
The Logistical Backbone of Photoreceptor Cell Function: Complementary Mechanisms of Dietary Vitamin A Receptors and Rhodopsin Transporters

 , , , ,
, , , ,
Abstract
:1. Introduction
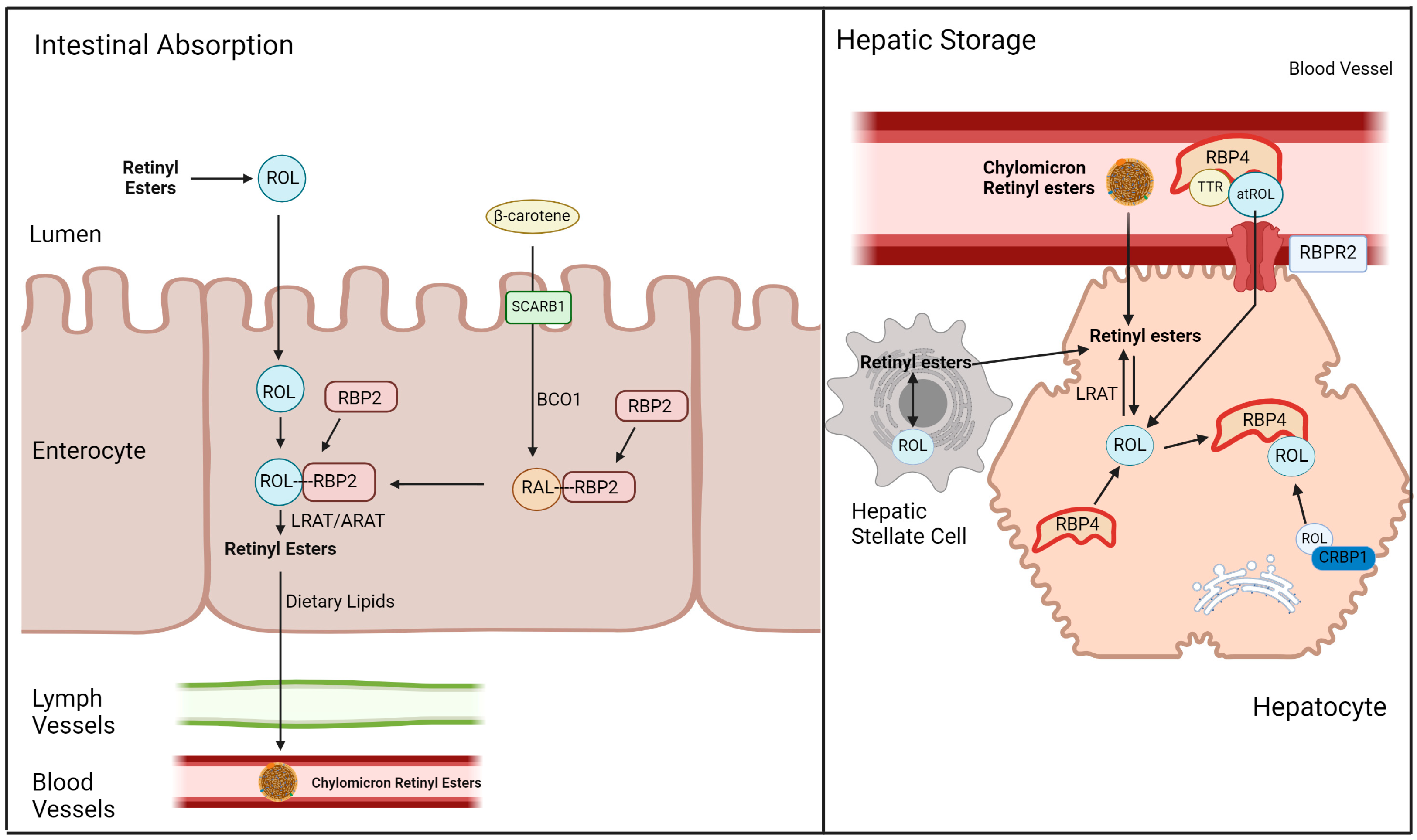
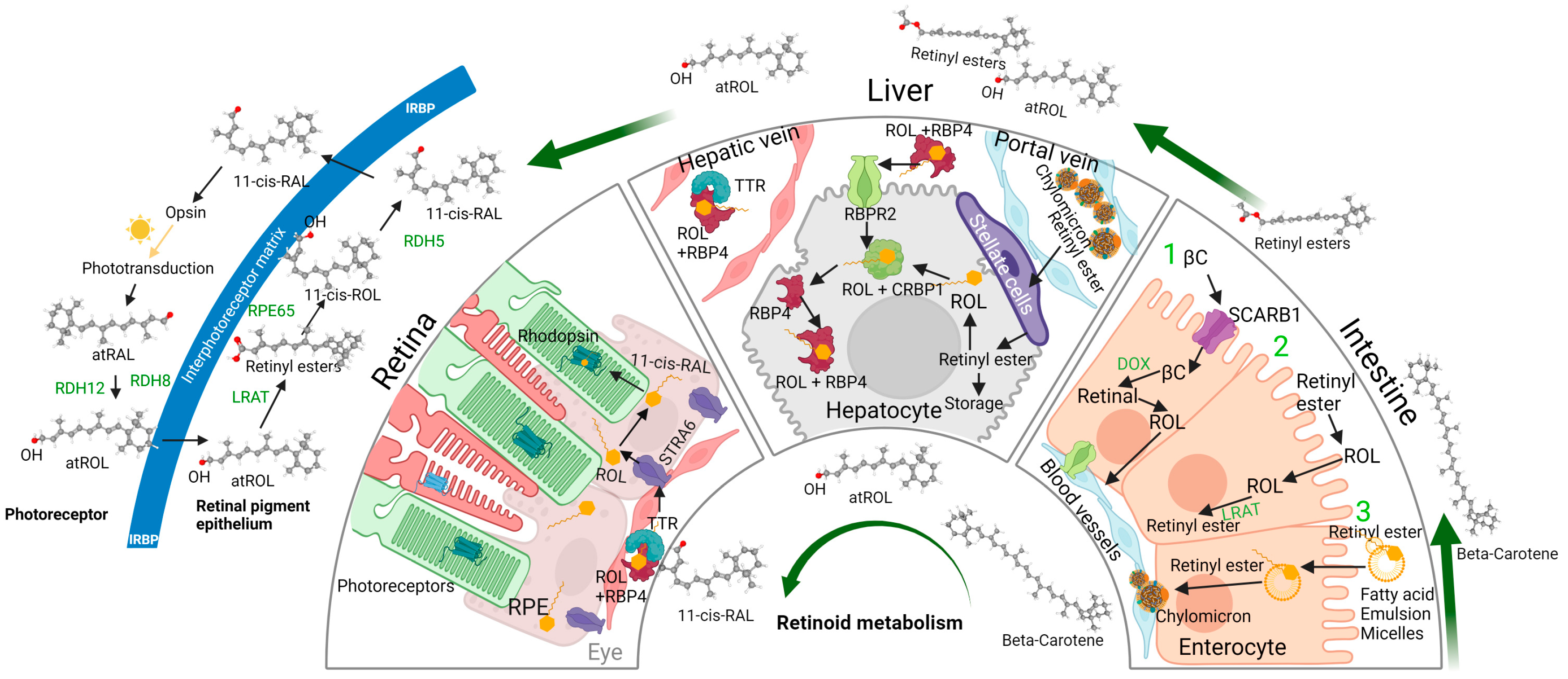
2. 11-cis Retinal, Vitamin A Metabolism, and Vitamin A Membrane Receptors
3. The Role of Vitamin A Membrane Receptors in Sustaining Visual Function
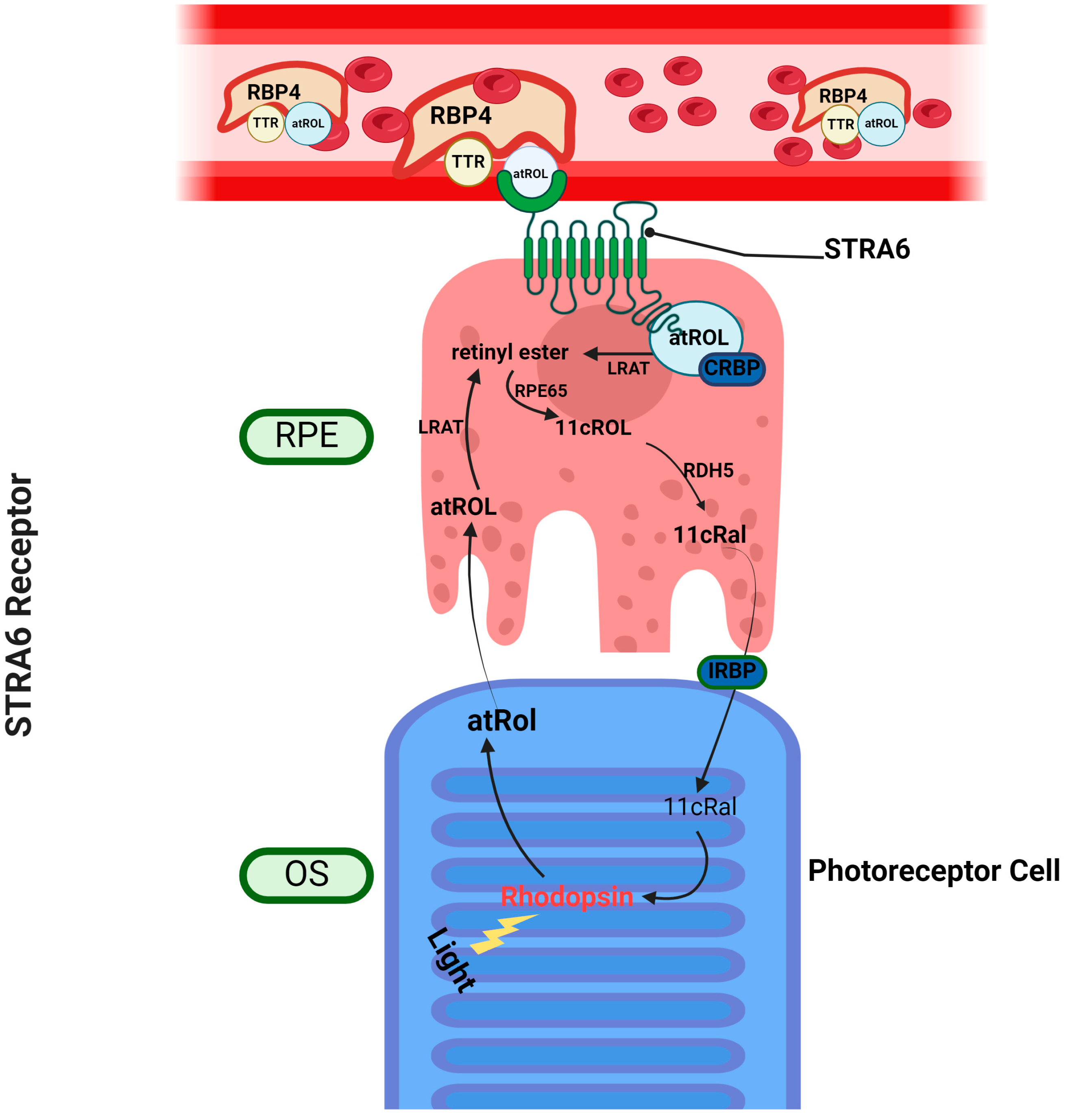
3.1. Stimulated by Retinoic Acid 6—STRA6
3.2. Retinol Binding Protein 4 Receptor 2—RBPR2
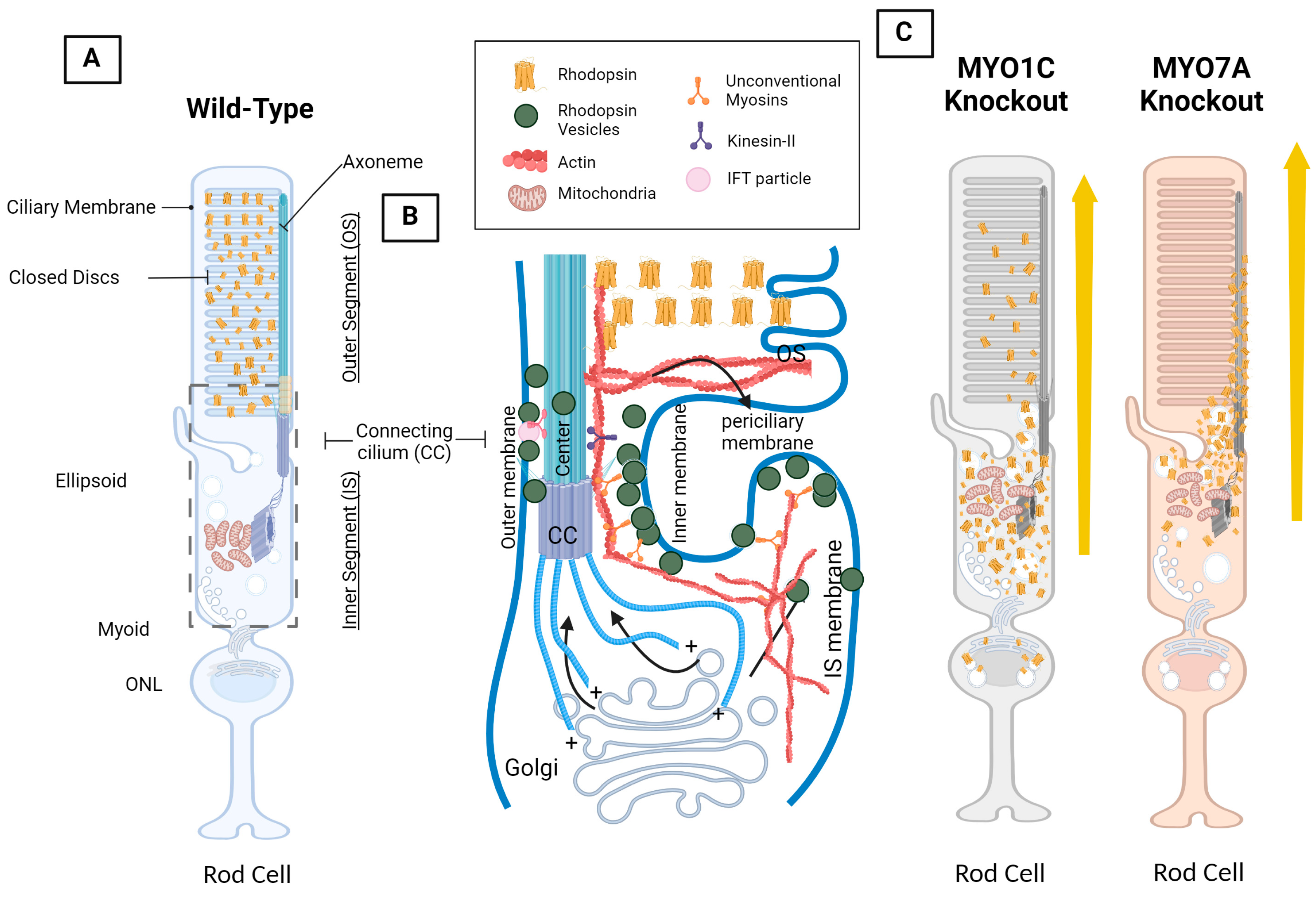
4. Opsin, Photoreceptor Morphology, Dynein and Kinesin Motor Proteins, and Unconventional Myosin Motor Proteins
4.1. The VxPx Domain and the Arf4 Complex: Post-Golgi Rhodopsin Transport through a Ciliary Targeting Complex
4.2. Cytoplasmic Dynein Motor Proteins and Tctex-1: Post-Golgi Rhodopsin Transport through Retrograde Microtubule Transport
4.3. Kinesin Motor Proteins and IFT: Through the Connecting Cilium to the Outer Segment
4.4. Myosin Motor Protein 7A: MYO7A
4.5. Unconventional Myosin Motor Protein 1C, MYO1C: A New Player in Understanding Rod Opsin Transport in Visual Function
5. The Visual Cycle and Phototransduction Cascade—Actions of Vitamin A Receptors and Myosin Motor Proteins Converge and Form the Retinylidene Protein
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Shichida, Y.; Matsuyama, T. Evolution of Opsins and Phototransduction. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Dronamraju, V.R.; Leung, M.; Gruesen, A.; Solanki, A.K.; Walterhouse, S.; Roehrich, H.; Song, G.; da Costa Monsanto, R.; Cureoglu, S.; et al. The Role of Motor Proteins in Photoreceptor Protein Transport and Visual Function. Ophthalmic Genet. 2022, 43, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Goletz, P.W.; Crouch, R.K. 11-Cis and All-Trans Retinols Can Activate Rod Opsin: Rational Design of the Visual Cycle. Biochemistry 2008, 47, 7567–7571. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E.; Wald, G. Vitamin A Deficiency and Night Blindness. Proc. Natl. Acad. Sci. USA 1958, 44, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E.; Wald, G. The Biological Function of Vitamin A Acid. Proc. Natl. Acad. Sci. USA 1960, 46, 587–608. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.R.; Cohen, G.B.; Zhukovsky, E.A.; Oprian, D.D. Constitutively Active Mutants of Rhodopsin. Neuron 1992, 9, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Amengual, J.; Palczewski, G.; Babino, D.; von Lintig, J. Carotenoid-Oxygenases: Key Players for Carotenoid Function and Homeostasis in Mammalian Biology. Biochim. Biophys. Acta 2012, 1821, 78–87. [Google Scholar] [CrossRef]
- Amengual, J.; Widjaja-Adhi, M.A.K.; Rodriguez-Santiago, S.; Hessel, S.; Golczak, M.; Palczewski, K.; von Lintig, J. Two Ca-rotenoid Oxygenases Contribute to Mammalian Provitamin A Metabolism. J. Biol. Chem. 2013, 288, 34081–34096. [Google Scholar] [CrossRef]
- Harrison, E.H. Mechanisms Involved in the Intestinal Absorption of Dietary Vitamin A and Provitamin A Carotenoids. Biochim. Biophys. Acta 2012, 1821, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H. Carotenoids, β-Apocarotenoids, and Retinoids: The Long and the Short of It. Nutrients 2022, 14, 1411. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wongsiriroj, N.; Blaner, W.S. The Multifaceted Nature of Retinoid Transport and Metabolism. Hepatobiliary Surg. Nutr. 2014, 3, 126–139. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and Retinyl Esters: Biochemistry and Physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Bok, D.; Heller, J. Transport of Retinol from the Blood to the Retina: An Autoradiographic Study of the Pigment Epithelial Cell Surface Receptor for Plasma Retinol-Binding Protein. Exp. Eye Res. 1976, 22, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Desmarchelier, C. Genetic Variations Associated with Vitamin A Status and Vitamin A Bioavailability. Nutrients 2017, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Heller, J. Uptake of Retinol and Retinoic Acid from Serum Retinol-Binding Protein by Retinal Pigment Epithelial Cells. J. Biol. Chem. 1977, 252, 5216–5221. [Google Scholar] [CrossRef] [PubMed]
- Gjøen, T.; Bjerkelund, T.; Blomhoff, H.K.; Norum, K.R.; Berg, T.; Blomhoff, R. Liver Takes up Retinol-Binding Protein from Plasma. J. Biol. Chem. 1987, 262, 10926–10930. [Google Scholar] [CrossRef]
- Blomhoff, R.; Norum, K.R.; Berg, T. Hepatic Uptake of [3H]Retinol Bound to the Serum Retinol Binding Protein Involves Both Parenchymal and Perisinusoidal Stellate Cells. J. Biol. Chem. 1985, 260, 13571–13575. [Google Scholar] [CrossRef]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-Binding Protein: The Transport Protein for Vitamin A in Human Plasma. J. Clin. Investig. 1968, 47, 2025–2044. [Google Scholar] [CrossRef]
- Kelly, M.; von Lintig, J. STRA6: Role in Cellular Retinol Uptake and Efflux. Hepatobiliary Surg. Nutr. 2015, 4, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Blaner, W.S.; Hamberger, L.; Novikoff, P.M.; Vogel, S.; Piantedosi, R.; Gottesman, M.E.; Colantuoni, V. The Role of Extrahepatic Retinol Binding Protein in the Mobilization of Retinoid Stores. J. Lipid Res. 2004, 45, 1975–1982. [Google Scholar] [CrossRef]
- Blaner, W.S. STRA6, a Cell-Surface Receptor for Retinol-Binding Protein: The Plot Thickens. Cell Metab. 2007, 5, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; Sapin, V.; Chazaud, C.; Messaddeq, N.; Décimo, D.; Dollé, P.; Chambon, P. Developmental Expression Pattern of Stra6, a Retinoic Acid-Responsive Gene Encoding a New Type of Membrane Protein. Mech. Dev. 1997, 63, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.; Kawaguchi, R.; Morrissey, M.; Sun, H.; McGettigan, P.; Nielsen, J.E.; Conroy, J.; Regan, R.; Kenny, E.; Cormican, P.; et al. First Implication of STRA6 Mutations in Isolated Anophthalmia, Microphthalmia, and Coloboma: A New Dimension to the STRA6 Phenotype. Hum. Mutat. 2011, 32, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood Syndrome Is Caused by Truncating Mutations in the Retinol-Binding Protein Receptor Gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Bok, D. A Specific Receptor for Retinol Binding Protein as Detected by the Binding of Human and Bovine Retinol Binding Protein to Pigment Epithelial Cells. Am. J. Ophthalmol. 1976, 81, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Ramkumar, S.; von Lintig, J. Genetic Tuning of β-Carotene Oxygenase-1 Activity Rescues Cone Photoreceptor Function in STRA6-Deficient Mice. Hum. Mol. Genet. 2023, 32, 798–809. [Google Scholar] [CrossRef]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirm-er-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 Cause a Broad Spectrum of Malformations Including Anophthalmia, Congenital Heart Defects, Diaphragmatic Hernia, Alveolar Capillary Dysplasia, Lung Hypoplasia, and Mental Retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef]
- Alapatt, P.; Guo, F.; Komanetsky, S.M.; Wang, S.; Cai, J.; Sargsyan, A.; Rodríguez Díaz, E.; Bacon, B.T.; Aryal, P.; Graham, T.E. Liver Retinol Transporter and Receptor for Serum Retinol-Binding Protein (RBP4). J. Biol. Chem. 2013, 288, 1250–1265. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Leung, M.; Roehrich, H.; Walterhouse, S.; Kondkar, A.A.; Fitzgibbon, W.; Biswal, M.R.; Lobo, G.P. Mice Lacking the Systemic Vitamin A Receptor RBPR2 Show Decreased Ocular Retinoids and Loss of Visual Function. Nutrients 2022, 14, 2371. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Martin Ask, N.; Leung, M.; Radhakrishnan, R.; Lobo, G.P. Vitamin A Transporters in Visual Function: A Mini Review on Membrane Receptors for Dietary Vitamin A Uptake, Storage, and Transport to the Eye. Nutrients 2021, 13, 3987. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yoshizawa, T.; Kamio, S.; Aoki, O.; Kawamata, Y.; Masushige, S.; Kato, S. Interactions of Transthyretin (TTR) and Retinol-Binding Protein (RBP) in the Uptake of Retinol by Primary Rat Hepatocytes. Exp. Cell Res. 1997, 234, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Leung, M.; Solanki, A.K.; Lobo, G.P. Mapping of the Extracellular RBP4 Ligand Binding Domain on the RBPR2 Receptor for Vitamin A Transport. Front. Cell Dev. Biol. 2023, 11, 1105657. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.K.; Kondkar, A.A.; Fogerty, J.; Su, Y.; Kim, S.-H.; Lipschutz, J.H.; Nihalani, D.; Perkins, B.D.; Lobo, G.P. A Functional Binding Domain in the Rbpr2 Receptor Is Required for Vitamin A Transport, Ocular Retinoid Homeostasis, and Pho-toreceptor Cell Survival in Zebrafish. Cells 2020, 9, 1099. [Google Scholar] [CrossRef]
- Shi, Y.; Obert, E.; Rahman, B.; Rohrer, B.; Lobo, G.P. The Retinol Binding Protein Receptor 2 (Rbpr2) Is Required for Photo-receptor Outer Segment Morphogenesis and Visual Function in Zebrafish. Sci. Rep. 2017, 7, 16207. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Zhang, N.; Kemerer, M.; Maeda, T.; Palczewski, K.; Von Lintig, J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum. Mol. Genet. 2014, 23, 5402–5417. [Google Scholar] [CrossRef] [PubMed]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef]
- Lobo, G.P.; Pauer, G.; Lipschutz, J.H.; Hagstrom, S.A. The Retinol-Binding Protein Receptor 2 (Rbpr2) Is Required for Pho-toreceptor Survival and Visual Function in the Zebrafish. In Proceedings of the Retinal Degenerative Diseases; Ash, J.D., Anderson, R.E., LaVail, M.M., Bowes Rickman, C., Hollyfield, J.G., Grimm, C., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 569–576. [Google Scholar]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a Glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef]
- Xiong, L.; Zhang, L.; Yang, Y.; Li, N.; Lai, W.; Wang, F.; Zhu, X.; Wang, T. ER Complex Proteins Are Required for Rhodopsin Biosynthesis and Photoreceptor Survival in Drosophila and Mice. Cell Death Differ. 2020, 27, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, D.; Feiner, L.; Lillo, C.; Teofilo, K.; Goldstein, L.S.B.; Pierce, E.A.; Williams, D.S. Analysis of Kinesin-2 Function in Photoreceptor Cells Using Synchronous Cre-loxP Knockout of Kif3a with RHO-Cre. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5039–5046. [Google Scholar] [CrossRef]
- Marszalek, J.R.; Liu, X.; Roberts, E.A.; Chui, D.; Marth, J.D.; Williams, D.S.; Goldstein, L.S.B. Genetic Evidence for Selective Transport of Opsin and Arrestin by Kinesin-II in Mammalian Photoreceptors. Cell 2000, 102, 175–187. [Google Scholar] [CrossRef]
- Chadha, A.; Volland, S.; Baliaouri, N.V.; Tran, E.M.; Williams, D.S. The Route of the Visual Receptor Rhodopsin along the Cilium. J. Cell Sci. 2019, 132, jcs229526. [Google Scholar] [CrossRef]
- Sung, C.H.; Makino, C.; Baylor, D.; Nathans, J. A Rhodopsin Gene Mutation Responsible for Autosomal Dominant Retinitis Pigmentosa Results in a Protein That Is Defective in Localization to the Photoreceptor Outer Segment. J. Neurosci. 1994, 14, 5818–5833. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, F.; Chen, J. Q344ter Mutation Causes Mislocalization of Rhodopsin Molecules That Are Catalytically Active: A Mouse Model of Q344ter-Induced Retinal Degeneration. PLoS ONE 2010, 5, e10904. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, F.; Mendez, A.; Chen, J. The Carboxyl-Terminal Domain Is Essential for Rhodopsin Transport in Rod Photore-ceptors. Vision Res. 2002, 42, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Deretic, D.; Schmerl, S.; Hargrave, P.A.; Arendt, A.; McDowell, J.H. Regulation of Sorting and Post-Golgi Trafficking of Rhodopsin by Its C-Terminal Sequence QVS(A)PA. Proc. Natl. Acad. Sci. USA 1998, 95, 10620–10625. [Google Scholar] [CrossRef]
- Deretic, D.; Williams, A.H.; Ransom, N.; Morel, V.; Hargrave, P.A.; Arendt, A. Rhodopsin C Terminus, the Site of Mutations Causing Retinal Disease, Regulates Trafficking by Binding to ADP-Ribosylation Factor 4 (ARF4). Proc. Natl. Acad. Sci. USA 2005, 102, 3301–3306. [Google Scholar] [CrossRef]
- Wang, J.; Morita, Y.; Mazelova, J.; Deretic, D. The Arf GAP ASAP1 Provides a Platform to Regulate Arf4- and Rab11–Rab8-mediated Ciliary Receptor Targeting. EMBO J. 2012, 31, 4057–4071. [Google Scholar] [CrossRef]
- Horgan, C.P.; Hanscom, S.R.; Jolly, R.S.; Futter, C.E.; McCaffrey, M.W. Rab11-FIP3 Links the Rab11 GTPase and Cytoplasmic Dynein to Mediate Transport to the Endosomal-Recycling Compartment. J. Cell Sci. 2010, 123, 181–191. [Google Scholar] [CrossRef]
- Horgan, C.P.; Hanscom, S.R.; Jolly, R.S.; Futter, C.E.; McCaffrey, M.W. Rab11-FIP3 Binds Dynein Light Intermediate Chain 2 and Its Overexpression Fragments the Golgi Complex. Biochem. Biophys. Res. Commun. 2010, 394, 387–392. [Google Scholar] [CrossRef]
- Kong, S.; Du, X.; Peng, C.; Wu, Y.; Li, H.; Jin, X.; Hou, L.; Deng, K.; Xu, T.; Tao, W. Dlic1 Deficiency Impairs Ciliogenesis of Photoreceptors by Destabilizing Dynein. Cell Res. 2013, 23, 835–850. [Google Scholar] [CrossRef]
- Tai, A.W.; Chuang, J.-Z.; Bode, C.; Wolfrum, U.; Sung, C.-H. Rhodopsin’s Carboxy-Terminal Cytoplasmic Tail Acts as a Membrane Receptor for Cytoplasmic Dynein by Binding to the Dynein Light Chain Tctex-1. Cell 1999, 97, 877–887. [Google Scholar] [CrossRef]
- Pearring, J.N.; San Agustin, J.T.; Lobanova, E.S.; Gabriel, C.J.; Lieu, E.C.; Monis, W.J.; Stuck, M.W.; Strittmatter, L.; Jaber, S.M.; Arshavsky, V.Y.; et al. Loss of Arf4 Causes Severe Degeneration of the Exocrine Pancreas but Not Cystic Kidney Disease or Retinal Degeneration. PLoS Genet. 2017, 13, e1006740. [Google Scholar] [CrossRef]
- Ying, G.; Gerstner, C.D.; Frederick, J.M.; Boye, S.L.; Hauswirth, W.W.; Baehr, W. Small GTPases Rab8a and Rab11a Are Dispensable for Rhodopsin Transport in Mouse Photoreceptors. PLoS ONE 2016, 11, e0161236. [Google Scholar] [CrossRef]
- Wang, J.; Fresquez, T.; Kandachar, V.; Deretic, D. The Arf GEF GBF1 and Arf4 Synergize with the Sensory Receptor Cargo, Rhodopsin, to Regulate Ciliary Membrane Trafficking. J. Cell Sci. 2017, 130, 3975–3987. [Google Scholar] [CrossRef]
- Deretic, D.; Lorentzen, E.; Fresquez, T. The Ins and Outs of the Arf4-Based Ciliary Membrane-Targeting Complex. Small GTPases 2019, 12, 1616355. [Google Scholar] [CrossRef]
- Baehr, W.; Hanke-Gogokhia, C.; Sharif, A.; Reed, M.; Dahl, T.; Frederick, J.M.; Ying, G. Insights into Photoreceptor Ciliogenesis Revealed by Animal Models. Prog. Retin. Eye Res. 2019, 71, 26–56. [Google Scholar] [CrossRef]
- Troutt, L.L.; Burnside, B. Microtubule Polarity and Distribution in Teleost Photoreceptors. J. Neurosci. 1988, 8, 2371–2380. [Google Scholar] [CrossRef]
- Krock, B.L.; Mills-Henry, I.; Perkins, B.D. Retrograde Intraflagellar Transport by Cytoplasmic Dynein-2 Is Required for Outer Segment Extension in Vertebrate Photoreceptors but Not Arrestin Translocation. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5463–5471. [Google Scholar] [CrossRef]
- Insinna, C.; Baye, L.M.; Amsterdam, A.; Besharse, J.C.; Link, B.A. Analysis of a Zebrafish Dync1h1 Mutant Reveals Multiple Functions for Cytoplasmic Dynein 1 during Retinal Photoreceptor Development. Neural Dev. 2010, 5, 12. [Google Scholar] [CrossRef]
- Dahl, T.M.; Reed, M.; Gerstner, C.D.; Ying, G.; Baehr, W. Effect of Conditional Deletion of Cytoplasmic Dynein Heavy Chain DYNC1H1 on Postnatal Photoreceptors. PLoS ONE 2021, 16, e0248354. [Google Scholar] [CrossRef]
- Ringo, D.L. Flagellar Motion and Fine Structure of the Flagellar Apparatus in Chlamydomonas. J. Cell Biol. 1967, 33, 543–571. [Google Scholar] [CrossRef]
- Richardson, T.M. Cytoplasmic and Ciliary Connections between the Inner and Outer Segments of Mammalian Visual Receptors. Vision Res. 1969, 9, 727–731. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A.; et al. Molecular Basis of Tubulin Transport Within the Cilium by IFT74 and IFT81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef]
- Hao, L.; Thein, M.; Brust-Mascher, I.; Civelekoglu-Scholey, G.; Lu, Y.; Acar, S.; Prevo, B.; Shaham, S.; Scholey, J.M. Intrafla-gellar Transport Delivers Tubulin Isotypes to Sensory Cilium Middle and Distal Segments. Nat. Cell Biol. 2011, 13, 790–798. [Google Scholar] [CrossRef]
- Marshall, W.F.; Rosenbaum, J.L. Intraflagellar Transport Balances Continuous Turnover of Outer Doublet Microtubules. J. Cell Biol. 2001, 155, 405–414. [Google Scholar] [CrossRef]
- Craft, J.M.; Harris, J.A.; Hyman, S.; Kner, P.; Lechtreck, K.F. Tubulin Transport by IFT Is Upregulated during Ciliary Growth by a Cilium-Autonomous Mechanism. J. Cell Biol. 2015, 208, 223–237. [Google Scholar] [CrossRef]
- Kocaoglu, O.P.; Liu, Z.; Zhang, F.; Kurokawa, K.; Jonnal, R.S.; Miller, D.T. Photoreceptor Disc Shedding in the Living Human Eye. Biomed. Opt. Express 2016, 7, 4554–4568. [Google Scholar] [CrossRef]
- Guillaud, L.; Wong, R.; Hirokawa, N. Disruption of KIF17–Mint1 Interaction by CaMKII-Dependent Phosphorylation: A Molecular Model of Kinesin–Cargo Release. Nat. Cell Biol. 2008, 10, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Pang, Y.; Wu, Q.; Hu, Z.; Han, X.; Xu, Y.; Deng, H.; Pan, J. FLA8/KIF3B Phosphorylation Regulates Kinesin-II Interaction with IFT-B to Control IFT Entry and Turnaround. Dev. Cell 2014, 30, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Scholey, J.M. Kinesin-2: A Family of Heterotrimeric and Homodimeric Motors with Diverse Intracellular Transport Functions. Annu. Rev. Cell Dev. Biol. 2013, 29, 443–469. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, T.; Katoh, Y.; Okazaki, M.; Sugawa, M.; Nakayama, K. Interaction of Heterotrimeric Kinesin-II with IFT-B–Connecting Tetramer Is Crucial for Ciliogenesis. J. Cell Biol. 2018, 217, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Rosenbaum, J.L. Chapter Two Intraflagellar Transport (IFT): Role in Ciliary Assembly, Resorption and Signaling. Curr. Top. Dev. Biol. Ciliary Funct. Mamm. Dev. 2008, 85, 23–61. [Google Scholar]
- Pedersen, L.B.; Geimer, S.; Rosenbaum, J.L. Dissecting the Molecular Mechanisms of Intraflagellar Transport in Chlamydo-monas. Curr. Biol. 2006, 16, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Grissom, P.M.; Vaisberg, E.A.; McIntosh, J.R. Identification of a Novel Light Intermediate Chain (D2LIC) for Mammalian Cytoplasmic Dynein 2. Mol. Biol. Cell 2002, 13, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Perrone, C.A.; Tritschler, D.; Taulman, P.; Bower, R.; Yoder, B.K.; Porter, M.E. A Novel Dynein Light Intermediate Chain Colocalizes with the Retrograde Motor for Intraflagellar Transport at Sites of Axoneme Assembly in Chlamydomonas and Mammalian Cells. Mol. Biol. Cell 2003, 14, 2041–2056. [Google Scholar] [CrossRef]
- Schafer, J.C.; Haycraft, C.J.; Thomas, J.H.; Yoder, B.K.; Swoboda, P. XBX-1 Encodes a Dynein Light Intermediate Chain Re-quired for Retrograde Intraflagellar Transport and Cilia Assembly in Caenorhabditis Elegans. Mol. Biol. Cell 2003, 14, 2057–2070. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar Transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef]
- Jiang, L.; Wei, Y.; Ronquillo, C.C.; Marc, R.E.; Yoder, B.K.; Frederick, J.M.; Baehr, W. Heterotrimeric Kinesin-2 (KIF3) Mediates Transition Zone and Axoneme Formation of Mouse Photoreceptors. J. Biol. Chem. 2015, 290, 12765–12778. [Google Scholar] [CrossRef]
- Jiang, L.; Tam, B.M.; Ying, G.; Wu, S.; Hauswirth, W.W.; Frederick, J.M.; Moritz, O.L.; Baehr, W. Kinesin Family 17 (Osmotic Avoidance Abnormal-3) Is Dispensable for Photoreceptor Morphology and Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4866–4880. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Arriaga, M.A.; Trzupek, K.M.; Lentz, J.J. Usher Syndrome Type I. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Watanabe, S.; Umeki, N.; Ikebe, R.; Ikebe, M. Impacts of Usher Syndrome Type IB Mutations on Human Myosin VIIa Motor Function. Biochemistry 2008, 47, 9505. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Vansant, G.; Udovichenko, I.P.; Wolfrum, U.; Williams, D.S. Myosin VIIa, the Product of the Usher 1B Syndrome Gene, Is Concentrated in the Connecting Cilia of Photoreceptor Cells. Cell Motil. 1997, 37, 240–252. [Google Scholar] [CrossRef]
- Hasson, T.; Heintzelman, M.B.; Santos-Sacchi, J.; Corey, D.P.; Mooseker, M.S. Expression in Cochlea and Retina of Myosin VIIa, the Gene Product Defective in Usher Syndrome Type 1B. Proc. Natl. Acad. Sci. USA 1995, 92, 9815–9819. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Udovichenko, I.P.; Brown, S.D.M.; Steel, K.P.; Williams, D.S. Myosin VIIa Participates in Opsin Transport through The Photoreceptor Cilium. J. Neurosci. 1999, 19, 6267–6274. [Google Scholar] [CrossRef] [PubMed]
- Lopes, V.S.; Gibbs, D.; Libby, R.T.; Aleman, T.S.; Welch, D.L.; Lillo, C.; Jacobson, S.G.; Radu, R.A.; Steel, K.P.; Williams, D.S. The Usher 1B Protein, MYO7A, Is Required for Normal Localization and Function of the Visual Retinoid Cycle Enzyme, RPE65. Hum. Mol. Genet. 2011, 20, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.; Azarian, S.M.; Lillo, C.; Kitamoto, J.; Klomp, A.E.; Steel, K.P.; Libby, R.T.; Williams, D.S. Role of Myosin VIIa and Rab27a in the Motility and Localization of RPE Melanosomes. J. Cell Sci. 2004, 117, 6473–6483. [Google Scholar] [CrossRef]
- Różanowski, B.; Burke, J.M.; Boulton, M.E.; Sarna, T.; Różanowska, M. Human RPE Melanosomes Protect from Photosensi-tized and Iron-Mediated Oxidation but Become Pro-Oxidant in the Presence of Iron upon Photodegradation. Investig. Oph-thalmol. Vis. Sci. 2008, 49, 2838–2847. [Google Scholar] [CrossRef]
- Solanki, A.K.; Biswal, M.R.; Walterhouse, S.; Martin, R.; Kondkar, A.A.; Knölker, H.-J.; Rahman, B.; Arif, E.; Husain, S.; Montezuma, S.R.; et al. Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function. Cells 2021, 10, 1322. [Google Scholar] [CrossRef]
- Hollingsworth, T.J.; Gross, A.K. Chapter One—Defective Trafficking of Rhodopsin and Its Role in Retinal Degenerations. Int. Rev. Cell Mol. Biol. 2012, 293, 1–44. [Google Scholar]
- Mazelova, J.; Astuto-Gribble, L.; Inoue, H.; Tam, B.M.; Schonteich, E.; Prekeris, R.; Moritz, O.L.; Randazzo, P.A.; Deretic, D. Ciliary Targeting Motif VxPx Directs Assembly of a Trafficking Module through Arf4. EMBO J. 2009, 28, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key Enzymes of the Retinoid (Visual) Cycle in Vertebrate Retina. Biochim. Biophys. Acta 2012, 1821, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Ripps, H.; Peachey, N.S.; Xu, X.; Nozell, S.E.; Smith, S.B.; Liou, G.I. The Rhodopsin Cycle Is Preserved in IRBP “Knockout” Mice despite Abnormalities in Retinal Structure and Function. Vis. Neurosci. 2000, 17, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Shichida, Y.; Morizumi, T. Mechanism of G-Protein Activation by Rhodopsin†. Photochem. Photobiol. 2007, 83, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Vinberg, F.; Wang, T.; De Maria, A.; Zhao, H.; Bassnett, S.; Chen, J.; Kefalov, V.J. The Na+/Ca2+, K+ Exchanger NCKX4 Is Required for Efficient Cone-Mediated Vision. eLife 2017, 6, e24550. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Vinberg, F.; Wang, T.; Chen, J.; Kefalov, V.J. The Na+/Ca2+, K+ Exchanger 2 Modulates Mammalian Cone Phototransduction. Sci. Rep. 2016, 6, 32521. [Google Scholar] [CrossRef] [PubMed]
- Reiländer, H.; Achilles, A.; Friedel, U.; Maul, G.; Lottspeich, F.; Cook, N.J. Primary Structure and Functional Expression of the Na/Ca,K-Exchanger from Bovine Rod Photoreceptors. EMBO J. 1992, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K. The Vertebrate Phototransduction Cascade: Amplification and Termination Mechanisms. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 978-3-540-32431-7. [Google Scholar]
- Donato, L.; Alibrandi, S.; Scimone, C.; Rinaldi, C.; Dascola, A.; Calamuneri, A.; D’Angelo, R.; Sidoti, A. The impact of modifier genes on cone-rod dystrophy heterogeneity: An explorative familial pilot study and a hypothesis on neurotransmission impairment. PLoS ONE 2022, 17, e0278857. [Google Scholar] [CrossRef]
- Chen, Y.; Clarke, O.B.; Kim, J.; Stowe, S.; Kim, Y.-K.; Assur, Z.; Cavalier, M.; Godoy-Ruiz, R.; von Alpen, D.C.; Manzini, C.; et al. Structure of the STRA6 Receptor for Retinol Uptake. Science 2016, 353, aad8266. [Google Scholar] [CrossRef]
- Costabile, B.K.; Kim, Y.-K.; Chen, Y.; Clarke, O.B.; Quadro, L.; Mancia, F. Sample Preparation for Structural and Functional Analyses of the STRA6 Receptor for Retinol-Binding Protein. Methods Enzymol. 2020, 637, 95–117. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Expression | Retinol Binding Protein Receptor 2 (RBPR2) | Stimulated by Retinoic Acid 6 (STRA6) |
|---|---|---|
| RPE/Eye | ||
| Brain | ||
| Liver | ||
| Intestine | ||
| Spleen | ||
| Kidney | ||
| Adipose | ||
| Lung | ||
| Zebrafish Expression | Retinol Binding protein Receptor 2 (RBPR2) | Stimulated by Retinoic Acid 6 (STRA6) |
| RPE | ||
| Brain | ||
| Liver | ||
| Intestine | ||
| Spleen | ||
| Kidney | ||
| Adipose | ||
| Pancreas |
| Protein | Type | Expression | Proposed Function |
|---|---|---|---|
| Myosin 1C | Unconventional motor protein | Widely distributed; Cytoplasmic as well as nuclear isoforms. Mouse Photoreceptors Upper tip link in the mouse ear inner hair cells | Transcription initiation; Intracellular vesicle transport to plasma membrane (in mice); may form adaptation motor complex; role in inner ear function; Opsin transport to Photoreceptor Outer Segments and Visual Function; Mouse Vitreous; Auditory function, hair cell adaption and Actin binding; Missense mutations reported in patients with sensorineural hearing loss |
| Myosin 7A | Unconventional motor protein | Retinal Pigmented Epithelium and Photoreceptor cilium | Rhodopsin transport; RPE65 localization in RPE |
| Kinesin | Motor | Photoreceptors | Microtubule-based anterograde intracellular transport |
| Kif3a | Motor | Photoreceptors | Rhodopsin transport |
| Dynein | Motor | Photoreceptors | Microtubule-based retrograde intracellular transport |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, M.; Steinman, J.; Li, D.; Lor, A.; Gruesen, A.; Sadah, A.; van Kuijk, F.J.; Montezuma, S.R.; Kondkar, A.A.; Radhakrishnan, R.; et al. The Logistical Backbone of Photoreceptor Cell Function: Complementary Mechanisms of Dietary Vitamin A Receptors and Rhodopsin Transporters. Int. J. Mol. Sci. 2024, 25, 4278. https://doi.org/10.3390/ijms25084278
Leung M, Steinman J, Li D, Lor A, Gruesen A, Sadah A, van Kuijk FJ, Montezuma SR, Kondkar AA, Radhakrishnan R, et al. The Logistical Backbone of Photoreceptor Cell Function: Complementary Mechanisms of Dietary Vitamin A Receptors and Rhodopsin Transporters. International Journal of Molecular Sciences. 2024; 25(8):4278. https://doi.org/10.3390/ijms25084278
Chicago/Turabian StyleLeung, Matthias, Jeremy Steinman, Dorothy Li, Anjelynt Lor, Andrew Gruesen, Ahmed Sadah, Frederik J. van Kuijk, Sandra R. Montezuma, Altaf A. Kondkar, Rakesh Radhakrishnan, and et al. 2024. "The Logistical Backbone of Photoreceptor Cell Function: Complementary Mechanisms of Dietary Vitamin A Receptors and Rhodopsin Transporters" International Journal of Molecular Sciences 25, no. 8: 4278. https://doi.org/10.3390/ijms25084278