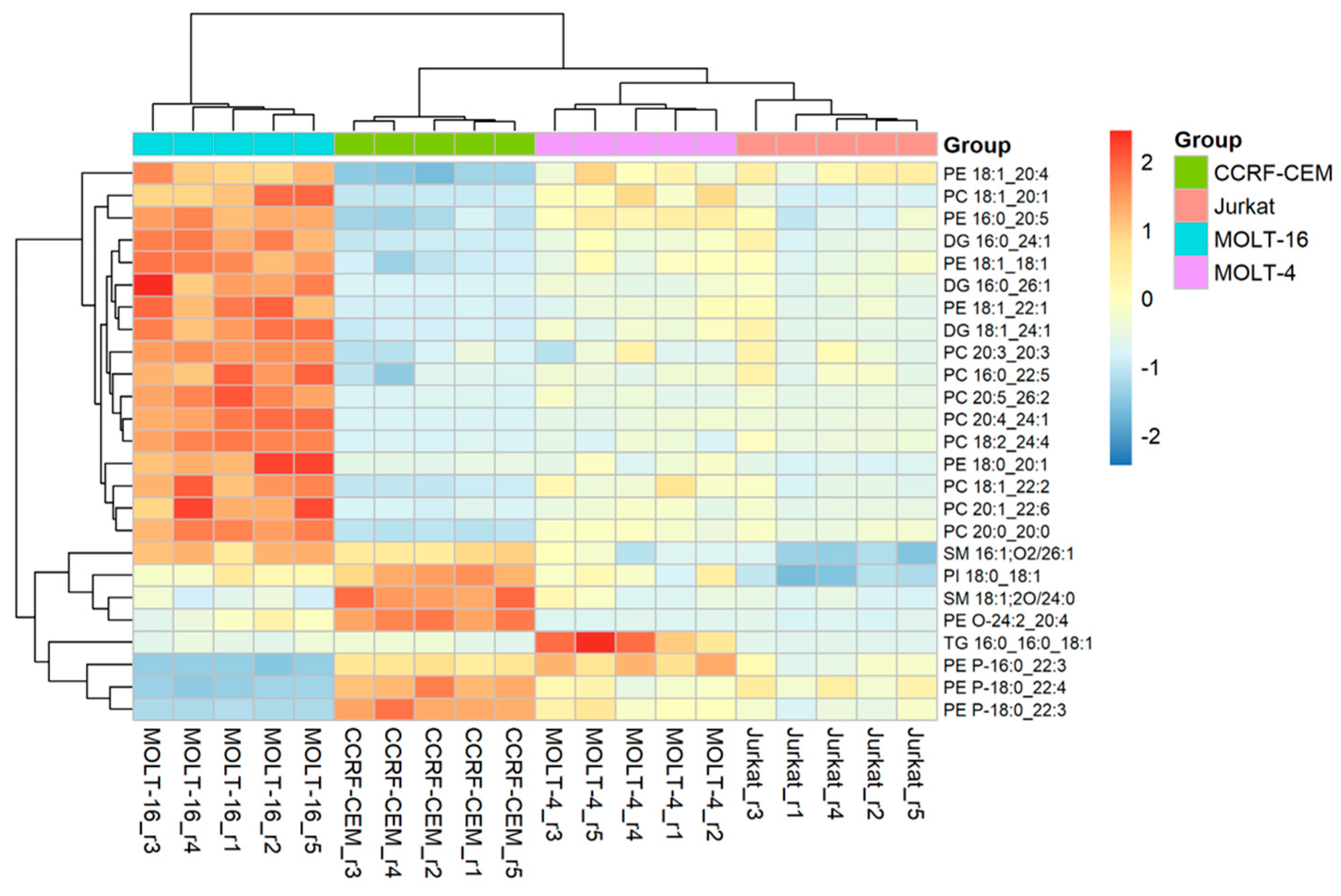
2.2.2. Analysis at the Level of Lipid Molecular Species
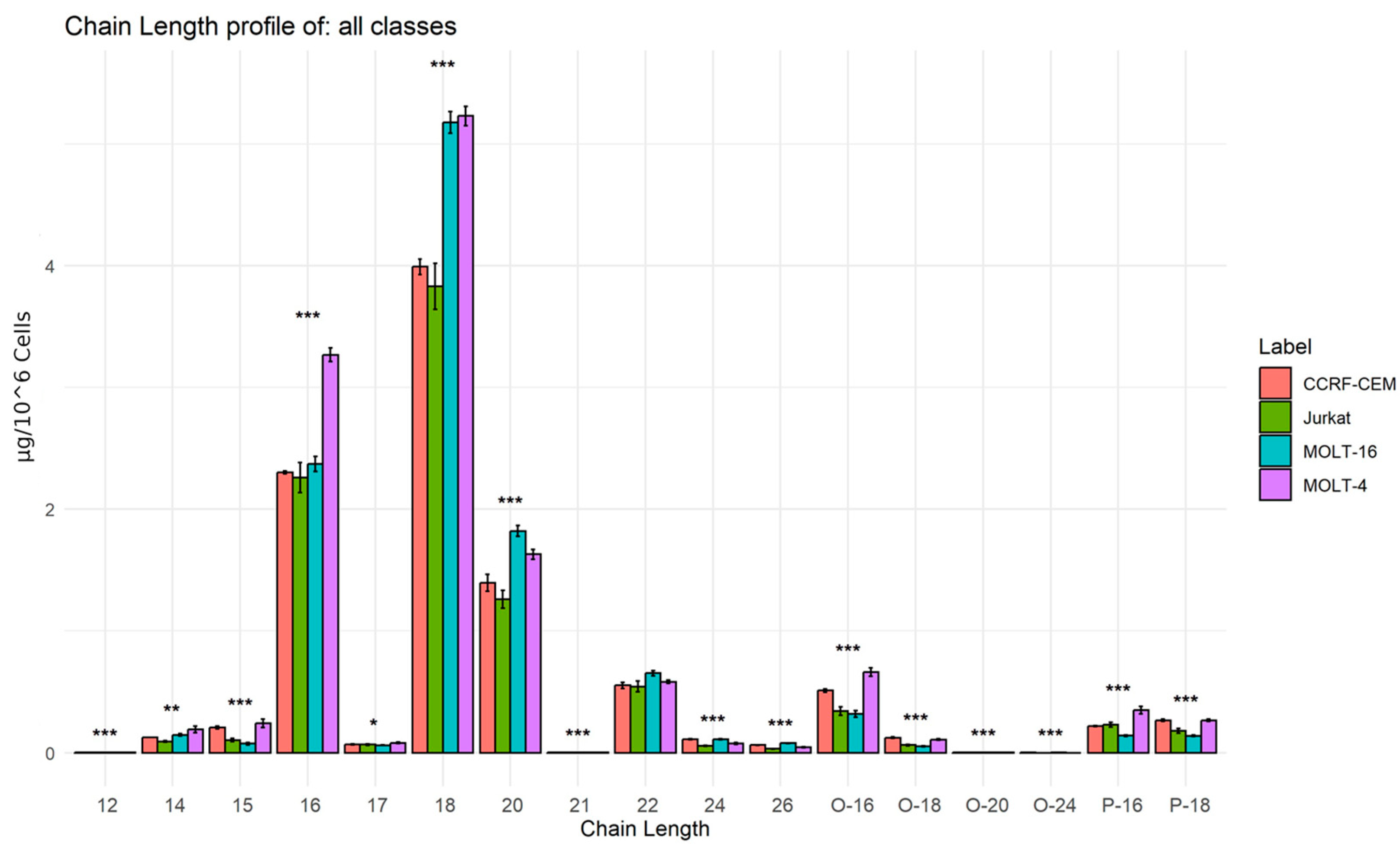
2.2.3. Analysis of Lipid Building Blocks
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Samples and Sample Preparation
4.2.1. Cell Lines
4.2.2. Preparation of Analytical Samples and Metabolites Extraction
4.3. LC/MS Analysis
4.3.1. Untargeted Polar Metabolomics
4.3.2. Untargeted Lipidomics
4.4. Raw Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Vlierberghe, P.; Ferrando, A. The Molecular Basis of T Cell Acute Lymphoblastic Leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R.; Ferrando, A.A. How I Treat T-Cell Acute Lymphoblastic Leukemia in Adults. Blood 2015, 126, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Karrman, K.; Johansson, B. Pediatric T-cell Acute Lymphoblastic Leukemia. Genes Chromos. Cancer 2017, 56, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Foà, R. T-Cell Acute Lymphoblastic Leukemia. Haematologica 2009, 94, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Puckett, Y.; Chan, O. Acute Lymphocytic Leukemia; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Ferrando, A.A.; Look, A.T. Gene Expression Profiling in T-Cell Acute Lymphoblastic Leukemia. Semin. Hematol. 2003, 40, 274–280. [Google Scholar] [CrossRef] [PubMed]
- La Starza, R.; Pierini, V.; Pierini, T.; Nofrini, V.; Matteucci, C.; Arniani, S.; Moretti, M.; Lema Fernandez, A.G.; Pellanera, F.; Di Giacomo, D.; et al. Design of a Comprehensive Fluorescence in Situ Hybridization Assay for Genetic Classification of T-Cell Acute Lymphoblastic Leukemia. J. Mol. Diagn. 2020, 22, 629–639. [Google Scholar] [CrossRef]
- Atak, Z.K.; Gianfelici, V.; Hulselmans, G.; De Keersmaecker, K.; Devasia, A.G.; Geerdens, E.; Mentens, N.; Chiaretti, S.; Durinck, K.; Uyttebroeck, A.; et al. Comprehensive Analysis of Transcriptome Variation Uncovers Known and Novel Driver Events in T-Cell Acute Lymphoblastic Leukemia. PLoS Genet. 2013, 9, e1003997. [Google Scholar] [CrossRef]
- Franciosa, G.; Smits, J.G.A.; Minuzzo, S.; Martinez-Val, A.; Indraccolo, S.; Olsen, J. V Proteomics of Resistance to Notch1 Inhibition in Acute Lymphoblastic Leukemia Reveals Targetable Kinase Signatures. Nat. Commun. 2021, 12, 2507. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, V.; Arniani, S.; Pierini, V.; Di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; La Starza, R. T-Cell Acute Lymphoblastic Leukemia: Biomarkers and Their Clinical Usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Pieters, R.; Beverloo, H.B.; Meijerink, J.P.P. Molecular-Genetic Insights in Paediatric T-Cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2008, 143, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.R.; Carvalho, M.; Aveiro, S.S.; Melo, T.; Domingues, M.R.; Macedo-Silva, C.; Coimbra, N.; Jerónimo, C.; Henrique, R.; Bastos, M.D.L.; et al. Comprehensive Metabolomics and Lipidomics Profiling of Prostate Cancer Tissue Reveals Metabolic Dysregulations Associated with Disease Development. J. Proteome Res. 2022, 21, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Armitage, E.G.; Southam, A.D. Monitoring Cancer Prognosis, Diagnosis and Treatment Efficacy Using Metabolomics and Lipidomics. Metabolomics 2016, 12, 146. [Google Scholar] [CrossRef]
- Wang, R.; Li, B.; Lam, S.M.; Shui, G. Integration of Lipidomics and Metabolomics for In-Depth Understanding of Cellular Mechanism and Disease Progression. J. Genet. Genom. 2020, 47, 69–83. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Han, X. Tutorial on Lipidomics. Anal. Chim. Acta 2019, 1061, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Hollywood, K.; Brison, D.R.; Goodacre, R. Metabolomics: Current Technologies and Future Trends. Proteomics 2006, 6, 4716–4723. [Google Scholar] [CrossRef] [PubMed]
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from Sample Preparation to Data Analysis: A Primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Harrieder, E.-M.; Kretschmer, F.; Böcker, S.; Witting, M. Current State-of-the-Art of Separation Methods Used in LC-MS Based Metabolomics and Lipidomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2022, 1188, 123069. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Pang, Z.; Lu, Y.; Ewald, J.; Xia, J. OmicsNet 2.0: A Web-Based Platform for Multi-Omics Integration and Network Visual Analytics. Nucleic Acids Res. 2022, 50, W527–W533. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.M.; Giulietti, M.; Alabed, H.B.R.; Buratta, S.; Urbanelli, L.; Piva, F.; Emiliani, C. LipidOne: User-Friendly Lipidomic Data Analysis Tool for a Deeper Interpretation in a Systems Biology Scenario. Bioinformatics 2022, 38, 1767–1769. [Google Scholar] [CrossRef] [PubMed]
- Gaud, C.; Sousa, B.C.; Nguyen, A.; Fedorova, M.; Ni, Z.; O’Donnell, V.B.; Wakelam, M.J.O.; Andrews, S.; Lopez-Clavijo, A.F. BioPAN: A Web-Based Tool to Explore Mammalian Lipidome Metabolic Pathways on LIPID MAPS. F1000Research 2021, 10, 4. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, S.-H.; Shin, M.-J.; Hwang, G.-S. Alteration in Metabolic Signature and Lipid Metabolism in Patients with Angina Pectoris and Myocardial Infarction. PLoS ONE 2015, 10, e0135228. [Google Scholar] [CrossRef]
- Kohno, S.; Keenan, A.L.; Ntambi, J.M.; Miyazaki, M. Lipidomic Insight into Cardiovascular Diseases. Biochem. Biophys. Res. Commun. 2018, 504, 590–595. [Google Scholar] [CrossRef]
- Alabed, H.B.R.; Gorello, P.; Pellegrino, R.M.; Lancioni, H.; La Starza, R.; Taddei, A.A.; Urbanelli, L.; Buratta, S.; Fernandez, A.G.L.; Matteucci, C.; et al. Comparison between Sickle Cell Disease Patients and Healthy Donors: Untargeted Lipidomic Study of Erythrocytes. Int. J. Mol. Sci. 2023, 24, 2529. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Ma, S.-X.; Chen, Y.-Y.; Chen, L.; Liu, B.-L.; Liu, Q.-Q.; Zhao, Y.-Y. Chronic Kidney Disease: Biomarker Diagnosis to Therapeutic Targets. Clin. Chim. Acta 2019, 499, 54–63. [Google Scholar] [CrossRef]
- Wood, P.L. Mass Spectrometry Strategies for Clinical Metabolomics and Lipidomics in Psychiatry, Neurology, and Neuro-Oncology. Neuropsychopharmacology 2014, 39, 24–33. [Google Scholar] [CrossRef]
- Li, Y.; Liang, L.; Deng, X.; Zhong, L. Lipidomic and Metabolomic Profiling Reveals Novel Candidate Biomarkers in Active Systemic Lupus Erythematosus. Int. J. Clin. Exp. Pathol. 2019, 12, 857–866. [Google Scholar]
- Farrokhi Yekta, R.; Rezaie Tavirani, M.; Arefi Oskouie, A.; Mohajeri-Tehrani, M.R.; Soroush, A.R. The Metabolomics and Lipidomics Window into Thyroid Cancer Research. Biomarkers 2016, 22, 595–603. [Google Scholar] [CrossRef]
- Burger, R.; Hansen-Hagge, T.E.; Drexler, H.G.; Gramatzki, M. Heterogeneity of T-Acute Lymphoblastic Leukemia (T-ALL) Cell Lines: Suggestion for Classification by Immunophenotype and T-Cell Receptor Studies. Leuk. Res. 1999, 23, 19–27. [Google Scholar] [CrossRef]
- Alarcon-Barrera, J.C.; von Hegedus, J.H.; Brouwers, H.; Steenvoorden, E.; Ioan-Facsinay, A.; Mayboroda, O.A.; Ondo-Mendez, A.; Giera, M. Lipid Metabolism of Leukocytes in the Unstimulated and Activated States. Anal. Bioanal. Chem. 2020, 412, 2353–2363. [Google Scholar] [CrossRef]
- Kwon, H.-Y.; Kumar Das, R.; Jung, G.T.; Lee, H.-G.; Lee, S.H.; Berry, S.N.; Tan, J.K.S.; Park, S.; Yang, J.-S.; Park, S.; et al. Lipid-Oriented Live-Cell Distinction of B and T Lymphocytes. J. Am. Chem. Soc. 2021, 143, 5836–5844. [Google Scholar] [CrossRef]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-Degrading Enzymes in Tumoral Immune Resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Cheng, C.-T.; Qi, Y.; Wang, Y.-C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.-R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine Starvation Kills Tumor Cells through Aspartate Exhaustion and Mitochondrial Dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef]
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-Dependent Purine Biosynthesis Affects Nucleolar Stress and Therapy Response in Prostate Cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef]
- Goswami, M.T.; Chen, G.; Chakravarthi, B.V.S.K.; Pathi, S.S.; Anand, S.K.; Carskadon, S.L.; Giordano, T.J.; Chinnaiyan, A.M.; Thomas, D.G.; Palanisamy, N.; et al. Role and Regulation of Coordinately Expressed de Novo Purine Biosynthetic Enzymes PPAT and PAICS in Lung Cancer. Oncotarget 2015, 6, 23445–23461. [Google Scholar] [CrossRef]
- Bahreyni, A.; Samani, S.S.; Rahmani, F.; Behnam-Rassouli, R.; Khazaei, M.; Ryzhikov, M.; Parizadeh, M.R.; Avan, A.; Hassanian, S.M. Role of Adenosine Signaling in the Pathogenesis of Breast Cancer. J. Cell Physiol. 2018, 233, 1836–1843. [Google Scholar] [CrossRef]
- Yamauchi, T.; Miyawaki, K.; Semba, Y.; Takahashi, M.; Izumi, Y.; Nogami, J.; Nakao, F.; Sugio, T.; Sasaki, K.; Pinello, L.; et al. Targeting Leukemia-Specific Dependence on the de Novo Purine Synthesis Pathway. Leukemia 2022, 36, 383–393. [Google Scholar] [CrossRef]
- Meana, C.; García-Rostán, G.; Peña, L.; Lordén, G.; Cubero, Á.; Orduña, A.; Győrffy, B.; Balsinde, J.; Balboa, M.A. The Phosphatidic Acid Phosphatase Lipin-1 Facilitates Inflammation-Driven Colon Carcinogenesis. JCI Insight 2018, 3, e97506. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, F.; Tay, L.W.R.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 Regulation of Phospholipid Synthesis Maintains Endoplasmic Reticulum Homeostasis and Is Critical for Triple-Negative Breast Cancer Cell Survival. FASEB J. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Zimmerman, T.; Gomez del Pulgar, T.; Moyer, M.P.; Lacal Sanjuan, J.C.; Cebrian, A. Choline Kinase Inhibition Induces Exacerbated Endoplasmic Reticulum Stress and Triggers Apoptosis via CHOP in Cancer Cells. Cell Death Dis. 2013, 4, e933. [Google Scholar] [CrossRef] [PubMed]
- Arora, G.K.; Palamiuc, L.; Emerling, B.M. Expanding Role of PI5P4Ks in Cancer: A Promising Druggable Target. FEBS Lett. 2022, 596, 3–16. [Google Scholar] [CrossRef]
- Yamashita, Y.; Nishiumi, S.; Kono, S.; Takao, S.; Azuma, T.; Yoshida, M. Differences in Elongation of Very Long Chain Fatty Acids and Fatty Acid Metabolism between Triple-Negative and Hormone Receptor-Positive Breast Cancer. BMC Cancer 2017, 17, 589. [Google Scholar] [CrossRef]
- Robichaud, P.-P.; Munganyiki, J.E.; Boilard, E.; Surette, M.E. Polyunsaturated Fatty Acid Elongation and Desaturation in Activated Human T-Cells: ELOVL5 Is the Key Elongase. J. Lipid Res. 2018, 59, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Stefanko, A.; Thiede, C.; Ehninger, G.; Simons, K.; Grzybek, M. Lipidomic Approach for Stratification of Acute Myeloid Leukemia Patients. PLoS ONE 2017, 12, e0168781. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mistriel-Zerbib, S.; Najar, R.A.; Engal, E.; Bentata, M.; Taqatqa, N.; Dahan, S.; Cohen, K.; Jaffe-Herman, S.; Geminder, O.; et al. Isoforms of the TAL1 Transcription Factor Have Different Roles in Hematopoiesis and Cell Growth. PLoS Biol. 2023, 21, e3002175. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O. Optimization of Mobile Phase Modifiers for Fast LC-MS-Based Untargeted Metabolomics and Lipidomics. Int. J. Mol. Sci. 2023, 24, 1987. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Goracci, L.; Cruciani, G. LC/MS Lipid Profiling from Human Serum: A New Method for Global Lipid Extraction. Anal. Bioanal. Chem. 2014, 406, 7937–7948. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, J.; Li, Q.; Fan, S.; Fan, L.; Ma, H.; Zhang, Y.; Zheng, L. Determination of 35 Free Amino Acids in Tea Using Ultra-Performance Liquid Chromatography Coupled with Quadrupole Time-of-Flight Mass Spectrometry. Front. Nutr. 2021, 8, 767801. [Google Scholar] [CrossRef] [PubMed]
- Alabed, H.B.R.; Del Grosso, A.; Bellani, V.; Urbanelli, L.; Carpi, S.; De Sarlo, M.; Bertocci, L.; Colagiorgio, L.; Buratta, S.; Scaccini, L.; et al. Untargeted Lipidomic Approach for Studying Different Nervous System Tissues of the Murine Model of Krabbe Disease. Biomolecules 2023, 13, 1562. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A Lipidome Atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS Classification, Nomenclature, and Shorthand Notation for MS-Derived Lipid Structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Gertner, D.S.; Violi, J.P.; Bishop, D.P.; Padula, M.P. Lipid Spectrum Generator: A Simple Script for the Generation of Accurate In Silico Lipid Fragmentation Spectra. Anal. Chem. 2023, 95, 2909–2916. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Class Abbreviation | Explained Class Name | Number of Annotated Molecules | Average ± Exp Er (Jurkat) | Average ± Exp Er (CCRF-CEM) | Average ± Exp Er (MOLT-4) | Average ± Exp Er (MOLT-16) | p-Value |
|---|---|---|---|---|---|---|---|
| CE | Cholesteryl ester | 6 | 0.54559 (±0.07061) | 0.89815 (±0.06599) | 0.69316 (±0.07075) | 1.13462 (±0.12362) | 0.001093 |
| CL | Cardiolipin | 9 | 0.03421 (±0.00316) | 0.03732 (±0.00131) | 0.04399 (±0.00265) | 0.02556 (±0.00065) | 0.000741 |
| Cer | Ceramide | 5 | 0.0371 (±0.00355) | 0.04498 (±0.00264) | 0.07195 (±0.01037) | 0.08302 (±0.00841) | 0.000205 |
| DG | Diacylglycerol | 9 | 0.08151 (±0.00735) | 0.07011 (±0.00243) | 0.10137 (±0.00789) | 0.09266 (±0.00154) | 0.006469 |
| HexCer | Hexosylceramide | 2 | 0.00399 (±0.00049) | 0.00274 (±0.00011) | 0.00754 (±0.00154) | 0.00554 (±0.0003) | 0.004946 |
| LPC | Lysophophatidylcholine | 3 | 0.05673 (±0.01429) | 0.03656 (±0.00711) | 0.04768 (±0.00934) | 0.08803 (±0.02083) | 0.094575 |
| LPE | Lysophosphatidylethanolamine | 1 | 0.09433 (±0.05043) | 0.03227 (±0.00886) | 0.0462 (±0.00317) | 0.0435 (±0.01015) | 0.37543 |
| PC | Phosphatidylcholine | 42 | 3.46958 (±0.21436) | 3.36706 (±0.03942) | 4.40161 (±0.06745) | 4.39338 (±0.07721) | 6.1 × |
| PC-O | Alkyl Ether-linked phosphatidylcholine | 10 | 0.59456 (±0.06462) | 1.03528 (±0.03938) | 1.25128 (±0.08139) | 0.58819 (±0.05143) | 1.01 × |
| PC-P | Vinyl Ether-linked phosphatidylcholine | 5 | 0.10567 (±0.01139) | 0.08346 (±0.00222) | 0.16357 (±0.00725) | 0.05577 (±0.00151) | 5.72 × |
| PE | Phosphatidylethanolamine | 20 | 0.92424 (±0.0747) | 0.86159 (±0.03958) | 1.20771 (±0.03715) | 1.2668 (±0.02376) | 2.27 × |
| PE-O | Alkyl Ether-linked phosphatidylethanolamine | 8 | 0.21023 (±0.02108) | 0.23111 (±0.00425) | 0.28497 (±0.0164) | 0.15191 (±0.0111) | 9.12 × |
| PE-P | Vinyl Ether-linked phosphatidylethanolamine | 18 | 0.71301 (±0.06548) | 0.88215 (±0.02568) | 1.06558 (±0.04709) | 0.49967 (±0.02099) | 7.29 × |
| PG | Phosphatidylglycerol | 2 | 0.04741 (±0.00487) | 0.0251 (±0.00093) | 0.07804 (±0.00477) | 0.04413 (±0.00145) | 1.42 × |
| PI | Phosphatidylinositol | 15 | 0.70036 (±0.08017) | 0.83635 (±0.01766) | 0.96282 (±0.04508) | 0.87907 (±0.03204) | 0.013254 |
| PI-O | Ether-linked phosphatidylinositol | 1 | 0.00489 (±0.00067) | 0.00513 (±0.0003) | 0.00523 (±0.00033) | 0.00634 (±0.00061) | 0.22212 |
| PS | Phosphatidylserine | 10 | 0.65673 (±0.05325) | 0.70552 (±0.01925) | 0.68516 (±0.01973) | 0.79491 (±0.02234) | 0.040757 |
| SM | Sphingomyelin | 9 | 0.65778 (±0.03399) | 0.58726 (±0.01193) | 0.87832(±0.04014) | 0.80118 (±0.02078) | 8.53 × |
| TG | Triacylglycerol | 42 | 0.12245 (±0.00637) | 0.19821 (±0.0283) | 0.73949 (±0.07466) | 0.19183 (±0.02192) | 3.39 × |
| Total | 217 | 9.06037 (±0.4956) | 9.94036 (±0.15381) | 12.73568 (±0.21066) | 11.14609 (±0.13639) | 7.45 × |
| Cell Line/Ratio | SFA/MUFA | SFA/PUFA |
|---|---|---|
| Jurkat | 0.9393 | 1.7644 |
| CCRF-CEM | 0.9926 | 1.8439 |
| Molt-4 | 0.9765 | 2.1925 |
| Molt-16 | 0.8479 | 1.4532 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alabed, H.B.R.; Pellegrino, R.M.; Buratta, S.; Lema Fernandez, A.G.; La Starza, R.; Urbanelli, L.; Mecucci, C.; Emiliani, C.; Gorello, P. Metabolic Profiling as an Approach to Differentiate T-Cell Acute Lymphoblastic Leukemia Cell Lines Belonging to the Same Genetic Subgroup. Int. J. Mol. Sci. 2024, 25, 3921. https://doi.org/10.3390/ijms25073921
Alabed HBR, Pellegrino RM, Buratta S, Lema Fernandez AG, La Starza R, Urbanelli L, Mecucci C, Emiliani C, Gorello P. Metabolic Profiling as an Approach to Differentiate T-Cell Acute Lymphoblastic Leukemia Cell Lines Belonging to the Same Genetic Subgroup. International Journal of Molecular Sciences. 2024; 25(7):3921. https://doi.org/10.3390/ijms25073921
Chicago/Turabian StyleAlabed, Husam B. R., Roberto Maria Pellegrino, Sandra Buratta, Anair Graciela Lema Fernandez, Roberta La Starza, Lorena Urbanelli, Cristina Mecucci, Carla Emiliani, and Paolo Gorello. 2024. "Metabolic Profiling as an Approach to Differentiate T-Cell Acute Lymphoblastic Leukemia Cell Lines Belonging to the Same Genetic Subgroup" International Journal of Molecular Sciences 25, no. 7: 3921. https://doi.org/10.3390/ijms25073921
APA StyleAlabed, H. B. R., Pellegrino, R. M., Buratta, S., Lema Fernandez, A. G., La Starza, R., Urbanelli, L., Mecucci, C., Emiliani, C., & Gorello, P. (2024). Metabolic Profiling as an Approach to Differentiate T-Cell Acute Lymphoblastic Leukemia Cell Lines Belonging to the Same Genetic Subgroup. International Journal of Molecular Sciences, 25(7), 3921. https://doi.org/10.3390/ijms25073921