
L-Asparaginase Conjugates from the Hyperthermophilic Archaea Thermococcus sibiricus with Improved Biocatalytic Properties

 ,
,  , , , and
, , , and 
Abstract
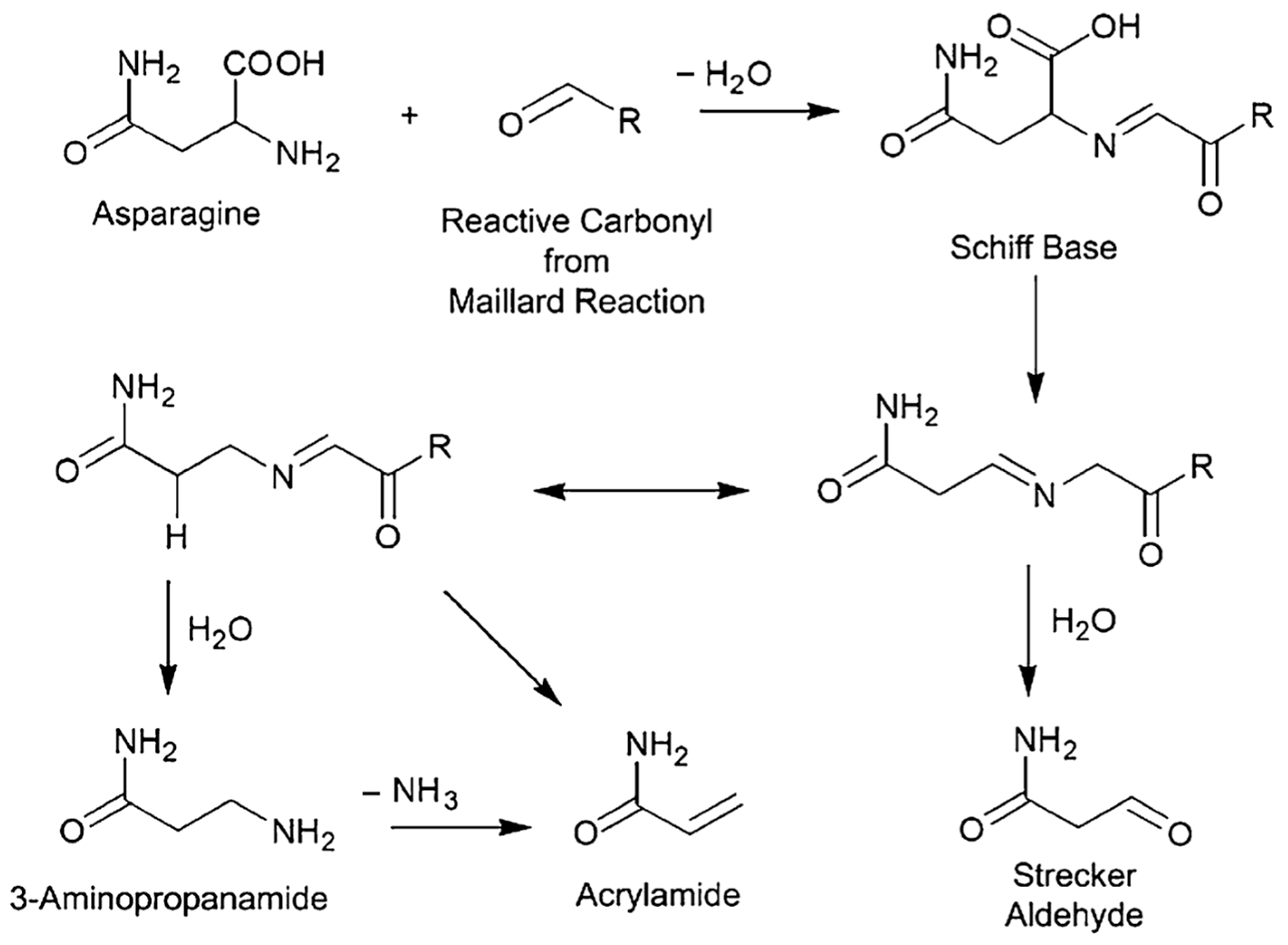
1. Introduction
2. Results
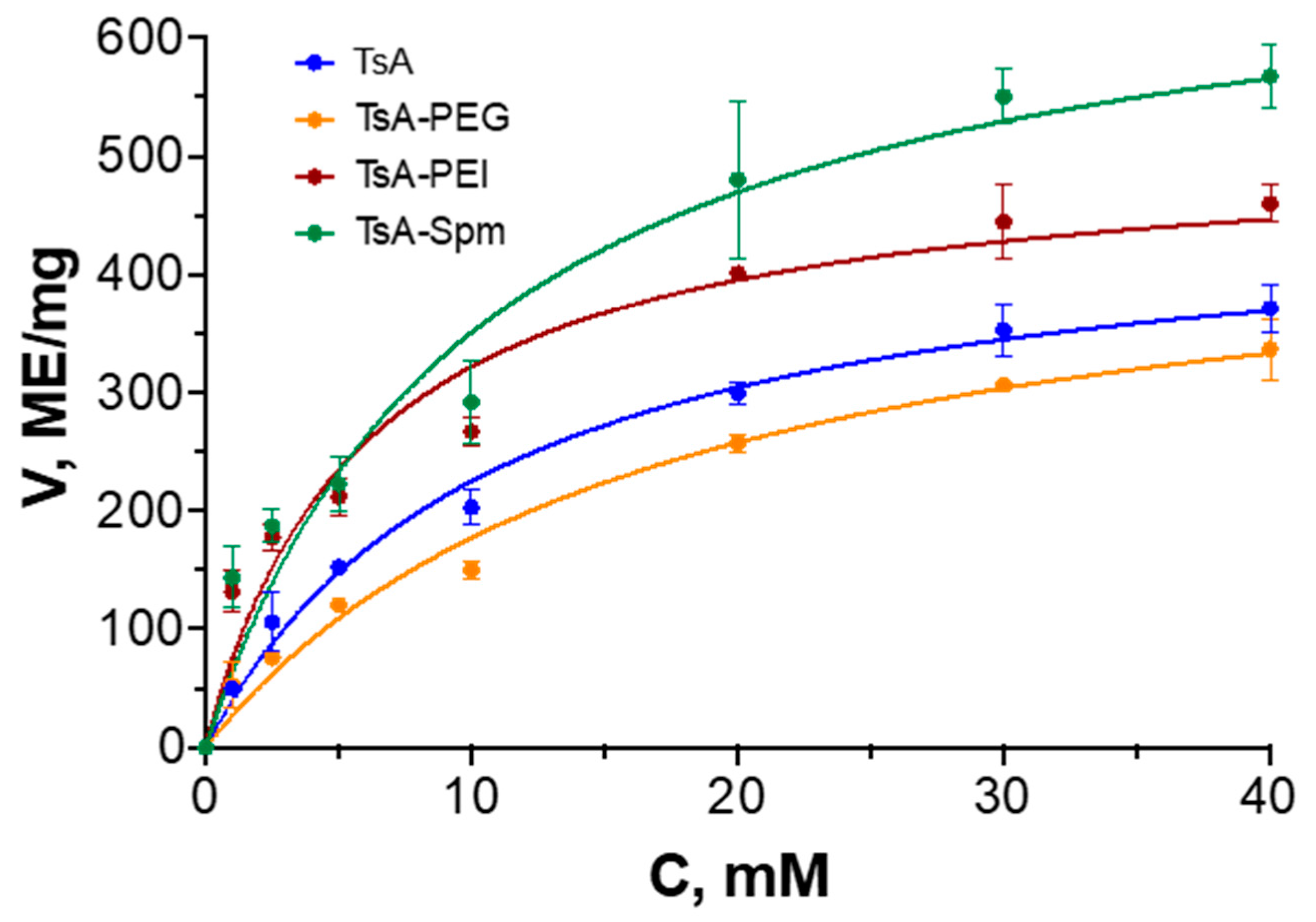
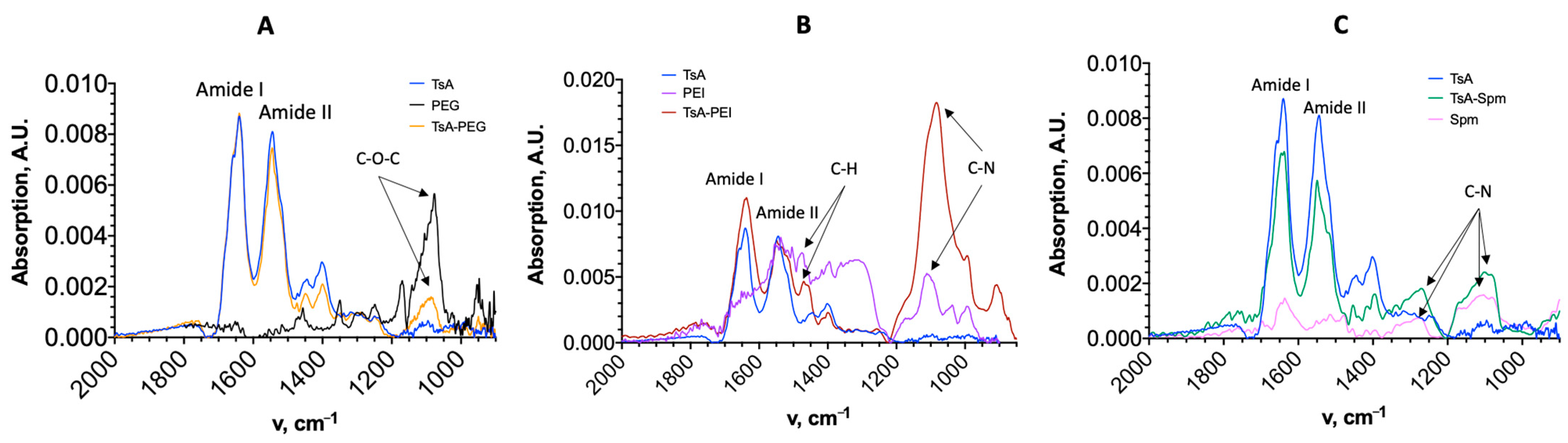
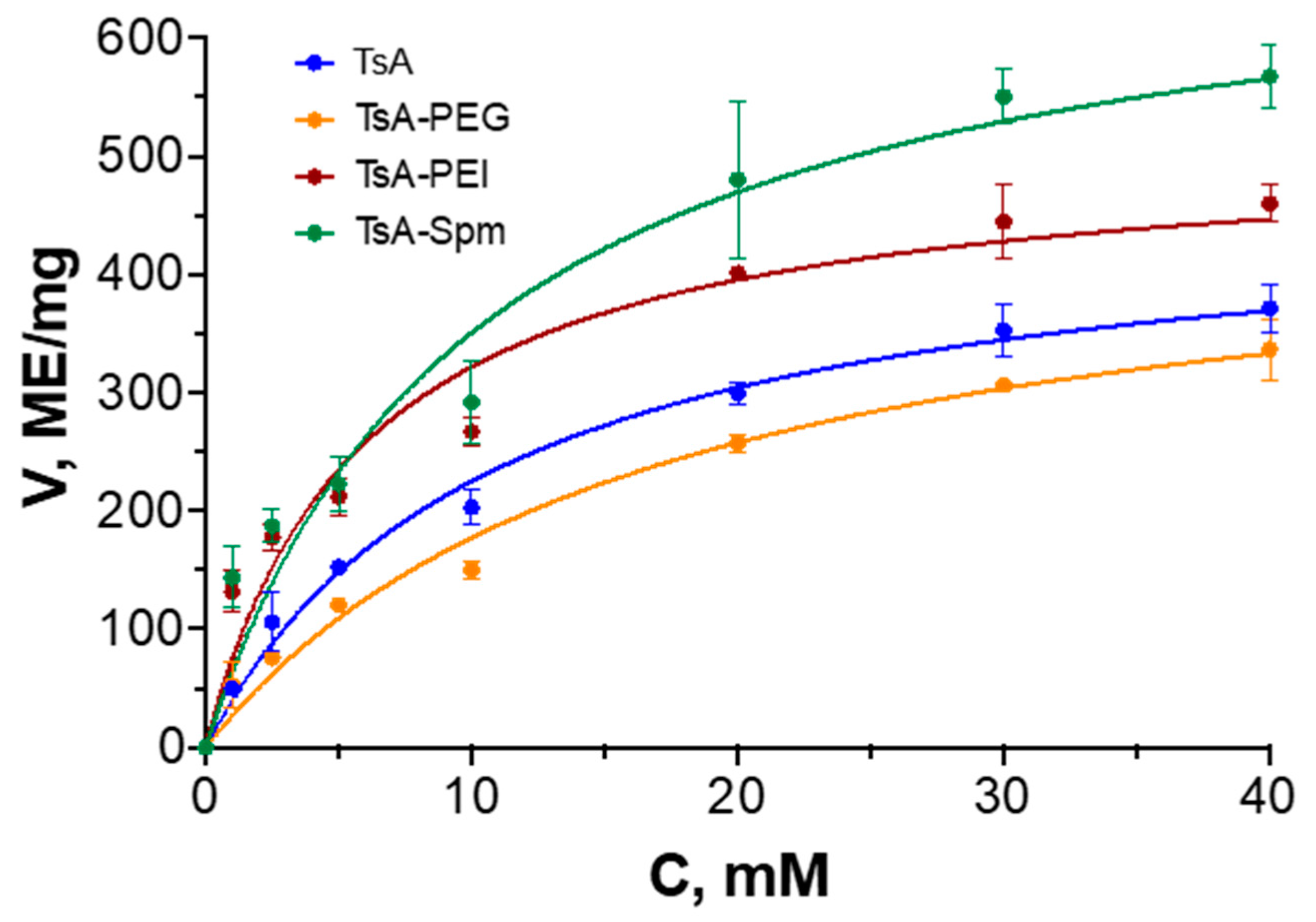
2.1. Characteristics of TsA Conjugates
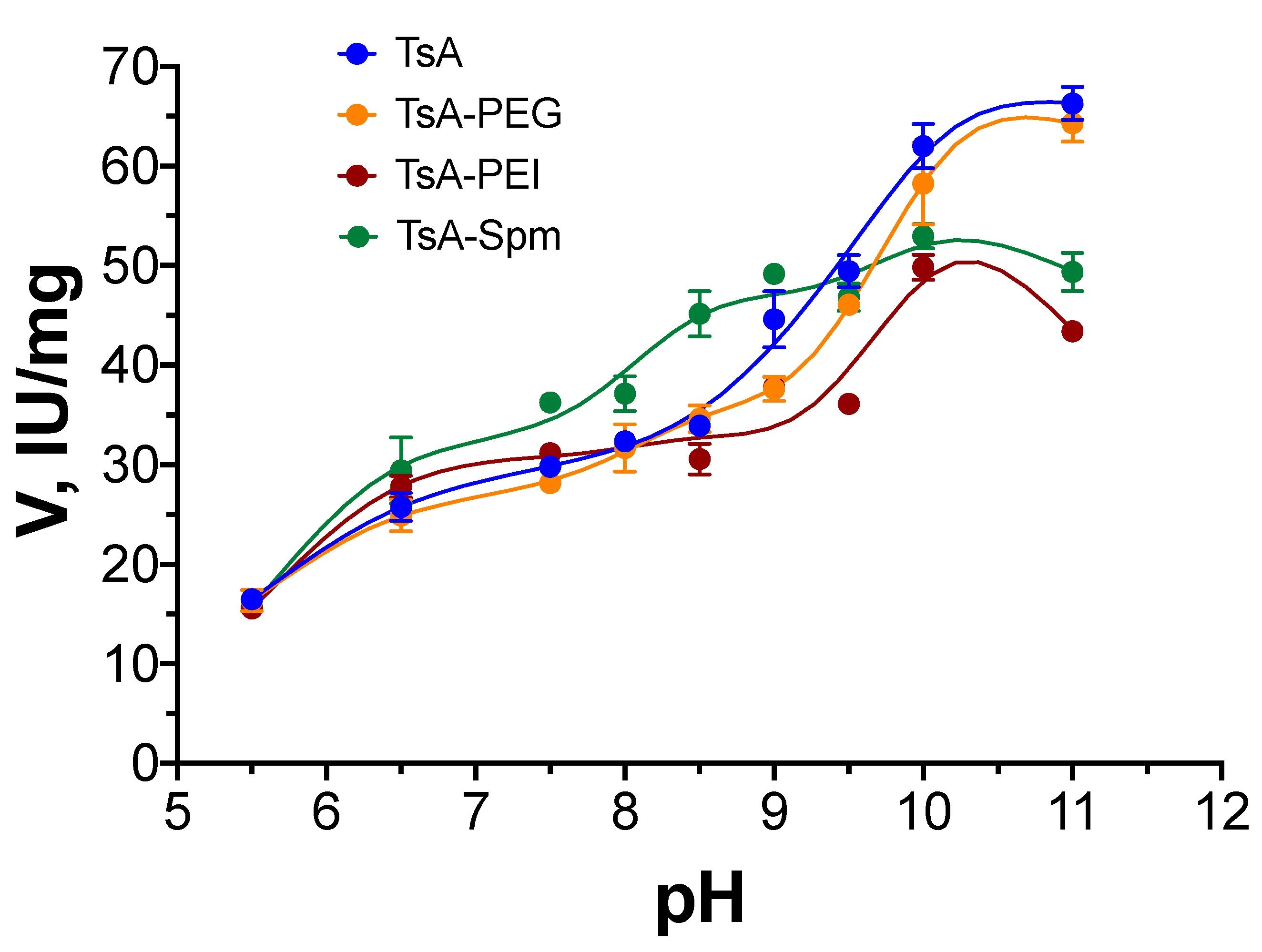
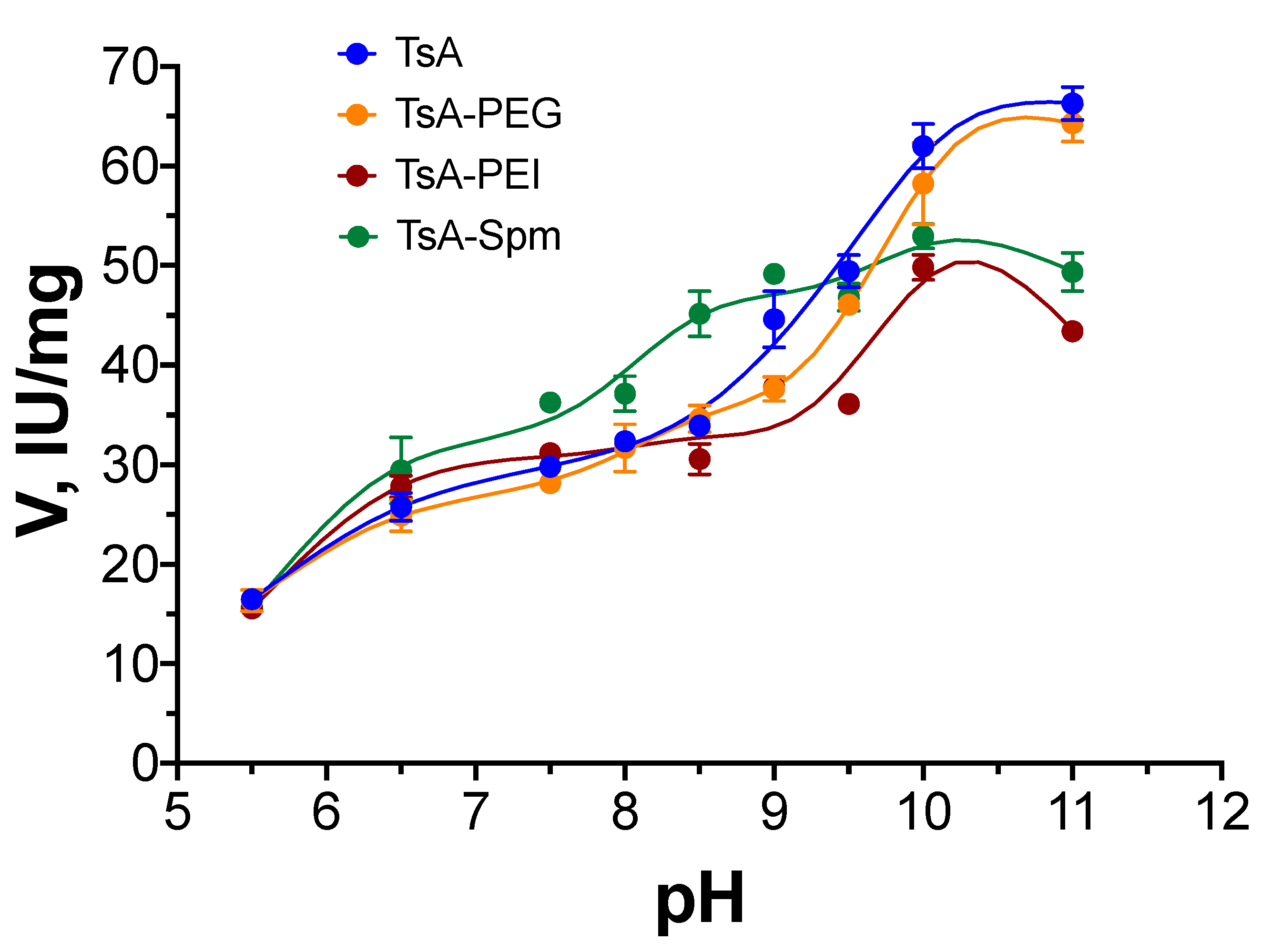
2.2. pH Dependence of TsA Conjugate Activity Compared to That of Native L-ASNase
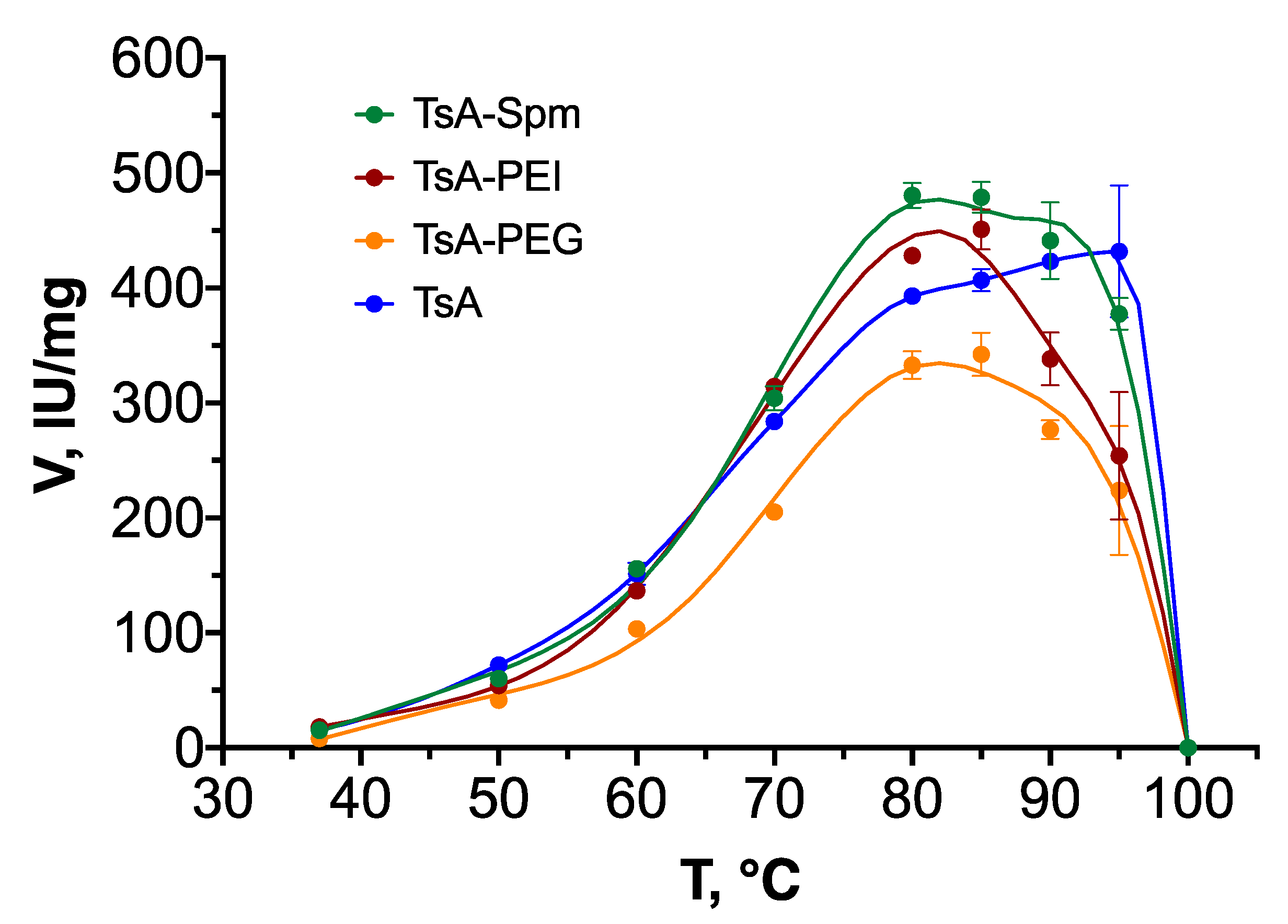
2.3. Temperature Dependence of the Activity
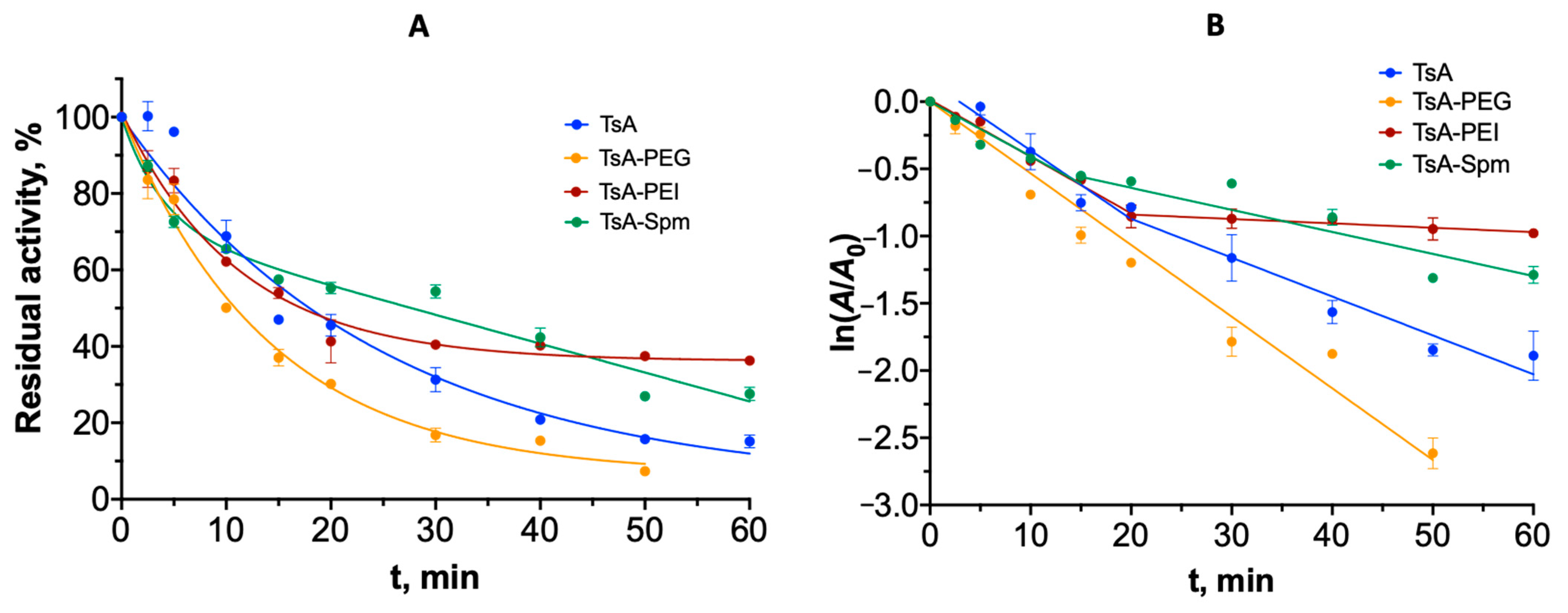
2.4. Thermal Inactivation Mechanism and Aggregation of TsA Conjugates
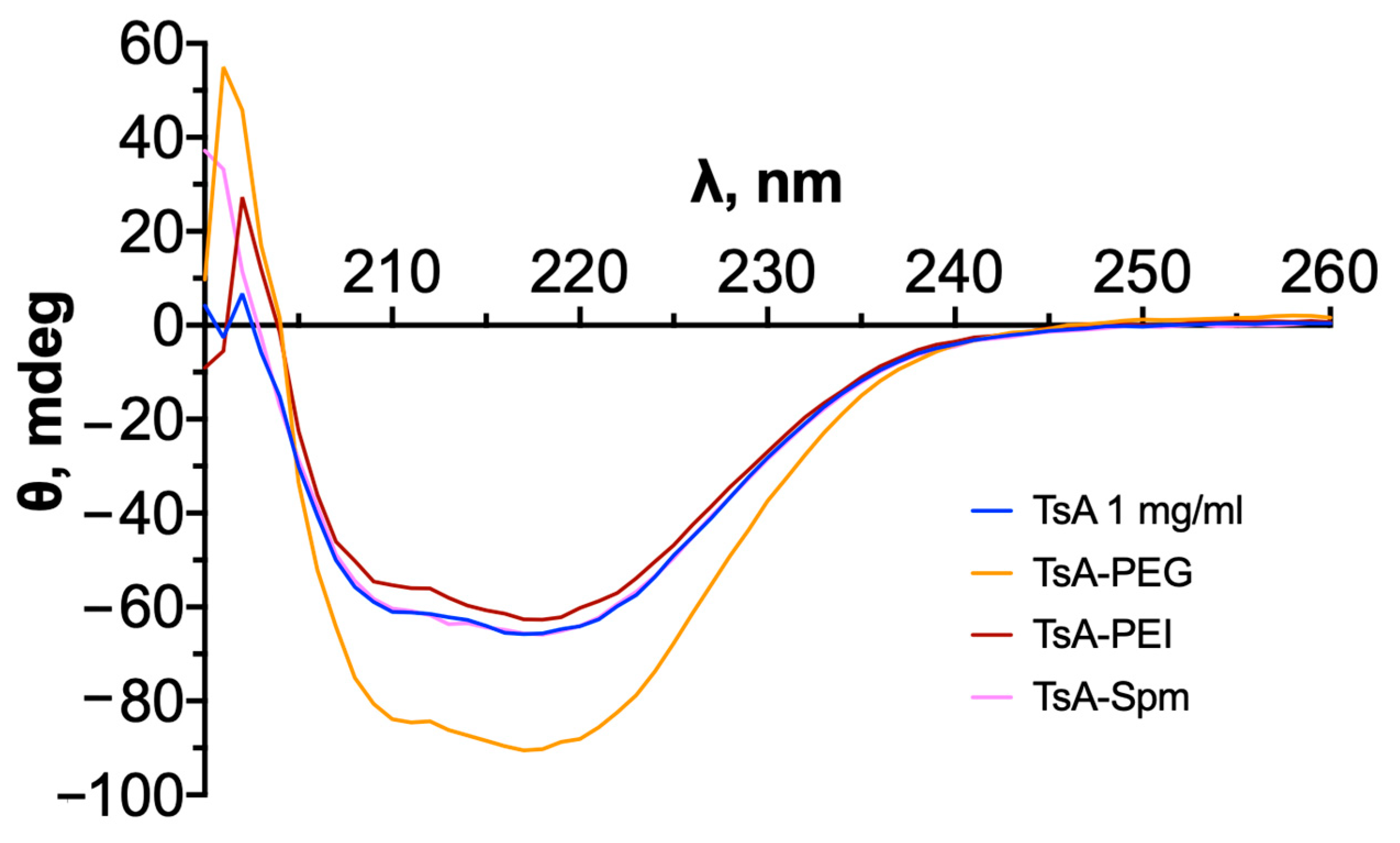
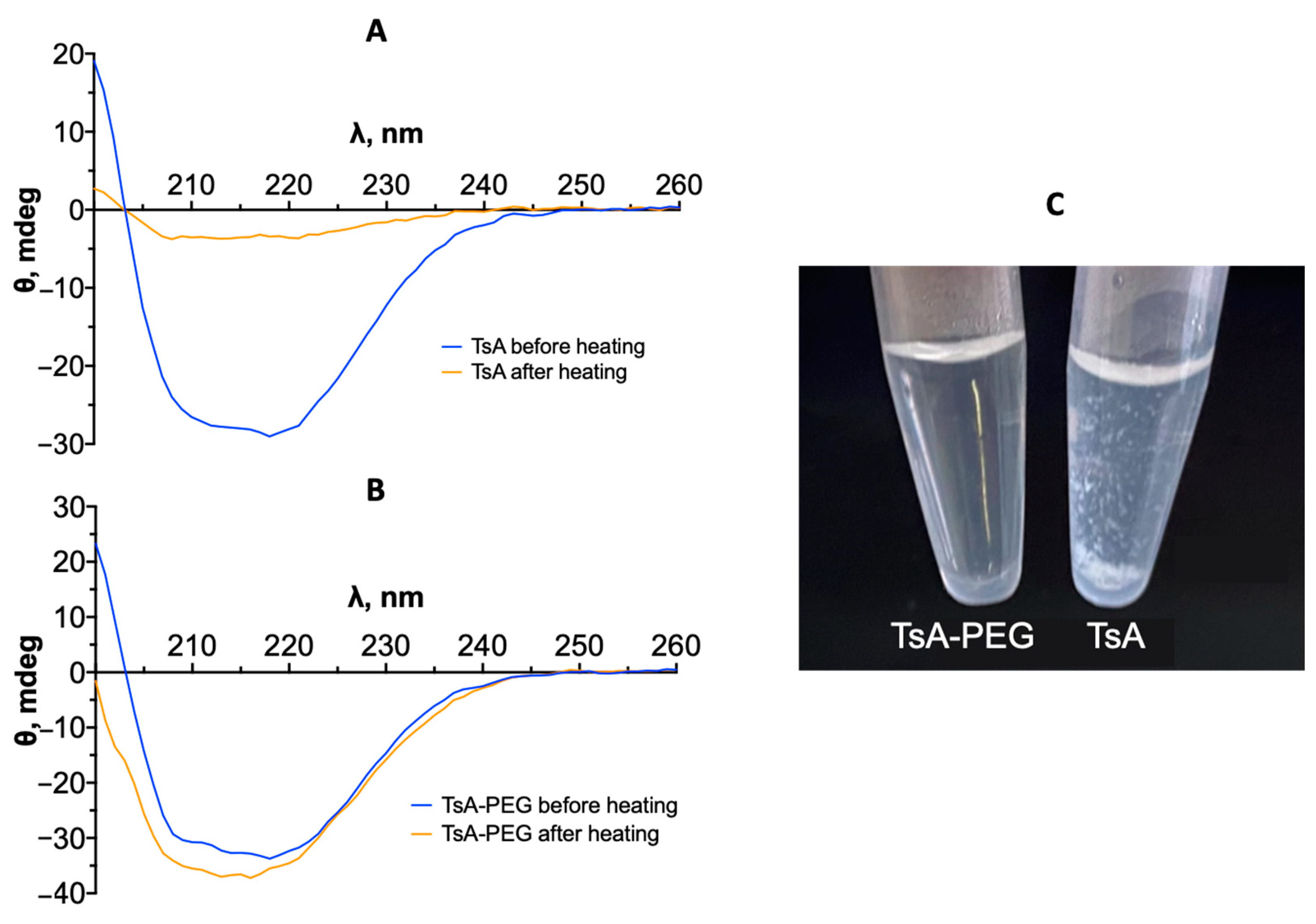
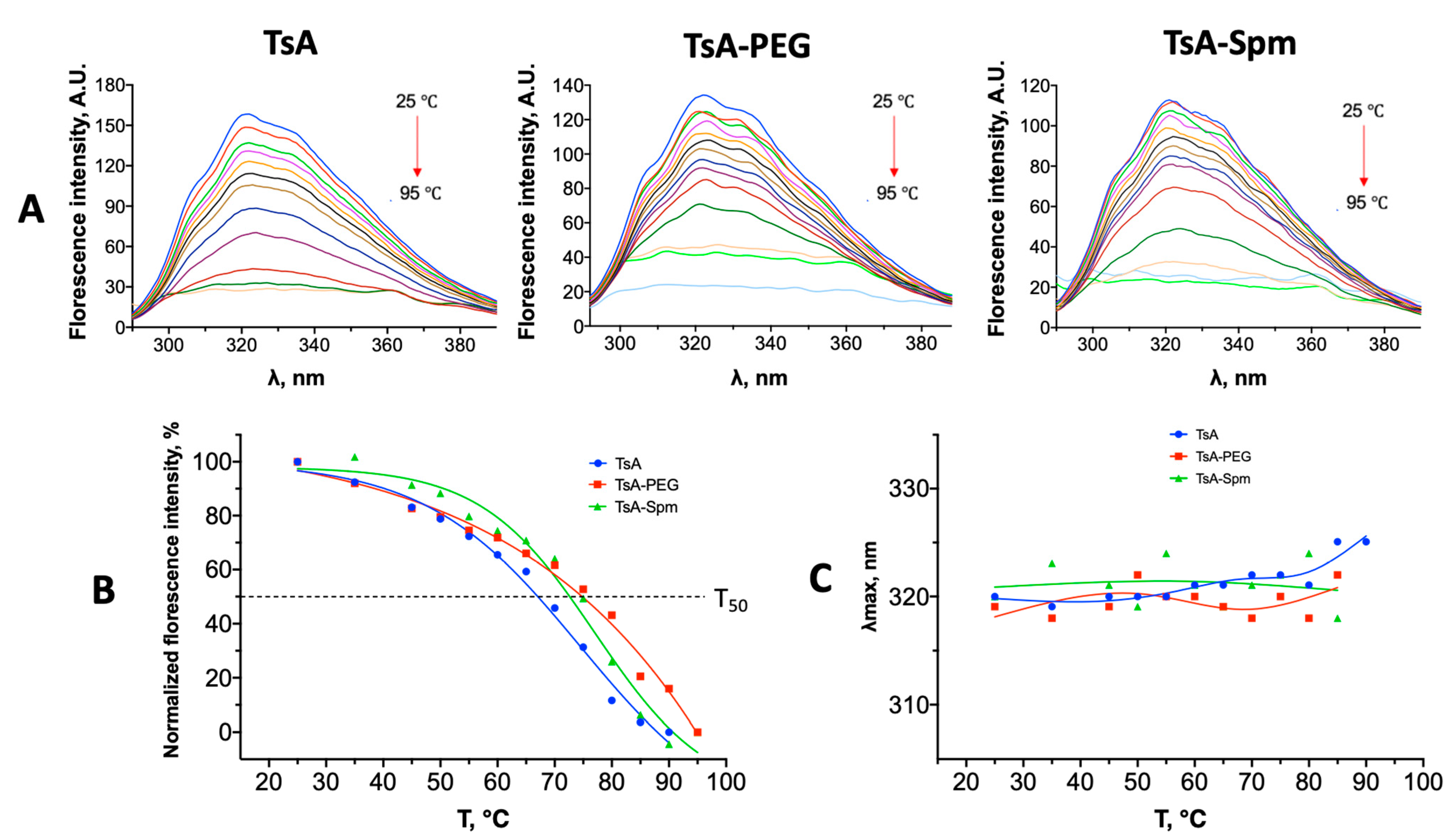
2.5. Changes in the Tertiary Structure of TsA and Conjugates during Thermal Denaturation
3. Discussion
4. Materials and Methods
4.1. Enzyme Preparation and Chemicals
4.2. Synthesis and Purification of L-Asparaginase Conjugates
4.3. L-Asparaginase Catalytic Activity Measurement
4.4. Registration of CD Spectra
4.5. Registration of IR Spectra
4.6. Registration of Fluorescence Spectra
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, R.; Wan, X.; Geng, X.; Xue, D.; Xie, Z.; Chen, C. Microbial L-Asparaginase for Application in Acrylamide Mitigation from Food: Current Research Status and Future Perspectives. Microorganisms 2021, 9, 1659. [Google Scholar] [CrossRef]
- Zuo, S.; Zhang, T.; Jiang, B.; Mu, W. Reduction of Acrylamide Level through Blanching with Treatment by an Extremely Thermostable L-Asparaginase during French Fries Processing. Extremophiles 2015, 19, 841–851. [Google Scholar] [CrossRef]
- Khan, M.; Razu, M.H. Method Development and Validation for the Quantification of Acrylamide in Potato Chips and Other Locally Available Food by LC-MS/MS in Bangladesh. Food Nutr. Sci. 2019, 10, 876–892. [Google Scholar] [CrossRef]
- Robert, F.; Hau, J.; Philippe, A.; Robert, M.; Riediker, S. Acrylamide Is Formed in the Maillard Reaction. Nature 2002, 419, 448–449. [Google Scholar]
- Liyanage, D.W.K.; Yevtushenko, D.P.; Konschuh, M.; Bizimungu, B.; Lu, Z.X. Processing Strategies to Decrease Acrylamide Formation, Reducing Sugars and Free Asparagine Content in Potato Chips from Three Commercial Cultivars. Food Control 2021, 119, 107452. [Google Scholar] [CrossRef]
- Pandey, A.K.; Ravi, N.; Chauhan, O.P. Quality Attributes of Vacuum Fried Fruits and Vegetables: A Review. J. Food Meas. Charact. 2020, 14, 1543–1556. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Bachas, L.G. Biosensor for Asparagine Using a Thermostable Recombinant Asparaginase from Archaeoglobus Fulgidus. Anal. Chem. 2002, 74, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Dumina, M.; Zhgun, A.; Pokrovskaya, M.; Aleksandrova, S.; Zhdanov, D.; Sokolov, N.; El’darov, M. A Novel L-Asparaginase from Hyperthermophilic Archaeon Thermococcus Sibiricus: Heterologous Expression and Characterization for Biotechnology Application. Int. J. Mol. Sci. 2021, 22, 9894. [Google Scholar] [CrossRef]
- Pereira, A.D.S.; Souza, C.P.L.; Moraes, L.; Fontes-Sant’ana, G.C.; Amaral, P.F.F. Polymers as Encapsulating Agents and Delivery Vehicles of Enzymes. Polymers 2021, 13, 4061. [Google Scholar] [CrossRef]
- Giri, P.; Pagar, A.D.; Patil, M.D.; Yun, H. Chemical Modification of Enzymes to Improve Biocatalytic Performance. Biotechnol. Adv. 2021, 53, 107868. [Google Scholar] [CrossRef]
- Li, R.; Zhang, Z.; Pei, X.; Xia, X. Covalent Immobilization of L-Asparaginase and Optimization of Its Enzyme Reactor for Reducing Acrylamide Formation in a Heated Food Model System. Front. Bioeng. Biotechnol. 2020, 8, 584758. [Google Scholar] [CrossRef]
- Tabandeh, M.R.; Aminlari, M. Synthesis, Physicochemical and Immunological Properties of Oxidized Inulin-l-Asparaginase Bioconjugate. J. Biotechnol. 2009, 141, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Chahardahcherik, M.; Ashrafi, M.; Ghasemi, Y.; Aminlari, M. Effect of Chemical Modification with Carboxymethyl Dextran on Kinetic and Structural Properties of L-Asparaginase. Anal. Biochem. 2020, 591, 113537. [Google Scholar] [CrossRef]
- Sannikova, E.P.; Bulushova, N.V.; Cheperegin, S.E.; Gubaydullin, I.I.; Chestukhina, G.G.; Ryabichenko, V.V.; Zalunin, I.A.; Kotlova, E.K.; Konstantinova, G.E.; Kubasova, T.S.; et al. The Modified Heparin-Binding l-Asparaginase of Wolinella Succinogenes. Mol. Biotechnol. 2016, 58, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Ates, B. Immobilization of l -Asparaginase on Carrier Materials: A Comprehensive Review. Bioconjug. Chem. 2017, 28, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Sukhoverkov, K.V.; Kudryashova, E.V. PEG-Chitosan and Glycol-Chitosan for Improvement of Biopharmaceutical Properties of Recombinant L-Asparaginase from Erwinia carotovora. Biochemistry 2015, 80, 113–119. [Google Scholar] [CrossRef]
- Dobryakova, N.V.; Zhdanov, D.D.; Sokolov, N.N.; Aleksandrova, S.S.; Pokrovskaya, M.V.; Kudryashova, E.V. Rhodospirillum rubrum L-Asparaginase Conjugates with Polyamines of Improved Biocatalytic Properties as a New Promising Drug for the Treatment of Leukemia. Appl. Sci. 2023, 13, 3373. [Google Scholar] [CrossRef]
- Dobryakova, N.V.; Kudryashova, E.V. Stabilization of Erwinia carotovora and Rhodospirillum rubrum L-Asparaginases in Complexes with Polycations. Appl. Biochem. Microbiol. 2023, 59, 1183–1191. [Google Scholar] [CrossRef]
- Egunova, O.R.; Reshetnikova, I.S.; German, S.V.; Kazimirova, K.O.; Khabibullin, V.R.; Zhelobitskaya, E.A.; Shtykov, S.N. Sorption-Fluorimetric Determination of Enrofloxacin with Magnetite Nanoparticles Modified by Polyethylenimine. Chem. Biol. Ecol. 2016, 16, 48–52. [Google Scholar] [CrossRef]
- Sakar Dasdan, D.; Tosun, G.; Karahan, Y. An Investigation of the PH Effect on the Particle Size and Zeta Potentials of Poly (Ethylene Glycol) and Poly Ehtylene-Block-Poly (Ethylene Glycol) with Various Molecular Weights. Bulg. Chem. Commun. 2018, 50, 5–11. [Google Scholar]
- Dobryakova, N.; Zhdanov, D.; Dumina, M.; Aleksandrova, S.; Pokrovskaya, M.; Genin, A.; Shishparenok, A.; Zhgun, A.; Kudryashova, E.V. Thermal Inactivation Mechanism and Structural Features Providing Enhanced Thermal Stability of Hyperthermophilic Thermococcus sibiricus L-Asparaginase in Comparison with Mesophilic and Thermophilic L-Asparaginases. Catalysts 2023, 13, 832. [Google Scholar] [CrossRef]
- Singh, K.; Shandilya, M.; Kundu, S.; Kayastha, A.M. Heat, Acid and Chemically Induced Unfolding Pathways, Conformational Stability and Structure-Function Relationship in Wheat α-Amylase. PLoS ONE 2015, 10, e0129203. [Google Scholar] [CrossRef]
- Yang, C.; Lu, D.; Liu, Z. How PEGylation Enhances the Stability and Potency of Insulin: A Molecular Dynamics Simulation. Biochemistry 2011, 50, 2585–2593. [Google Scholar] [CrossRef]
- Mu, Q.; Hu, T.; Yu, J. Molecular Insight into the Steric Shielding Effect of PEG on the Conjugated Staphylokinase: Biochemical Characterization and Molecular Dynamics Simulation. PLoS ONE 2013, 8, e68559. [Google Scholar] [CrossRef]
- Matteo, C.; Colombini, A.; Bettini, L.R.; Porcu, L.; Barzaghi, S.; Ceruti, T.; Silvestri, D.; Amoroso, A.; Dell’Acqua, F.; Gotti, G.; et al. Pharmacological and Clinical Monitoring in Children with Acute Lymphoblastic Leukemia Treated with a Biogeneric PEG-L-Asparaginase Product. Pediatr. Blood Cancer 2022, 69, e29753. [Google Scholar] [CrossRef]
- Meneguetti, G.P.; Santos, J.H.P.M.; Obreque, K.M.T.; Vaz Barbosa, C.M.; Monteiro, G.; Farsky, S.H.P.; De Oliveira, A.M.; Angeli, C.B.; Palmisano, G.; Ventura, S.P.M.; et al. Novel Site-Specific PEGylated L-Asparaginase. PLoS ONE 2019, 14, e0211951. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Paz, J.; Saxena, M.; Delinois, L.J.; Joaquín-Ovalle, F.M.; Lin, S.; Chen, Z.; Rojas-Nieves, V.A.; Griebenow, K. Thiol-Maleimide Poly(Ethylene Glycol) Crosslinking of L-Asparaginase Subunits at Recombinant Cysteine Residues Introduced by Mutagenesis. PLoS ONE 2018, 13, e0197643. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Pozdyshev, D.V.; Semenyuk, P.I. Polyelectrolytes for Enzyme Immobilization and the Regulation of Their Properties. Polymers 2022, 14, 4204. [Google Scholar] [CrossRef]
- Kurinomaru, T.; Tomita, S.; Hagihara, Y.; Shiraki, K. Enzyme Hyperactivation System Based on a Complementary Charged Pair of Polyelectrolytes and Substrates. Langmuir 2014, 30, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Yasutake, Y.; Morita, H.; Tanaka, I. Structure of the Type I L-Asparaginase from the Hyperthermophilic Archaeon Pyrococcus Horikoshii at 2.16 Å Resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 294–301. [Google Scholar] [CrossRef]
- Guo, J.; Coker, A.R.; Wood, S.P.; Cooper, J.B.; Chohan, S.M.; Rashid, N.; Akhtar, M. Structure and Function of the Thermostable L-Asparaginase from Thermococcus Kodakarensis. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Xue, D.; Zhang, T.; Jiang, B.; Mu, W. Biochemical Characterization of an Extremely Thermostable L-Asparaginase from Thermococcus Gammatolerans EJ3. J. Mol. Catal. B Enzym. 2014, 109, 122–129. [Google Scholar] [CrossRef]
- Luo, J.; Ying, K.; Bai, J. Savitzky-Golay Smoothing and Differentiation Filter for Even Number Data. Signal Process. 2005, 85, 1429–1434. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | MM, Da | Origins | Structure | ζ-Potential, mV (at pH 7.0) | Ref. |
|---|---|---|---|---|---|
| Polyethylenimine | 2000 | Synthetic | Linear | 20 | [19] |
| Spermine | 202 | Natural | Linear | - | |
| Polyethyleneglycol | 5000 | Synthetic | Linear | −(4–6) | [20] |
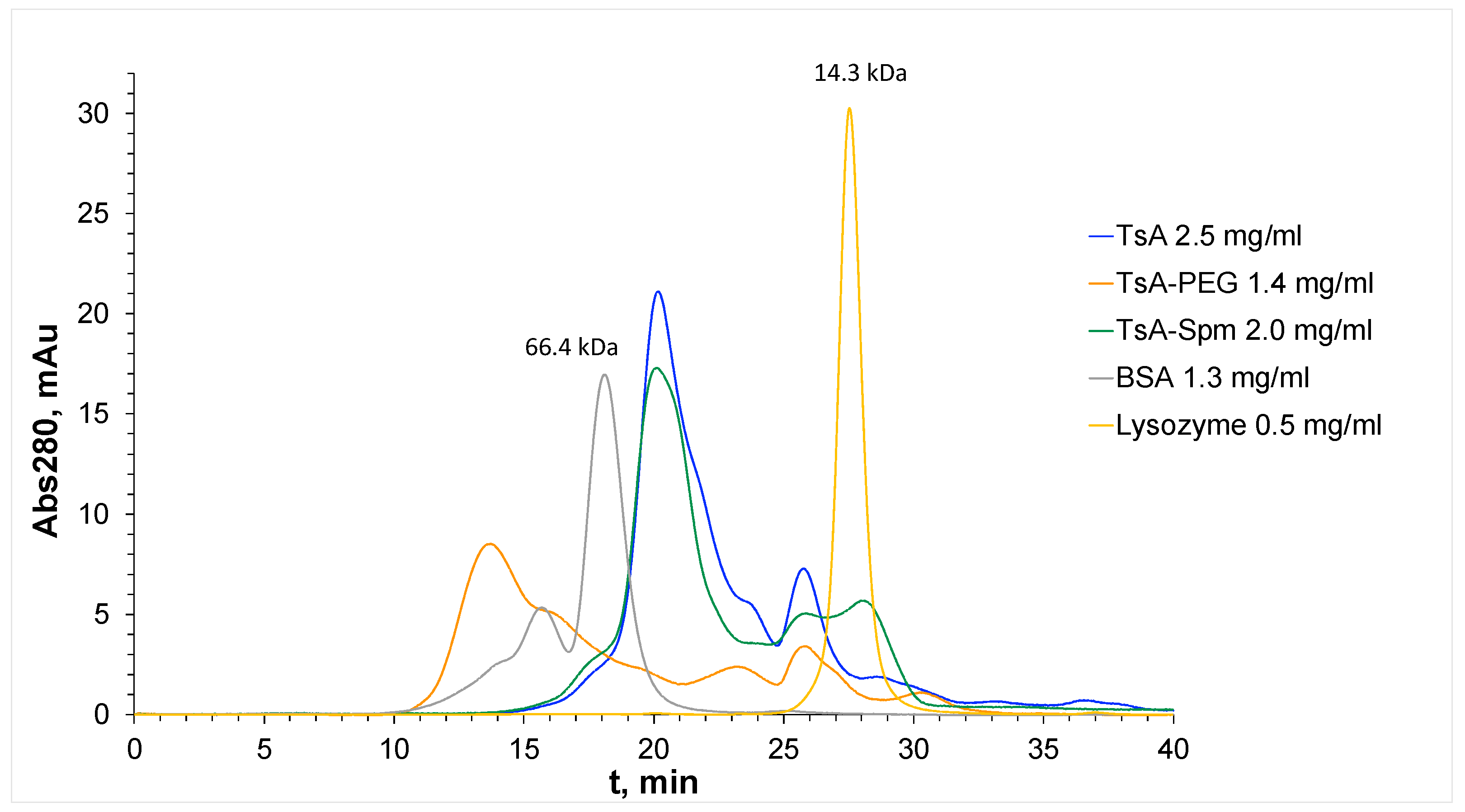
| Samples | tR, min | MM, kDa |
|---|---|---|
| Lysozyme | 27.6 | 14.3 |
| BSA | 18.2 | 66.4 |
| TsA | 20.2 | 37.5 |
| TsA-Spm | 19.5 | 40.4 |
| TsA-PEG | 13.9 | 90 |
| Secondary Structures | TsA | TsA-PEG | TsA-PEI | TsA-Spm |
|---|---|---|---|---|
| Helix | 33.4 ± 1.6 | 32.2 ± 1.8 | 33.6 ± 2.1 | 34.3 ± 0.5 |
| Antiparallel | 8.2 ± 0.7 | 8.3 ± 0.2 | 8.2 ± 0.8 | 8.0 ± 0.3 |
| Parallel | 8.8 ± 0.4 | 9.3 ± 0.8 | 8.8 ± 0.6 | 8.7 ± 0.2 |
| Beta-Turn | 16.8 ± 0.3 | 16.9 ± 0.2 | 16.7 ± 0.3 | 16.6 ± 0.1 |
| Rndm. Coil | 32.8 ± 0.7 | 34.2 ± 1.6 | 33.0 ± 1.3 | 32.6 ± 0.3 |
| KM, mM | Vmax, IU/mg | Vmax/KM | |
|---|---|---|---|
| TsA | 6.1 ± 1.3 | 470 ± 40 | 77 |
| TsA-PEG | 6.3 ± 0.8 | 460 ± 20 | 73 |
| TsA-PEI | 4.1 ± 0.7 | 520 ± 20 | 126 |
| TsA-Spm | 6.2 ± 1.6 | 710 ± 30 | 115 |
| kin, min−1 (0–20 min) | kin, min−1 (20–60 min) | |
|---|---|---|
| TsA | 0.052 ± 0.002 | 0.029 ± 0.002 |
| TsA-PEG | 0.053 ± 0.002 | 0.053 ± 0.002 |
| TsA-PEI | 0.042 ± 0.003 | 0.003 ± 0.001 |
| TsA-Spm | 0.036 ± 0.001 * | 0.019 ± 0.001 * |
| Secondary Structures | TsA | TsA-PEG | ||
|---|---|---|---|---|
| Before Heating | After * Heating | Before Heating | After Heating | |
| Helix | 33.4 ± 1.6 | 8.8 ± 1.2 | 32.2 ± 1.8 | 32.4 ± 1.6 |
| Antiparallel | 8.2 ± 0.7 | 15.2 ± 0.3 | 8.3 ± 0.2 | 8.6 ± 0.8 |
| Parallel | 8.8 ± 0.4 | 15.7 ± 0.5 | 9.3 ± 0.8 | 9.0 ± 0.3 |
| Beta-Turn | 16.8 ± 0.3 | 17.4 ± 0.4 | 16.9 ± 0.2 | 17.1 ± 0.4 |
| Rndm. Coil | 32.8 ± 0.7 | 43.2 ± 1.1 | 34.2 ± 1.5 | 33.1 ± 0.3 |
| Sample | T50, °C |
|---|---|
| TsA | 68 |
| TsA-PEG | 76 |
| TsA-Spm | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobryakova, N.V.; Dumina, M.V.; Zhgun, A.A.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Zhdanov, D.D.; Kudryashova, E.V. L-Asparaginase Conjugates from the Hyperthermophilic Archaea Thermococcus sibiricus with Improved Biocatalytic Properties. Int. J. Mol. Sci. 2024, 25, 4174. https://doi.org/10.3390/ijms25084174
Dobryakova NV, Dumina MV, Zhgun AA, Pokrovskaya MV, Aleksandrova SS, Zhdanov DD, Kudryashova EV. L-Asparaginase Conjugates from the Hyperthermophilic Archaea Thermococcus sibiricus with Improved Biocatalytic Properties. International Journal of Molecular Sciences. 2024; 25(8):4174. https://doi.org/10.3390/ijms25084174
Chicago/Turabian StyleDobryakova, Natalia V., Maria V. Dumina, Alexander A. Zhgun, Marina V. Pokrovskaya, Svetlana S. Aleksandrova, Dmitry D. Zhdanov, and Elena V. Kudryashova. 2024. "L-Asparaginase Conjugates from the Hyperthermophilic Archaea Thermococcus sibiricus with Improved Biocatalytic Properties" International Journal of Molecular Sciences 25, no. 8: 4174. https://doi.org/10.3390/ijms25084174
APA StyleDobryakova, N. V., Dumina, M. V., Zhgun, A. A., Pokrovskaya, M. V., Aleksandrova, S. S., Zhdanov, D. D., & Kudryashova, E. V. (2024). L-Asparaginase Conjugates from the Hyperthermophilic Archaea Thermococcus sibiricus with Improved Biocatalytic Properties. International Journal of Molecular Sciences, 25(8), 4174. https://doi.org/10.3390/ijms25084174