

A Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer

 ,
,  and
and 
Abstract
1. Introduction
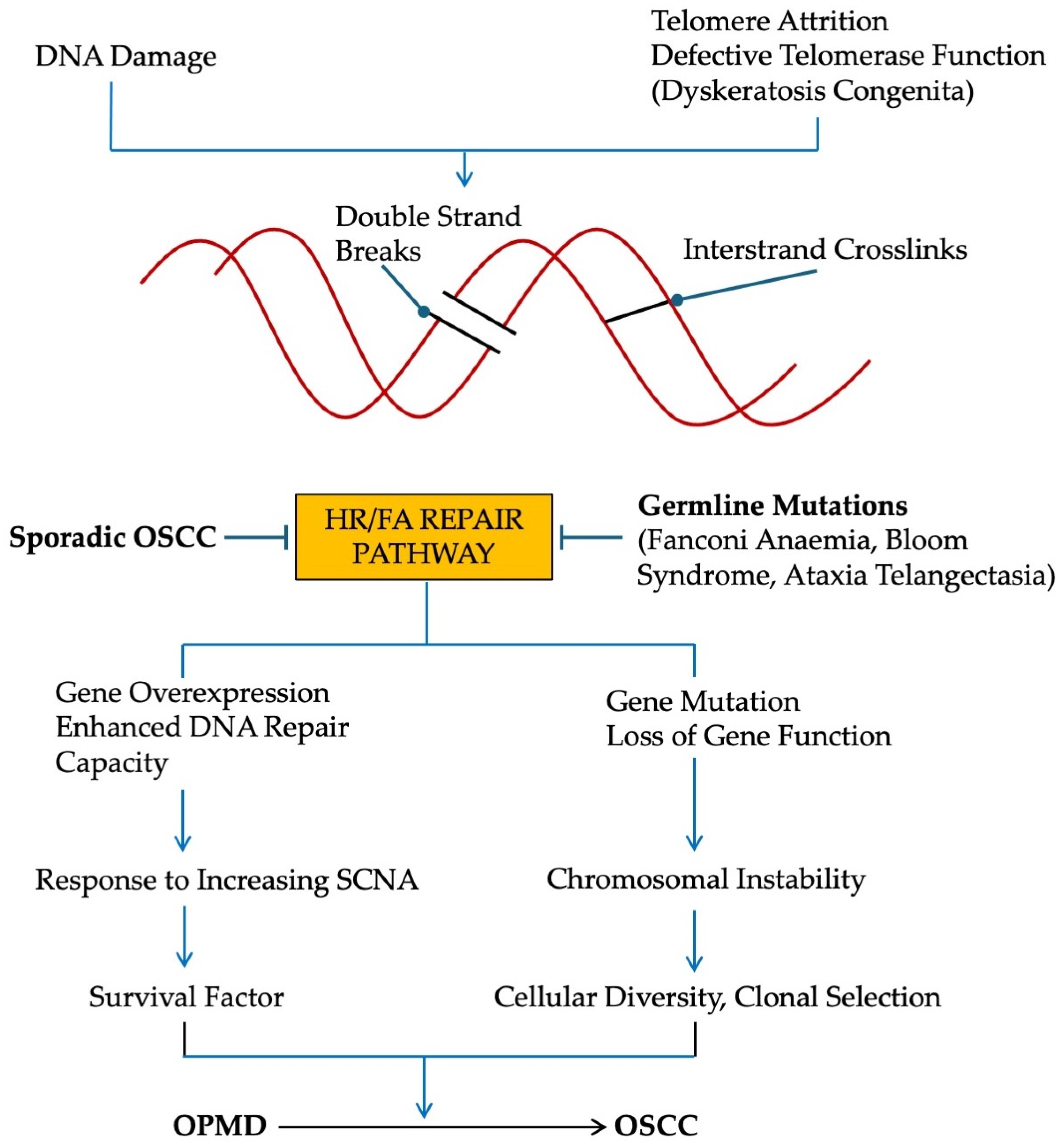
2. DNA Damage
3. Causes of DSBs
4. Consequences of DSBs
- (1)
- Ageing
- (2)
- Programmed cell death
- (3)
- Cellular senescence
- (4)
- Cancer
5. DNA Damage Response
5.1. Response
5.2. Repair
6. Response and Repair of DSBs in OPMD
6.1. Response
6.2. Repair
6.3. Single Nucleotide Polymorphisms in DSB Repair Genes
7. Conditions That Predispose to OSCC
7.1. Germline Mutations of HR/FA Pathway [93]
7.2. Germline Mutations of NHEJ Pathway
7.3. Defects in Telomere Maintenance
8. OSCC in Young Individuals
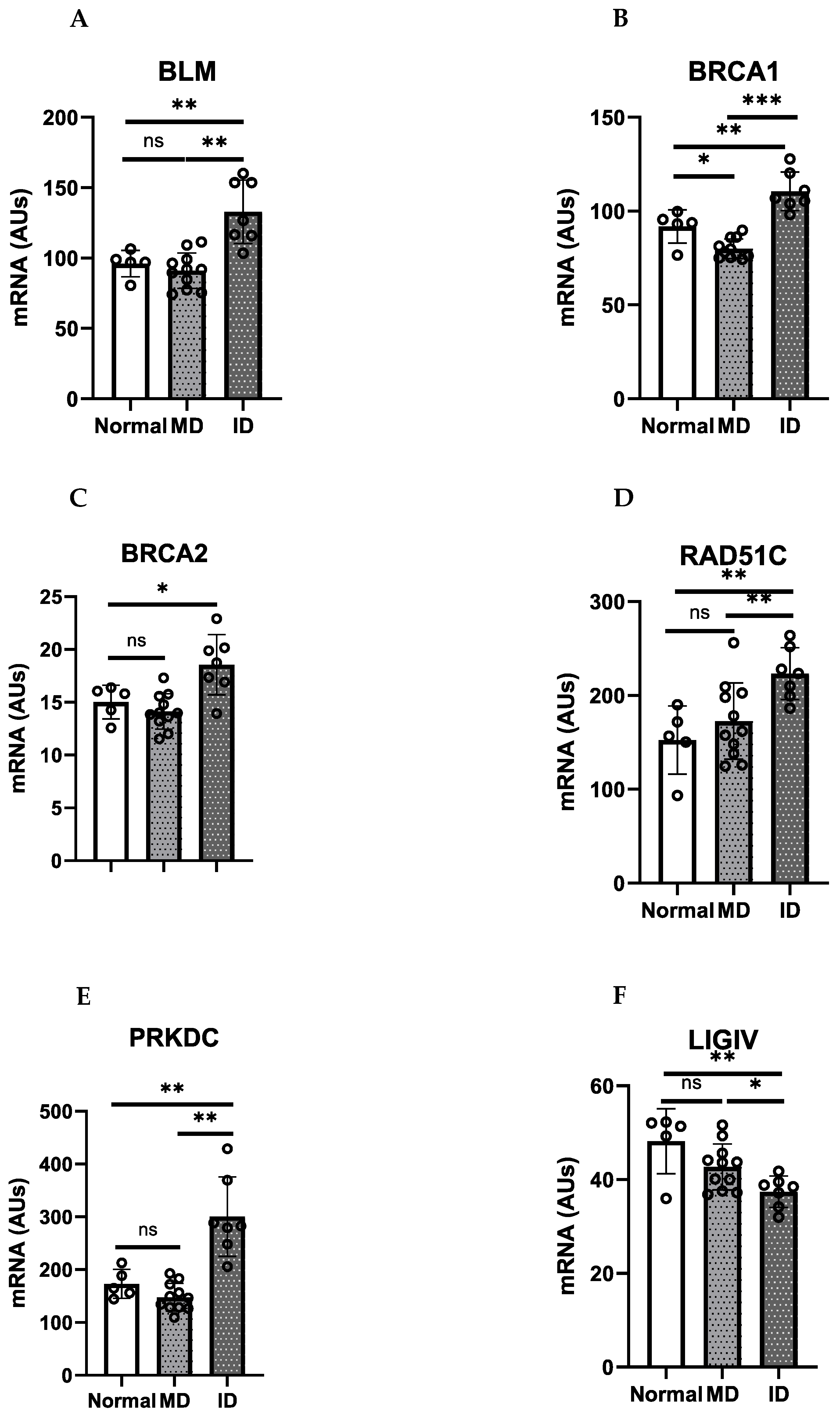
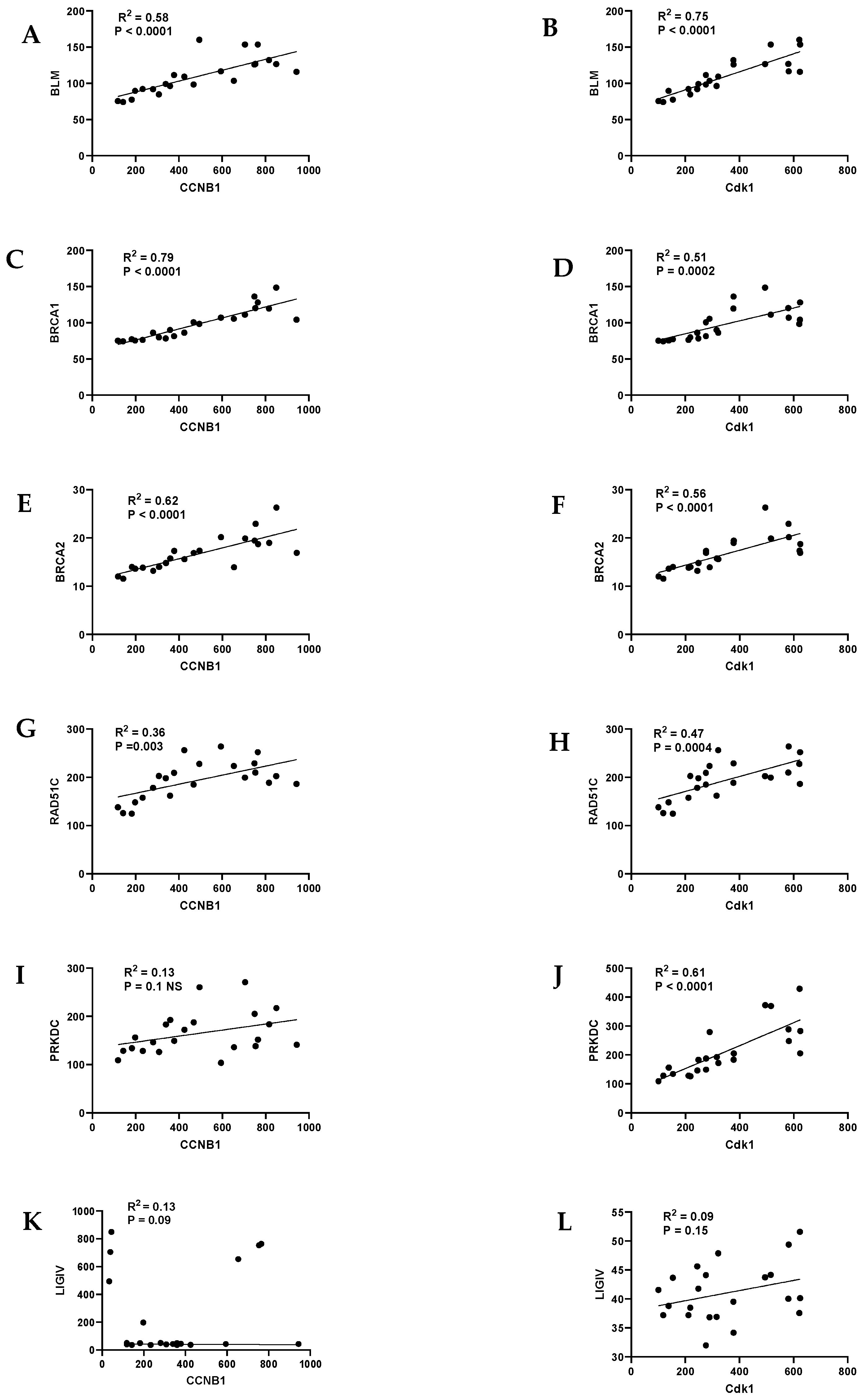
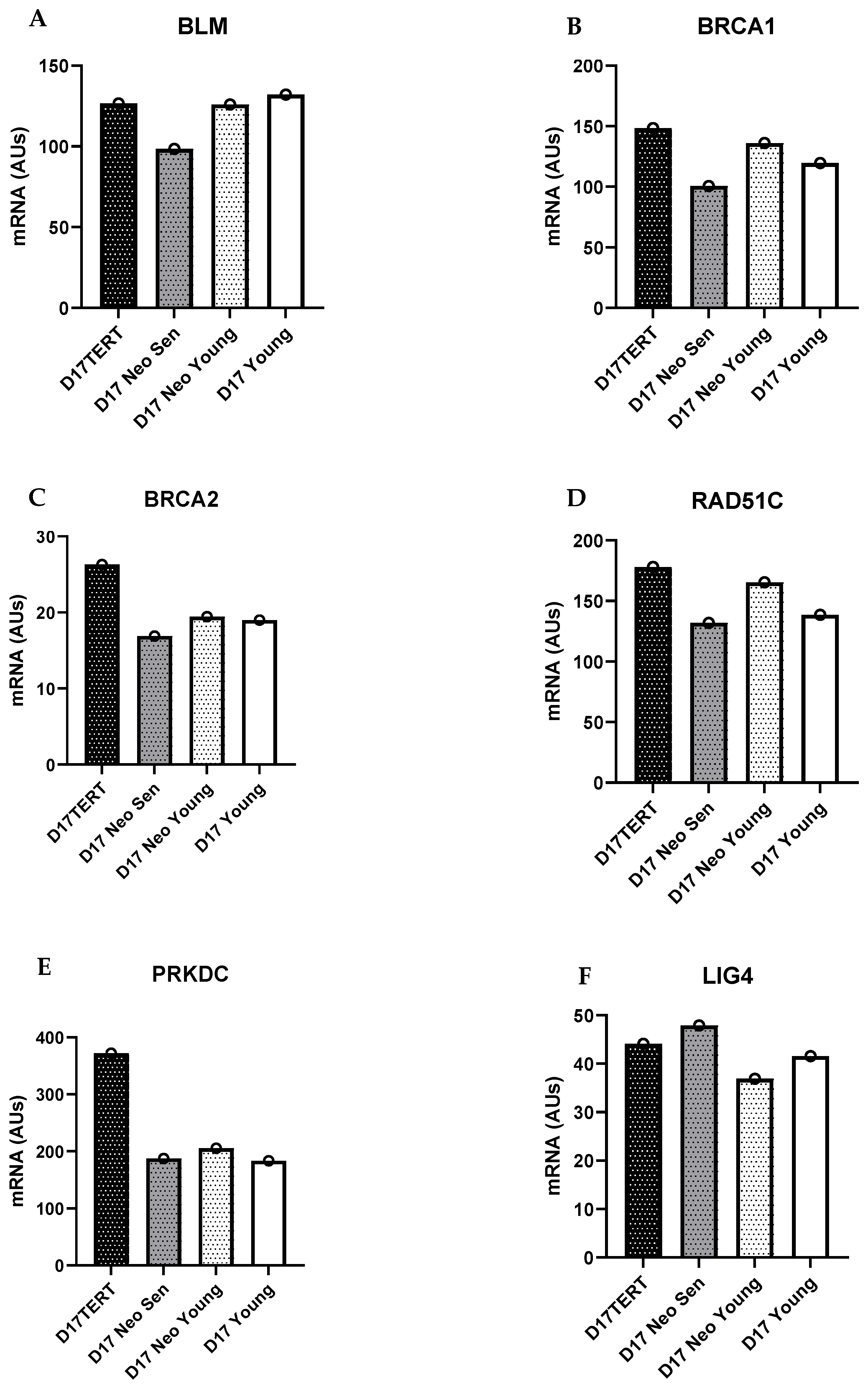
9. HR/FA Genes Are Upregulated in OPMD-Derived Keratinocyte Cultures That Have Bypassed Crisis
10. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomantarum, I.; Jemal, A.; Bray, F. Global statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- GBD 2019 Lip, Oral and Pharyngeal Cancer Collaborators. The global, regional, and national burden of adult lip, oral, and pharyngeal cancer in 204 countries and territories: A systematic analysis for the global burden of disease study 2019. JAMA Oncol. 2023, 9, 1401–1416. [Google Scholar] [CrossRef] [PubMed]
- Auperin, A. Epidemiology of head and neck cancers: An update. Curr. Opin. Oncol. 2020, 32, 178–186. [Google Scholar] [CrossRef]
- Costa, R.F.E.; Leao, M.L.B.; Sant’Ana, M.S.P.; Mesquita, R.A.; Gomez, R.S.; Santos-Silva, A.R.; Khurram, S.L.; Tailor, A.; Schouwstra, C.-M.; Robinson, L.; et al. Oral squamous cell carcinoma frequency in young patients from referral centers around the world. Head Neck Pathol. 2022, 16, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.R.; Netterville, J.L.; Sinard, R.J.; Mannion, K.; Rohde, S.L.; Langerman, A.; Kim, Y.J.; Lewis, J.S., Jr.; Lang Kuhs, K.A. Early onset oral tongue cancer in the United States: A literature review. Oral Oncol. 2018, 87, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.H.; Iyer, N.G.; Tan, M.H.; Edgren, G. Changing epidemiology of oral squamous cell carcinoma of the tongue. A global study. Head Neck 2017, 39, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Napier, S.S.; Speight, P.M. Natural history of potentially malignant oral lesions and conditions: An overview of the literature. J. Oral Pathol. Med. 2008, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.M.; Khurram, S.A.; Kujan, O. Oral potentially malignant disorders: Risk of progression to malignancy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.W.; Risk, J.M.; Woolgar, J.A.; Field, E.A.; Field, J.K.; Steele, J.C.; Rajlawat, B.P.; Triantafyllou, A.; Rogers, S.N.; Lowe, D.; et al. The clinical determinants of malignant transformation in oral epithelial dysplasia. Oral Oncol. 2012, 48, 969–976. [Google Scholar] [CrossRef]
- Warnakulasuriya, S.; Kujan, O.; Aguirre-Urizar, J.M.; Bagan, J.V.; Gonzalez-Moles, M.A.; Kerr, A.R.; Lodi, G.; Mello, F.W.; Monteiro, L.; Ogden, G.R.; et al. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by WHO Collaborating Centre for Oral Cancer. Oral Dis. 2021, 27, 1862–1880. [Google Scholar] [CrossRef]
- Prime, S.S.; Cirillo, N.; Cheong, S.C.; Prime, M.S.; Parkinson, E.K. Targeting the genetic landscape of oral potentially malignant disorders has the potential as a preventative strategy in oral cancer. Cancer Lett. 2021, 518, 102–114. [Google Scholar] [CrossRef]
- Prime, S.S.; Cirillo, N.; Parkinson, E.K. Chromosome instability is associated with escape from cellular senescence in oral premalignancy. Biology 2023, 12, 103. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudsen, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Ishida, T.; Tashiro, S.; Uchida, H.; Sakai, C.; Hironobe, N.; Miura, K.; Hashimoto, Y.; Arihiro, K.; Chayama, K.; et al. Smoking cessation reverses DNA double-strand breaks in human mononuclear cells. PLoS ONE 2014, 9, e103993. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, F.; Chen, S.; Li, X.; Kong, L.; Zhang, W. Potential role of host microbiome in areca-nut-associated carcinogenesis and addiction. Molecules 2022, 27, 8171. [Google Scholar] [CrossRef]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Kim, H. The effect of oral bacterial infection on DNA damage response in host cells. Am. J. Cancer Res. 2023, 13, 3157–3168. [Google Scholar]
- Sukmana, B.I.; Saleh, R.O.; Najim, M.A.; Al-Ghamdi, H.S.; Achmad, H.; Al-Hamdani, M.M.; Taher, A.A.; Alsalamy, A.; Khaledi, M.; Javadi, K. Oral microbiota and oral squamous cell carcinoma: A review of their relation and carcinogenic mechanisms. Front. Oncol. 2024, 14, 1319777. [Google Scholar] [CrossRef]
- Barash, H.; Gross, E.R.; Edrei, Y.; Ella, E.; Israel, A.; Cohen, I.; Corchia, N.; Ben-Moshe, T.; Pappo, O.; Pikarsky, E.; et al. Accelerated carcinogenesis following liver regeneration is associated with chronic inflammation-induced double-strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 2207–2212. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayapan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J.; et al. Exome sequencing in head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Kanemaru, Y.; Kamoshita, N.; Suzuki, T.; Arakawa, T.; Honma, M. Tracing the fates of site-specifically introduced DNA adducts in the human genome. DNA Repair 2014, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Eppard, M.; Passos, J.F.; Victorelli, S. Telomeres, cellular senescence, and aging: Past and future. Biogerontology 2024, 25, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barratt, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuesterfeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of premalignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression profile of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescent-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzman, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.K.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Niklander, S.E.; Lambert, D.W.; Hunter, K.D. Senescent cell in cancer: Wanted and unwanted citizens. Cells 2021, 10, 3315. [Google Scholar] [CrossRef]
- Yoshioka, K.I.; Katsumoto-Matsuo, R.; Matsuno, Y.; Ishiai, M. Genome instability and cancer risk associated with erroneous DNA repair. Int. J. Mol. Sci. 2021, 22, 12254. [Google Scholar] [CrossRef]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, K. Gamma-irradiated quiescent cells repair directly induced double strand breaks but accumulate persistent double strand breaks during subsequent DNA replication. Genes Cells 2016, 21, 789–797. [Google Scholar] [CrossRef]
- Matsuno, Y.; Hyodo, M.; Suzuki, M.; Tanaka, Y.; Horikoshi, Y.; Murakami, Y.; Torigoe, H.; Mano, H.; Tashiro, S.; Yoshioka, K. Replication-stress-associated DSBs induced by ionizing radiation risk genomic destabilization and associated clonal evolution. Science 2021, 24, 102313. [Google Scholar] [CrossRef]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022, 36, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-H.; Kang, M.K.; Kim, R.H.; Kameta, A.; Baluda, M.A.; Park, N.-H. Abnormal DNA end-joining activity in human head and neck cancer. Int. J. Mol. Med. 2006, 17, 917–924. [Google Scholar] [CrossRef]
- Werbrouck, J.; De Ruyck, K.; Duprez, F.; Van Eijeren, M.; Rietzschel, E.; Bekaert, S.; Vral, A.; De Neve, W.; Thierens, H. Single-nucleotide polymorphisms in DNA double strand break repair genes: Association with head and neck cancer and interaction with tobacco use and alcohol consumption. Mutat. Res. 2008, 656, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.-T.; Lin, C.-C.; Chiu, C.-F.; Tsai, M.-H. Role of nonhomologous end-joining in oral cancer and personalized pharmacogenomics. Biomedicine 2012, 2, 41–47. [Google Scholar] [CrossRef]
- Joshi, J.S.; Vora, H.H.; Ghosh, N.R.; Tankshali, R.N.; Jetly, D.H.; Trivedi, T.I. Nonhomologous end joining repair pathway molecules as predictive biomarkers for patients with oral squamous cell carcinoma. J. Cancer Res. Ther. 2021, 17, 1031–1038. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA checkpoints and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2020, 319, 1352–1355. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef]
- Psyrri, A.; Gkotzamanidou, M.; Papaxoinis, G.; Krikoni, L.; Economopoulou, P.; Kotsantis, I.; Anastasiou, M.; Souliotis, V.L. The DNA damage response network in the treatment of head and neck squamous cell carcinoma. ESMO Open 2021, 6, 100075. [Google Scholar] [CrossRef]
- Krais, J.J.; Johnson, N. BRCA 1 mutations in cancer: Coordinating deficiencies in homologous recombination with tumorigenesis. Cancer Res. 2020, 80, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Shi, L.; Xiao, X.; Wu, W.; Zhou, Z. Expression of DNA double strand repair proteins in oral leukoplakia and the risk of malignant transformation. Oncol. Lett. 2018, 15, 9827–9835. [Google Scholar] [PubMed]
- Nikitakis, N.G.; Rassidakis, G.Z.; Tasoulas, J.; Gkouveris, I.; Kamperos, G.; Daskaloupos, A.; Sklavounou, A. Alterations in the expression of DNA damage response-related molecules in potentially preneoplastic oral epithelial lesions. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tsai, H.J.; Gordon, M.R.; Li, R. Cellular stress associated with aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-J.; Alawi, F. Expression of DNA damage response biomarkers during oral carcinogenesis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2011, 111, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Karen-Ng, L.P.; Ahmad, U.S.; Gomes; Hunter, K.D.; Wan, H.; Hagi-Pavli, E.; Parkinson, E.K. Extracellular prostaglandins E1 and E2 and inflammatory cytokines are regulated by the senescence program in potentially malignant oral keratinocytes. Cancers 2022, 14, 2636. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- De Boer, D.V.; Brink, A.; Buijze, M.; Stigter-van Walsum, M.; Hunter, K.D.; Yistra, B.; Bloemena, E.; Leemans, C.R.; Brakenhoff, R.H. Establishment and genetic landscape of precancer cell model systems from the head and neck mucosal lining. Mol. Cancer Res. 2019, 17, 120–130. [Google Scholar] [CrossRef]
- Wright, W.E.; Pereira-Smith, O.M.; Shay, J.W. Reversible cellular senescence: Implications for immortalization of normal human diploid fibroblasts. Mol. Cell Biol. 1989, 9, 3088–3092. [Google Scholar] [PubMed]
- Counter, C.M.; Avilion, A.A.; Le Feuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Andera, L.; Opattova, A.; Vodickova, L. The interactions of DNA repair, telomere homeostasis, and p53 mutational status in solid cancers: Risk, prognosis, and prediction. Cancers 2021, 13, 479. [Google Scholar] [CrossRef] [PubMed]
- Andersson, N.; Saba, K.H.; Magnusson, L.; Nilsson, J.; Karlsson, J.; Nord, K.H.; Gisselsson, D. Inactivation of RB1, CDKN2A, and TP53 have distinct effects on genomic stability at side-by-side comparison in karyotypically normal cells. Genes Chromosomes Cancer 2022, 62, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Dentro, S.; Ong, S.H.; Sood, D.; Fernandez-Antoran, D.; Herms, A.; Kostiou, V.; Abnizova, I.; Hall, B.A.; Gerstung, M.; et al. p53 mutation in normal esophagus promotes multiple stages of carcinogenesis but is constrained by clonal competition. Nat. Commun. 2022, 13, 6206. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; He, A.; Jiang, Y.; Ruan, M.; Han, N. Targeting the DNA damage response as a potential therapeutic strategy for head and neck squamous cell carcinoma. Front. Oncol. 2022, 12, 1031944. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; He, F.; Shao, X.; Urano, M.; Shen, L.; Kim, D. Adenovirus mediated heat-activated antisense Ku70 expression radiosensitizes tumor cells in vitro and in vivo. Cancer Res. 2003, 63, 3268–3274. [Google Scholar] [PubMed]
- Feng, S.; Rabii, R.; Liang, G.; Song, C.; Chen, W.; Guo, M.; Wei, X.; Messadi, D.; Hu, S. the expression levels of XLF and mutant p53 are inversely correlated in head and neck cancer cells. J. Cancer 2016, 7, 1374–1382. [Google Scholar] [CrossRef]
- Marsit, C.J.; Liu, M.; Nelson, H.H.; Posner, M.; Suzuki, M.; Kelsey, K.T. Inactivation of the Fanconi anaemia/BRCA pathway in lung and oral cancers: Implications for treatment and survival. Oncogene 2004, 23, 1000–1004. [Google Scholar] [CrossRef]
- Sparano, A.; Quesnelle, K.M.; Kumar, M.S.; Wang, Y.; Sylvester, A.J.; Feldman, M.; Sewell, D.A.; Weinstein, G.S.; Brose, M.S. Genome-wide profiling of oral squamous cell carcinoma by array-based comparative genome hybridization. Laryngoscope 2006, 116, 735–741. [Google Scholar] [CrossRef]
- Morris, L.G.T.; Chandramohan, R.; West, L.; Zehir, A.; Chakravarty, D.; Pfister, D.G.; Wong, R.J.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; et al. The molecular landscape of recurrent and metastatic head and neck cancers: Insights from a precision oncology sequencing platform. JAMA Oncol. 2017, 3, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, X.; Lin, J.; Gao, X.; Xiong, J.; Liu, J.; Fei, Z.; Chen, C. A high homologous recombination deficiency score is associated with poor survival and a non-inflamed tumor microenvironment in head and neck squamous cell carcinoma patients. Oral Oncol. 2022, 128, 105860. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.S.; Jesssri, M.; Bennett, N.C.; Dalley, A.J.; Shearston, K.D.; Fox, S.A. Exome sequencing of oral leukoplakia and oral squamous cell carcinoma implicates DNA damage repair gene defects in malignant transformation. Oral Oncol. 2019, 96, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.W.; Ryan, M.P.; Gupta, J.; Triantafyllou, A.; Risk, J.; Shaw, R.J.; Wilson, J.B. Loss of FANCD2 and related proteins may predict malignant transformation in oral epithelial dysplasia. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2022, 133, 77–387. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Das, C.; Ghose, S.; Maitra, A.; Roy, B.; Majumder, P.P.; Biswas, N.K. Integrative analysis of genomic and transcriptomic data of normal, tumour, and co-occurring leukoplakia tissue triads drawn from patients with gingivobuccal oral cancer identifies signatures of tumour initiation and progression. J. Pathol. 2022, 257, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.T.; Tseng, H.C.; Wang, C.H.; Chiu, C.F.; Hua, C.H.; Wu, C.N.; Liang, S.Y.; Wang, C.L.; Tsai, M.H. Oral cancer and genetic polymorphism of DNA double strand break gene Ku70 in Taiwan. Oral Oncol. 2008, 44, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.F.; Tsai, M.H.; Tseng, H.C.; Wang, C.L.; Wang, C.H.; Wu, C.N.; Lin, C.C.; Bau, D.T. A novel single nucleotide polymorphism in XRCC4 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncol. 2008, 44, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Tsai, M.H.; Chiu, C.F.; Wang, C.H.; Chang, N.W.; Huang, C.Y.; Tsai, C.W.; Liang, S.Y.; Wang, C.L.; Bau, D.T. Association of XRCC4 codon 247 polymorphism in oral cancer susceptibility in Taiwan. Anticancer Res. 2008, 28, 1687–1691. [Google Scholar]
- Hsu, C.F.; Tseng, H.C.; Chiu, C.F.; Liang, S.Y.; Tsai, C.W.; Tsai, M.H.; Bau, D.T. Association between DNA double strand break gene Ku80 polymorphisms and oral cancer susceptibility. Oral Oncol. 2009, 45, 789–793. [Google Scholar] [CrossRef]
- Al-Hadyan, K.S.; Al-Harbi, N.M.; Al-Qahtani, S.S.; Alsbeih, G.A. Involvement of single nucleotide polymorphisms in predisposition to head and neck cancer in Saudia Arabia. Genet. Test. Mol. Biomark. 2012, 16, 95–101. [Google Scholar] [CrossRef]
- Yang, H.; Lippman, S.M.; Huang, M.; Jack Lee, J.; Wang, W.; Spitz, M.R.; Wu, X. Genetic polymorphisms in double-strand break DNA repair genes associated with risk of oral premalignant lesions. Eur. J. Cancer 2008, 44, 1603–1611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sliwinski, T.; Walczak, A.; Przybylowska, K.; Rusin, P.; Pietruszewska, W.; Zielinska-Blizniewska, H.; Olszewski, J.; Morawiec-Sztandera, A.; Jendrzejczyk, S.; Mlynarski, W.; et al. Polymorphisms of the XRCC3 C722T and the RAD51 G135C genes and the risk of head and neck cancer in a Polish population. Exp. Mol. Pathol. 2010, 89, 358–366. [Google Scholar] [CrossRef]
- Gresner, P.; Gromadzinska, J.; Polanska, K.; Twardowska, E.; Jurewicz, J.; Wasowicz, W. Genetic variability Xrcc3 Rad51 modulates risk of head and neck cancer. Gene 2012, 504, 166–174. [Google Scholar] [CrossRef]
- Kong, F.; Wu, J.; Hu, L.; Du, Y.; Pan, Y. Association between RAD51 polymorphisms and susceptibility of head and neck cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 6412–6419. [Google Scholar]
- Majumder, M.; Sikdar, N.; Ghosh, S.; Roy, B. Polymorphisms at XPD and XRCC1 DNA repair loci and increased risk of oral leukoplakia and cancer among NAT2 slow acetylators. Int. J. Cancer 2007, 120, 2148–2156. [Google Scholar] [CrossRef]
- Yadav, B.K.; Kaur, J.; Srivastava, A.; Ralhan, R. Effect of polymorphisms in XRCC1, CCND1 and GSTM1 and tobacco exposure as risk modifier for oral leukoplakia. Int. J. Bio Mark. 2009, 24, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Silva Barros, C.C.; Almeida Freitas, R.; Costa Miguel, M.C.; Silveira, E.J.D. DNA damage through oxidative stress is an important event in oral leukoplakia. Arch. Oral Biol. 2022, 135, 105359. [Google Scholar] [CrossRef]
- Tan, H. The association between SNPs and cancer predisposition: Correlation or causality? EBioMedicine 2017, 16, 8–9. [Google Scholar] [CrossRef]
- Zhao, H.; Albino, A.P.; Jorgensen, E.; Traganos, F.; Darzynkiewicz, Z. DNA damage response induced by tobacco smoke in normal human bronchial epithelial and A549 pulmonary adenocarcinoma cells assessed by laser scanning cytometry. Cytom. A 2009, 75, 840–847. [Google Scholar] [CrossRef]
- Mondal, P.; Datta, S.; Maiti, G.P.; Baral, A.; Jha, G.N.; Panda, C.K.; Chowdhury, S.; Ghosh, S.; Roy, B.; Roychoudhury, S. Comprehensive SNP scan of DNA repair and DNA damage response genes reveal multiple susceptibility loci conferring risk to tobacco associated leukoplakia and oral cancer. PLoS ONE 2013, 8, e56952. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Miller, M.W. Ethanol-induced DNA repair in neural stem cells is transforming growth factor β1-dependent. Exp. Neurol. 2019, 317, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Laimins, L. High-risk human papillomaviruses and DNA repair. Recent. Results Cancer Res. 2021, 217, 141–155. [Google Scholar] [PubMed]
- Sharma, R.; Lewis, S.; Wlodarski, M.W. DNA repair syndromes and cancer: Insights into genetics and phenotype patterns. Front. Pediatr. 2020, 8, 570084. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Cancer in Fanconi anaemia, 1927–2021. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Amenabar, J.M.; Torres-Pereira, C.C.; Tang, K.D.; Punyadeera, C. Two enemies, one fight: An update of oral cancer in patients with Fanconi anaemia. Cancer 2019, 125, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Peake, J.D.; Noguchi, E. Fanconi anaemia: Current insights regarding epidemiology, cancer, and DNA repair. Hum. Genet. 2022, 141, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- German, J.; Ellis, N.A. Bloom syndrome. In The Genetic Basis of Human Cancer; Vogelstein, B., Kinzler, K.W., Eds.; McGraw-Hill: New York, NY, USA, 1998; pp. 301–315. [Google Scholar]
- Hecht, F.; Hecht, B.K. Cancer in ataxia telangiectasia. Cancer Genet. Cytogenet. 1990, 46, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-L.; Xia, L.; Zhao, C.; Yao, J. ATM rs189037 (G > A) polymorphism increased the risk of cancer: An updated meta-analysis. BMC Med. Genet. 2019, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.D.; Winter, M.R.; Nogueira, C.P.; Larson, P.S.; Finnemore, E.M.; Dolan, R.W.; Fuleihan, N.; Chakravarti, A.; Zeitman, A.; Rosenbury, C.L. Loss of heterozygosity at 11q23 in squamous cell carcinoma of the head and neck is associated with recurrent disease. Clin. Cancer Res. 1998, 4, 2787–2793. [Google Scholar]
- Ai, L.; Vo, Q.N.; Zuo, C.; Li, L.; Ling, W.; Suen, J.Y.; Hanne, E.; Brown, K.D. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: Promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol. Biomark. Prev. 2004, 13, 150–156. [Google Scholar] [CrossRef]
- Dos Santos, E.S.; Santos-Silva, A.R. Lack of association between oral squamous cell carcinoma and Li Fraumeni syndrome. Head Neck Pathol. 2023, 17, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef]
- Sahm, V.; Maurer, C.; Baumeister, T.; Anand, A.; Strangmann, J.; Schmid, R.M.; Wang, T.C.; Quante, M. Telomere shortening accelerates tumor initiation in the L2-IL1B mouse model of Barrett esophagus and emerges as a possible biomarker. Oncotarget 2022, 13, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Aida, J.; Kobayashi, T.; Saku, T.; Yamaguchi, M.; Shimomura, N.; Nakamura, K.-I.; Ishikawa, N.; Maruyama, S.; Cheng, J.; Poon, S.S.; et al. Short telomeres in an oral precancerous lesion: Q-FISH analysis of leukoplakia. J. Oral Pathol. Med. 2012, 41, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Pal, J.; Raiput, Y.; Shrivastava, S.; Gahine, R.; Mungutwar, V.; Baradiya, T.; Chandraker, A.; Ramakrishnar, P.P.; Mishra, S.S.; Banjara, H.; et al. A standalone approach to utilize telomere length measurement as a surveillance tool in oral leukoplakia. Mol. Oncol. 2021, 16, 1650–1660. [Google Scholar] [CrossRef]
- Doksani, Y.; de Lange, T. The role of double strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- McGregor, F.; Muntoni, A.; Fleming, J.; Brown, J.; Felix, D.H.; MacDonald, D.G.; Parkinson, E.K.; Harrison, P.R. Molecular changes associated with oral dysplasia progression and acquisition of immortality: Potential for its reversal by 5-azacytidine. Cancer Res. 2002, 62, 4757–4766. [Google Scholar] [PubMed]
- Bell, R.J.; Rube, H.T.; Xavier-Magalhaes, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT promoter mutations: A common path to immortality. Mol. Cancer Res. 2016, 14, 315323. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mut. Res. Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef]
- Chang, K.P.; Wang, C.I.; Pickering, C.R.; Huang, Y.; Tsai, C.N.; Tsang, N.M.; Kao, H.K.; Chang, M.H.; Myers, J.N. Prevalence of promoter mutations in the TERT gene in oral cavity squamous cell carcinoma. Head Neck 2017, 39, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; El-Naggar, A.K.; Fan, Y.H.; Lee, J.S.; Lippman, S.M.; Kayser, S.; Lotan, R.; Hong, W.K. Telomerase activity in head and neck squamous cell carcinoma and adjacent tissues. Cancer Res. 1996, 56, 5600–5604. [Google Scholar] [PubMed]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, A.; Darzacq, X.; Bastion, B.C.; Hockmeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Zhang, S.; Chen, B.P.C. DNA-dependent protein kinase in telomere maintenance and protection. Cell Mol. Biol. Lett. 2020, 25, 2. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Samper, E.; Flores, J.M.; Blasco, M.A. Telomere-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 2000, 26, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Schiemann, W.P. Telomerase in cancer: Function, regulation and clinical translation. Cancers 2022, 14, 808. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.M.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hou, K.; Zhang, K.; Jia, S. Regulation of replication stress in alternative lengthening of telomeres by Fanconi anaemia protein. Genes 2022, 13, 180. [Google Scholar] [CrossRef]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Picket, H.A.; Tam, P.P.L.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef]
- Bojovic, B.; Booth, R.E.; Jin, Y.; Zhou, X.; Crowe, D.L. Alternative lengthening of telomeres in cancer stem cell in vivo. Oncogene 2015, 34, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, R.; Walker, T.; Revil, T.; De Weck, A.; Badescu, D.; O’Sullivan, J.; Higgins, C.; Elliott, L.; Liloglou, T.; Risk, J.M.; et al. Analysis of head and neck cancer progression reveals novel and relevant stage-specific changes associated with immortalisation and malignancy. Sci. Rep. 2019, 9, 11912. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Walne, A.; Dokal, I. The biology and management of dyskeratosis congenita and related disorders of telomeres. Expert. Rev. Haematol. 2022, 15, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Tummula, H.; Dokal, A.D.; Walne, A.; Ellison, A.; Cardoso, S.; Amirthasigamanipillai, S.; Kirwan, M.; Browne, I.; Sidhu, J.K.; Rajeeve, V.; et al. Genome instability is a consequence of transcription deficiency in patients with bone marrow failure harboring ERCC variants. Proc. Natl. Acad. Sci. USA 2018, 115, 7777–7782. [Google Scholar] [CrossRef] [PubMed]
- Walne, A.J.; Vulliamy, T.; Bewicke-Copley, F.; Wang, J.; Alnajar, J.; Bridger, M.G.; Ma, B.; Tummala, H.; Dokal, I. Genome-wide whole blood transcriptome profiling across inherited bone marrow failure subtypes. Blood Adv. 2021, 5, 5360–5371. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.E.; Gibson, F.M.; Rizzo, S.; Tooze, J.A.; Marsh, J.C.; Gordon-Smith, E.C. Progressive telomere shortening in aplastic anaemia. Blood 1998, 91, 3582–3592. [Google Scholar] [CrossRef]
- Brummendorf, T.H.; Maciejewski, J.P.; Mak, J.; Young, N.S.; Lansdorp, P.M. Telomere length in leukocyte subpopulations of patients with aplastic anaemia. Blood 2001, 97, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ferreira dos Santos-Costa, S.F.; Brennan, P.A.; Gomez, R.S.; Fregnani, E.R.; Santos-Silva, A.R.; Martins, M.D.; de Castro-Junior, G.; Fonseca, F.P. Molecular basis of oral squamous cell carcinoma in young patients: Is it any different from older patients? J. Oral Pathol. Med. 2018, 47, 541–546. [Google Scholar] [CrossRef]
- Kolegova, E.S.; Palysheva, M.R.; Larionova, I.V.; Fedorova, I.K.; Kulbakin, D.E.; Choinzonov, E.L.; Denisov, E.V. Early-onset oral cancer as a clinical entity: Aetiology and pathogenesis. Int. J. Oral Maxillofac. Surg. 2022, 51, 1497–1509. [Google Scholar] [CrossRef]
- Cury, S.S.; Miranda, P.M.; Marchi, F.A.; Canto, L.M.; Chulam, T.C.; Petersen, A.H.; Aagaard, M.M.; Pinto, C.A.L.; Kowalski, L.P.; Rogatto, S.R. Germline variants in DNA repair genes are associated with young-onset head and neck cancer. Oral Oncol. 2021, 122, 105545. [Google Scholar] [CrossRef]
- Tremblay, S.; Dos Reis, P.P.; Bradley, G.; Galloni, N.N.; Perez-Ordonez, B.; Freeman, J.; Brown, D.; Gilbert, R.; Gullane, P.; Irish, J.; et al. Young patients with oral squamous cell carcinoma: Study of GSTP1 and deregulation of the Fanconi anaemia genes. Arch. Otolaryngol. Head Neck Surg. 2006, 132, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, S.C.; Chinn, S.B.; Donavan, F.X.; Chowdhury, N.I.; Kamat, A.; Adeyemo, A.A.; Thomas, J.W.; Vemulapalli, M.; Hussey, C.S.; Reid, H.H.; et al. Assessing the spectrum of germline variation of Fanconi anaemia genes among patients with head and neck cancer before age 50. Cancer 2017, 123, 3943–3954. [Google Scholar] [CrossRef]
- Edington, K.G.; Loughran, O.P.; Berry, I.J.; Parkinson, E.K. Cellular immortality: A late event in the progression of human squamous cell carcinoma of the head and neck associated with p53 alteration and a high frequency of allele loss. Mol. Carcinog. 1995, 13, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Loughran, O.; Malliri, A.; Owens, D.; Gallimore, P.H.; Stanley, M.A.; Ozanne, B.; Frame, M.C.; Parkinson, E.K. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: Relationship of dysfunction to immortalization and neoplasia. Oncogene 1996, 13, 561–568. [Google Scholar] [PubMed]
- Munro, J.; Stott, F.J.; Vousden, K.H.; Peters, G.; Parkinson, E.K. Role of the alternative INK4A proteins in human keratinocyte senescence: Evidence for the specific inactivation of p16INK4A upon immortalization. Cancer Res. 1999, 59, 2516–2521. [Google Scholar]
- Hunter, K.D.; Thurlow, J.K.; Fleming, J.; Drake, P.J.; Vass, K.; Kalna, G.; Higham, D.J.; Herzyk, P.; Macdonald, D.G.; Parkinson, E.K.; et al. Divergent routes to oral cancer. Cancer Res. 2006, 66, 7405–7413. [Google Scholar] [CrossRef]
- Karen-Ng, L.P.; James, E.L.; Stephen, A.; Bennett, M.H.; Mycielska, M.E.; Parkinson, E.K. The extracellular metabolome stratifies low and high risk potentially malignant oral keratinocytes and identifies citrate as a potential non-invasive marker of tumour progression. Cancers 2021, 13, 4212. [Google Scholar] [CrossRef]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits lifespan become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef]
- Muntoni, A.; Fleming, J.; Gordon, K.E.; Hunter, K.; McGregor, F.; Parkinson, E.K.; Harrison, P.R. Senescing oral dysplasias are not immortalized by ectopic expression of hTERT alone without other molecular changes, such as loss of INK4A and/or retinoic acid receptor-beta, but p53 mutations are not necessarily required. Oncogene 2003, 22, 7804–7808. [Google Scholar] [CrossRef][Green Version]
- Rieckmann, T.; Kriegs, M.; Nitsch, L.; Hoffer, K.; Rohaly, G.; Kocher, S.; Petersen, C.; Dikomey, E.; Dornreiter, I.; Dahm-Daphi, J. p53 modulates homologous recombination at I-SceI-induced double-strand breaks through cell-cycle regulation. Oncogene 2013, 32, 968–975. [Google Scholar] [CrossRef]
- Bertrand, P.; Saintigny, Y.; Lopez, B.S. p53’s double life: Transactivation-independent repression of homologous recombination. Trends Genet. 2004, 20, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Rouillard, D.; Boulet, A.; Levalois, C.; Soussi, T.; Lopez, B.S. Increase of spontaneous intrachromosomal homologous recombination in mammalian cells expressing mutant p53. Oncogene 1997, 14, 1117–1122. [Google Scholar] [CrossRef][Green Version]
- Liu, R.; Hu, J.J.; Wu, X.; Hu, Q.; Jiang, W.; Zhao, Z.; Xia, F.; Lou, X. Precisely detecting the telomerase activities by an AIEgen probe with dual signal outputs after cell cycle synchronization. Anal. Chem. 2022, 94, 4874–4880. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, D.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting replication stress in cancer therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E. Why your new cancer biomarker may never work: Recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anaemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 39. [Google Scholar] [CrossRef]
- Hoskins, E.E.; Gunawardeena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Well, S.I. Coordinate regulation of Fanconi anaemia gene expression occurs through the Rb/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.E.; Franceschi, S.; Barra, S.; Talamini, R.; Vecchia, C. A comparison of the joint effects of alcohol and smoking on the risk of cancer in the upper aerodigestive tract. Cancer Epidemiol. Biomark. Prev. 1993, 2, 519–523. [Google Scholar]
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–191. [Google Scholar] [CrossRef]
- Sari, E.F.; Prayogo, G.P.; Loo, Y.T.; Zhang, P.; McCullough, M.J.; Cirillo, N. Distinct phenolic, alkaloid and antioxidant profile in betel quids from four regions of Indonesia. Sci. Rep. 2020, 10, 16254. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; Vossen, D.M.; Borgmann, K.; Hageman, F.; Grenman, R.; Verwijs-Janssen, M.; Mout, L.; Kluin, R.J.C.; Nieuwland, M.; Severson, T.M.; et al. Fanconi anaemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget 2018, 9, 18198–18213. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, N.; Morgan, D.J.; Pedicillo, M.C.; Celentano, A.; Lo Muzio, L.; McCullough, M.J.; Prime, S.S. Characterisation of the cancer-associated glucocorticoid system: Key role of 11β-hydroxysteroid dehydrogenase type 2. Br. J. Cancer 2017, 117, 984–993. [Google Scholar] [CrossRef]
- Ritter, H.D.; Antonova, L.; Mueller, C.R. The unliganded glucocorticoid receptor positively regulates the tumor suppressor gene BRCA1 through GABP beta. Mol. Cancer Res. 2012, 10, 558–569. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homologous Recombination (HR) | Non-Homologous End Joining (NHEJ) |
|---|---|
| RAD51: encodes RAD51 | XRCC5: encodes Ku80 XRCC6: encodes KU70 |
| XRCC2: encodes XRCC2 XRCC3: encodes XRCC3 | PRKDC and XRCC7: encode DNA-dependent protein kinase (PK) catalytic subunits |
| TP53: encodes p53 | DCLRE 1C: encodes Artemis protein which acts as an endonuclease |
| RPA1: encodes Replication Protein A (RPA) | POLM: encodes DNA polymerase Pol μ POLM: encodes DNA polymerase Pol λ |
| BRCA1 BRCA2 | XRCC4/LIGIV: encodes Ligase IV |
| BLM: encodes DNA helicase RecQ | |
| MUS81: encodes endonuclease enzyme |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prime, S.S.; Darski, P.; Hunter, K.D.; Cirillo, N.; Parkinson, E.K. A Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer. Int. J. Mol. Sci. 2024, 25, 4092. https://doi.org/10.3390/ijms25074092
Prime SS, Darski P, Hunter KD, Cirillo N, Parkinson EK. A Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer. International Journal of Molecular Sciences. 2024; 25(7):4092. https://doi.org/10.3390/ijms25074092
Chicago/Turabian StylePrime, Stephen S., Piotr Darski, Keith D. Hunter, Nicola Cirillo, and E. Kenneth Parkinson. 2024. "A Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer" International Journal of Molecular Sciences 25, no. 7: 4092. https://doi.org/10.3390/ijms25074092
APA StylePrime, S. S., Darski, P., Hunter, K. D., Cirillo, N., & Parkinson, E. K. (2024). A Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer. International Journal of Molecular Sciences, 25(7), 4092. https://doi.org/10.3390/ijms25074092