
Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape


Abstract
:1. Introduction
2. Results and Discussion
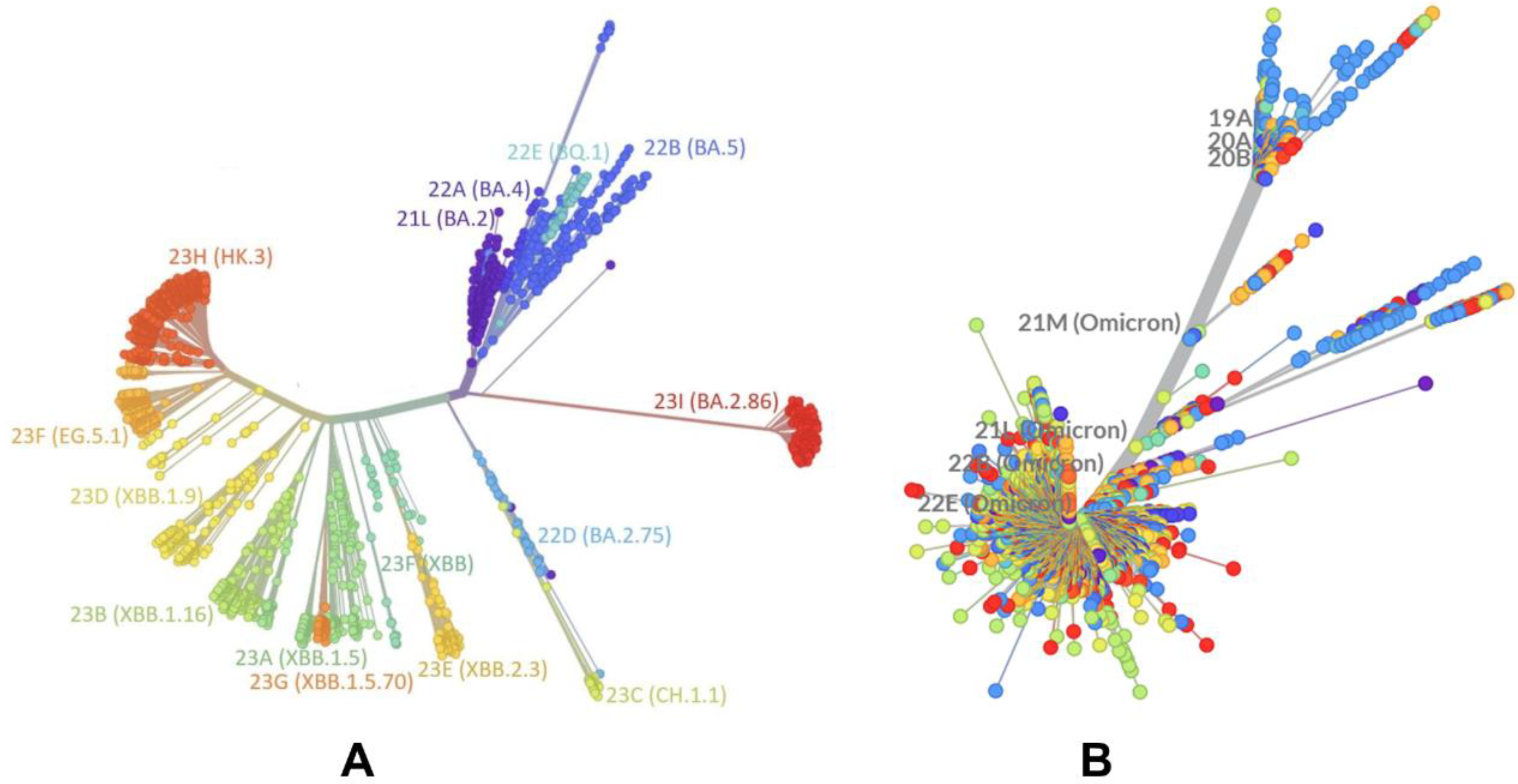
2.1. Evolutionary and Phylogenetic Analysis of Differences between BA.2 and XBB.1.5 Lineages
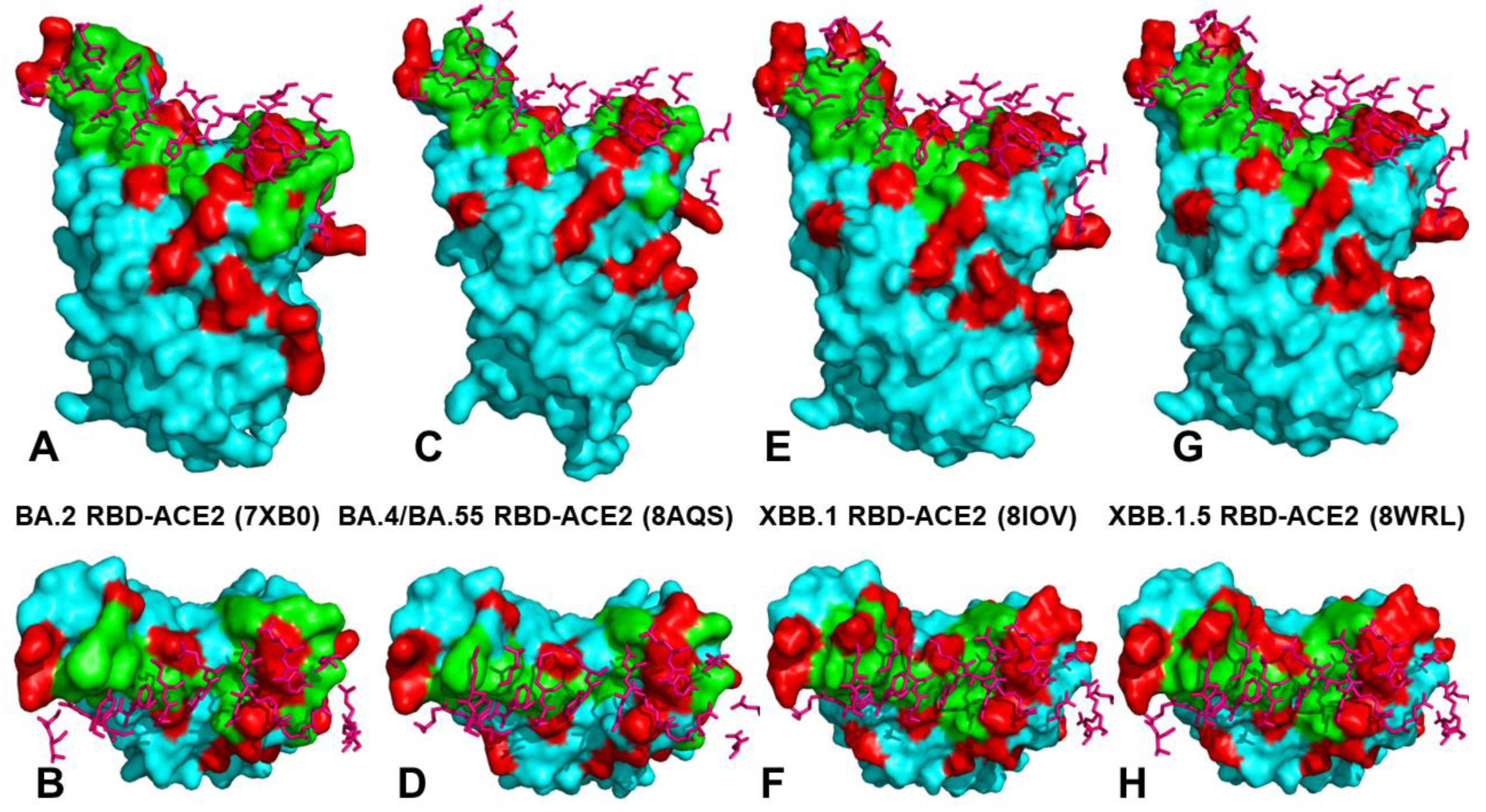
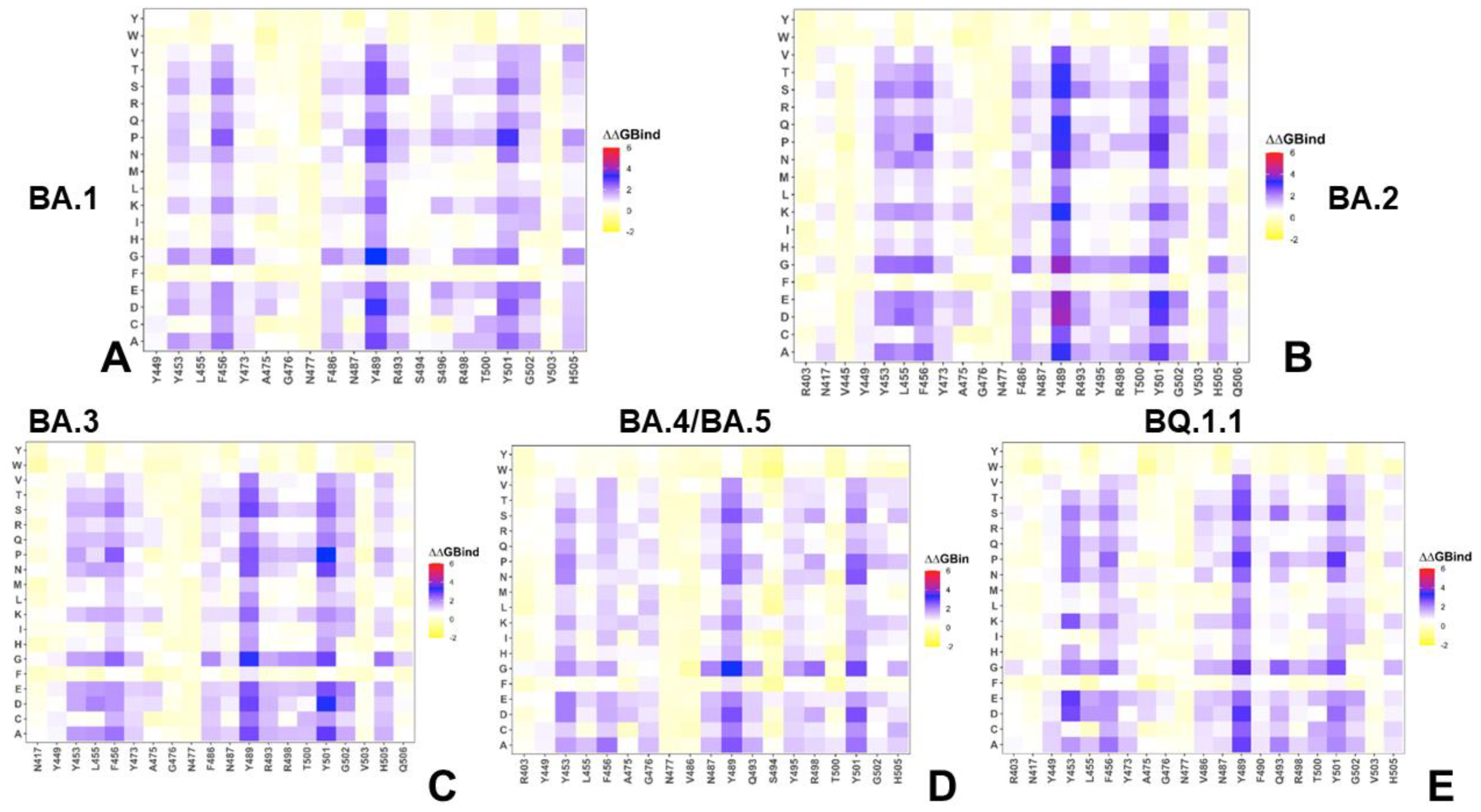
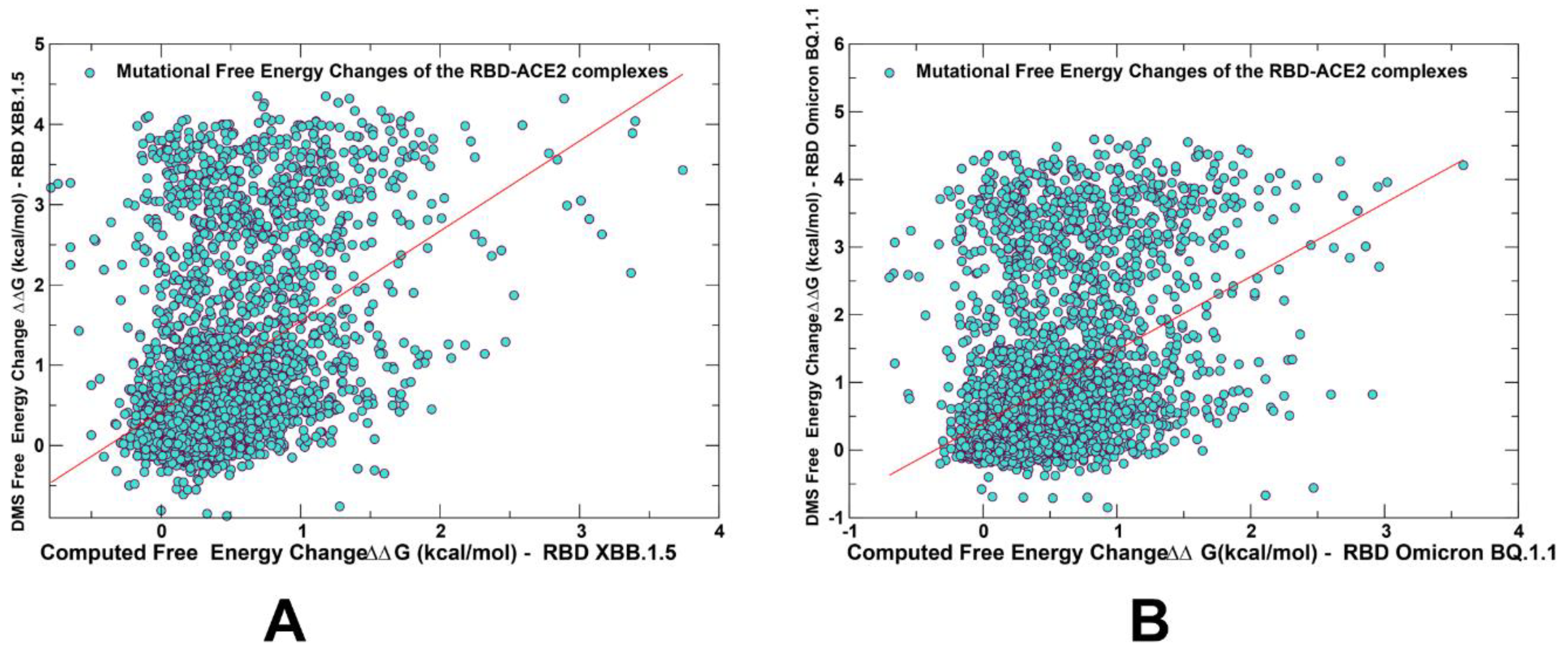
2.2. Mutational Profiling Analysis of Omicron Variants Identifies Key Identifies Universal and Variant-Specific Structural Stability and Binding Affinity Hotspots in the SARS-CoV-2 RBD Complexes with ACE2
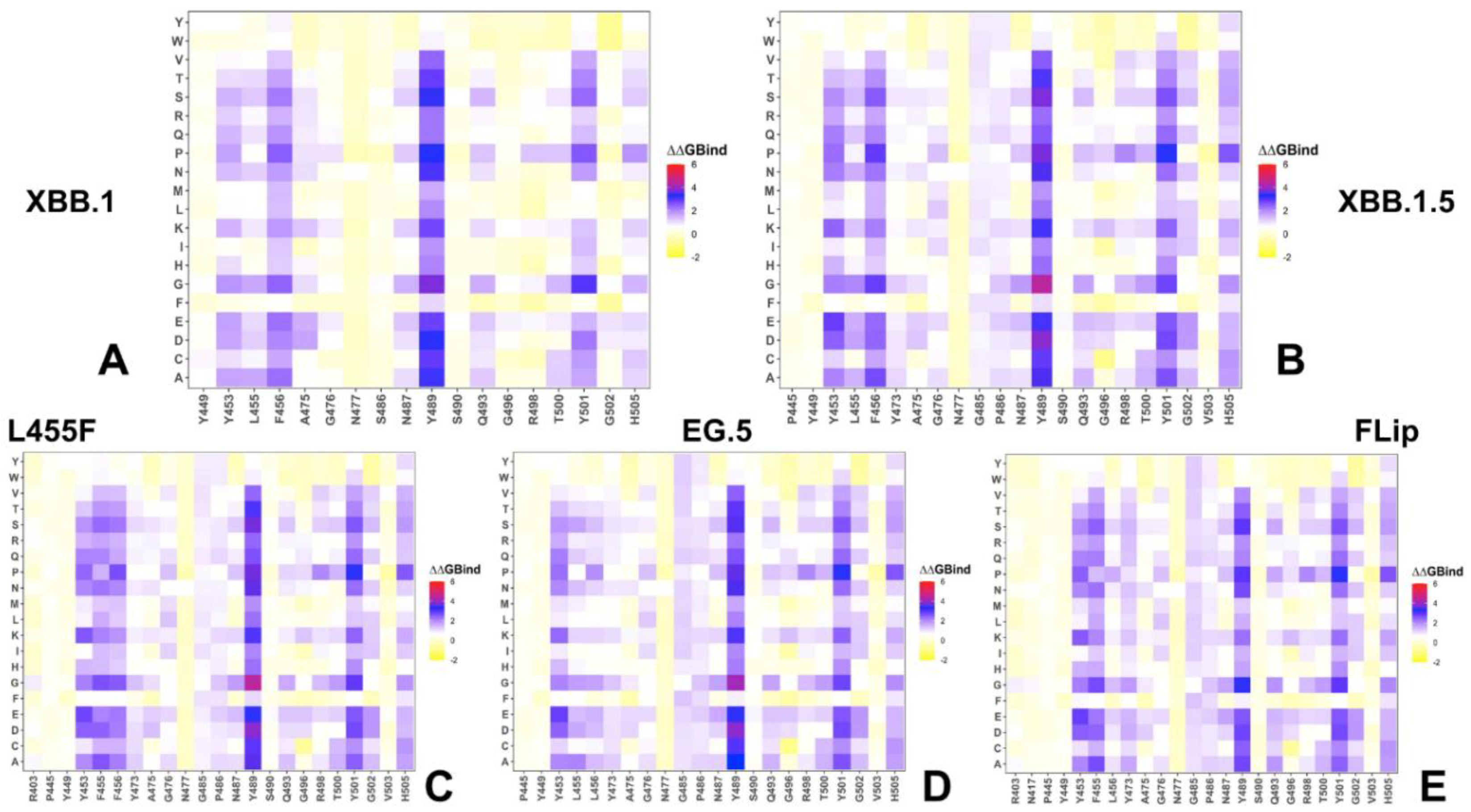
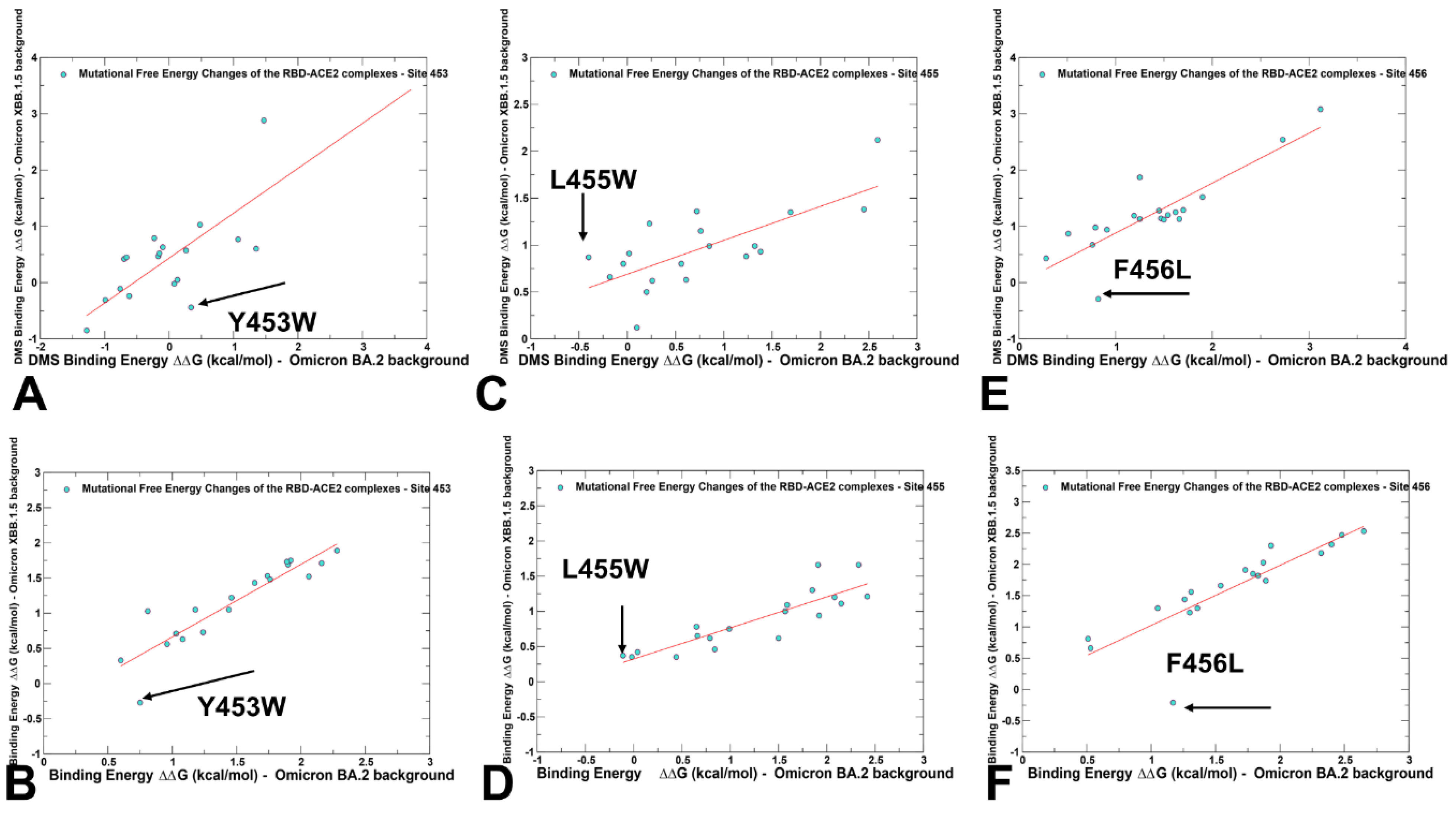
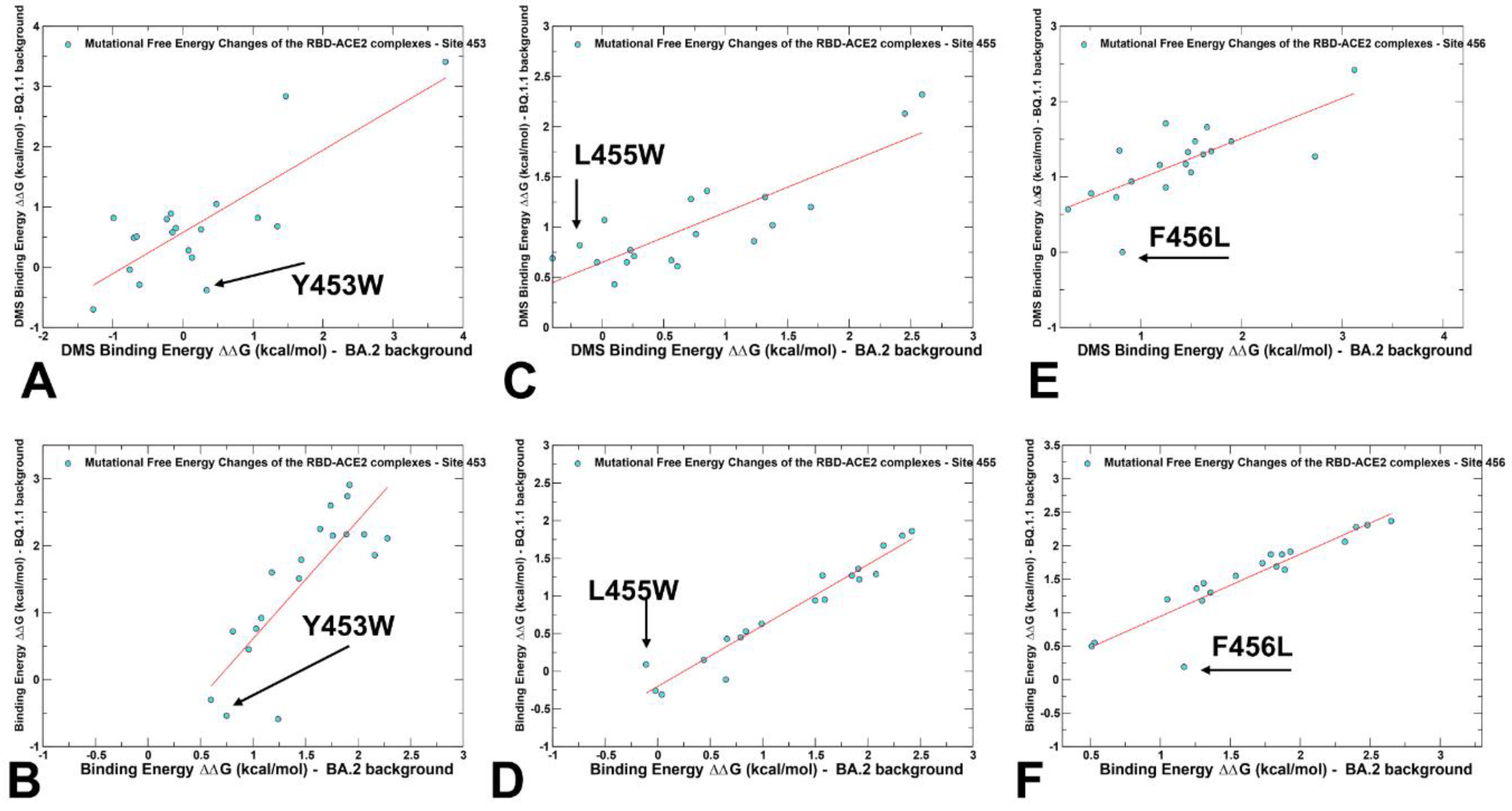
2.3. Probing Epistatic Relationships in the XBB.1.5 and BQ.1.1 Variants Using Mutational Scanning
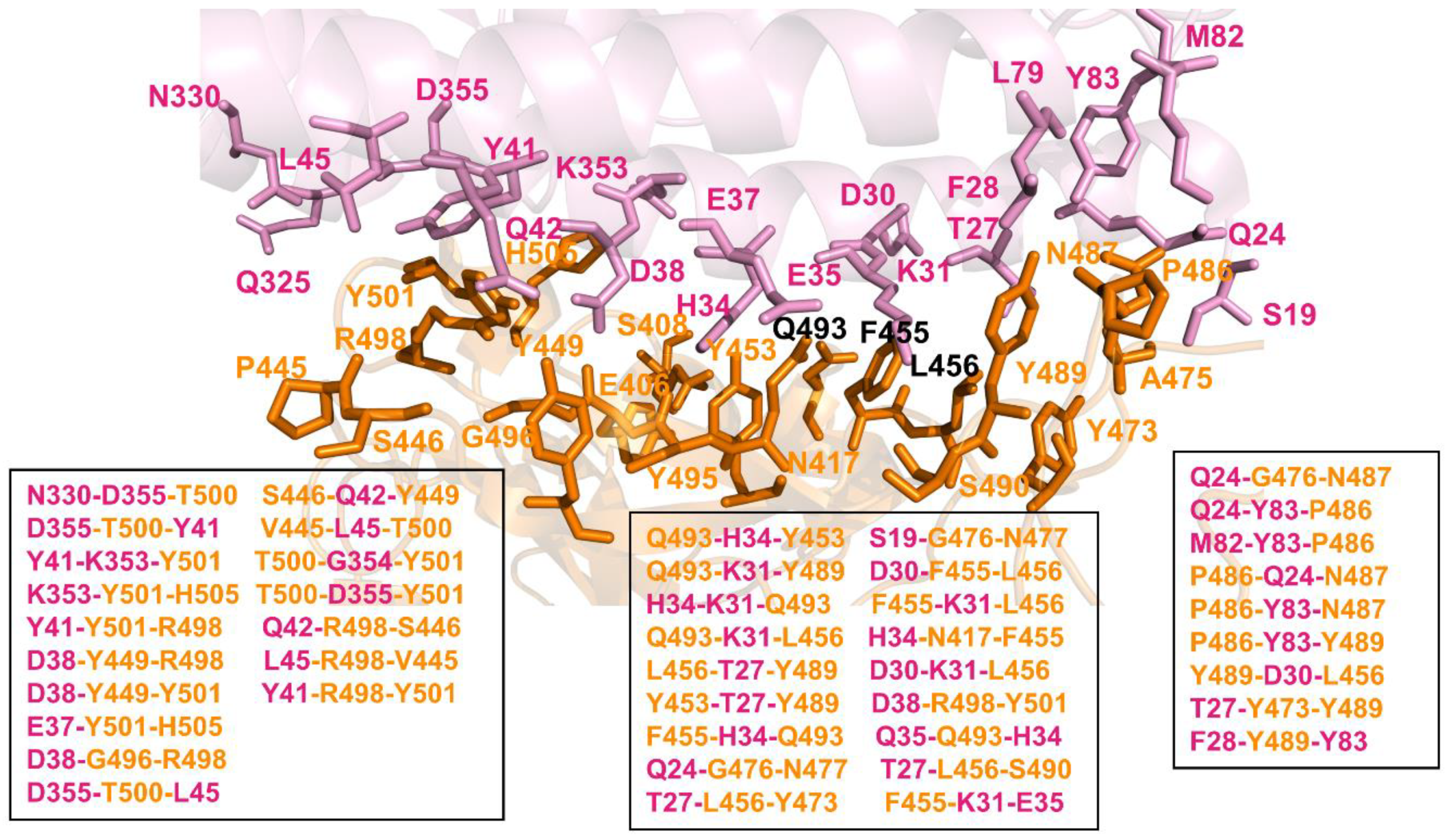
2.4. Network-Based Community Analysis of Epistatic Couplings in the RBD–ACE2 Complexes
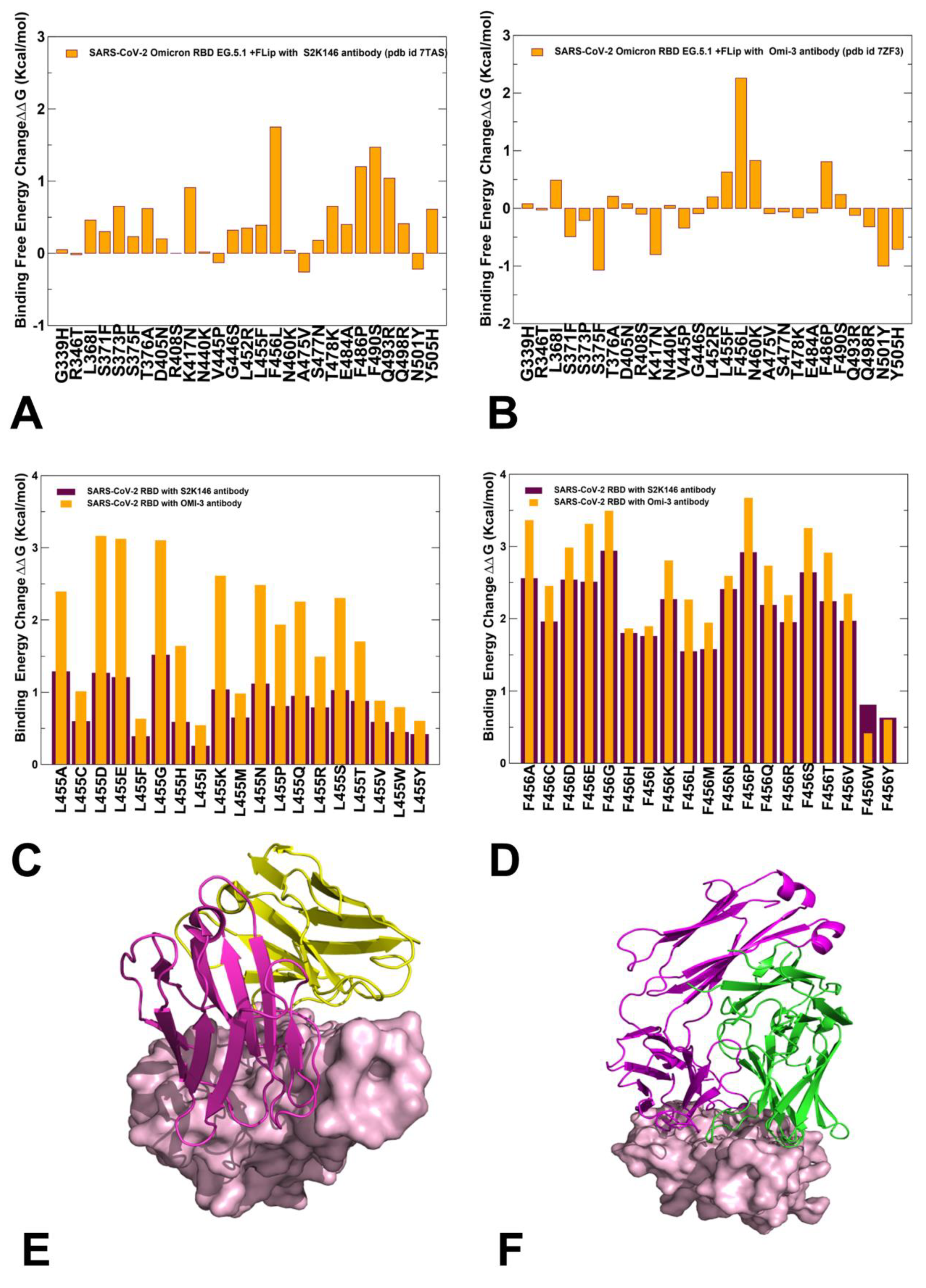
2.5. Mutational Profiling of Spike Protein Binding with Monoclonal RBD Antibodies: Revealing Central Role of F456L and F486P Mutations in Immune Evasion
3. Materials and Methods
3.1. Structure Preparation and Molecular Dynamics Simulations
3.2. Mutational Scanning and Binding Free Energy Computations
3.3. Network-Based Community Analysis and Clique-Based Model of Epistatic Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022, 27, 229–238. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Turoňová, B.; Sikora, M.; Schuerman, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. Elife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Li, X.; Peng, Q.; Su, C.; Huang, B.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular basis of receptor binding and antibody neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol Cell. 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Bowen, J.E.; Addetia, A.; Dang, H.V.; Stewart, C.; Brown, J.T.; Sharkey, W.K.; Sprouse, K.R.; Walls, A.C.; Mazzitelli, I.G.; Logue, J.K.; et al. Omicron spike function and neutralizing activity elicited by a comprehensive panel of vaccines. Science 2022, 377, 890–894. [Google Scholar] [CrossRef]
- Huo, J.; Dijokaite-Guraliuc, A.; Liu, C.; Zhou, D.; Ginn, H.M.; Das, R.; Supasa, P.; Selvaraj, M.; Nutalai, R.; Tuekprakhon, A.; et al. A Delicate Balance between Antibody Evasion and ACE2 Affinity for Omicron BA.2.75. Cell Rep. 2023, 42, 111903. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y.; et al. Characterization of the Enhanced Infectivity and Antibody Evasion of Omicron BA.2.75. Cell Host Microbe 2022, 30, 1527–1539.e5. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Tamura, T.; Zahradnik, J.; Deguchi, S.; Tabata, K.; Anraku, Y.; Kimura, I.; Ito, J.; Yamasoba, D.; Nasser, H.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2.75 Variant. Cell Host Microbe 2022, 30, 1540–1555.e15. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Coronavirus variant XBB.1.5 rises in the United States—Is it a global threat? Nature 2023, 613, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 Binding and Antibody Evasion in Enhanced Transmissibility of XBB.1.5. Lancet Infect. Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Nehlmeier, I.; Kempf, A.; Cossmann, A.; Schulz, S.R.; Morillas Ramos, G.; Manthey, L.A.; Jäck, H.-M.; Behrens, G.M.N.; et al. Profound Neutralization Evasion and Augmented Host Cell Entry Are Hallmarks of the Fast-Spreading SARS-CoV-2 Lineage XBB.1.5. Cell Mol Immunol. 2023, 20, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Yamasoba, D.; Uriu, K.; Plianchaisuk, A.; Kosugi, Y.; Pan, L.; Zahradnik, J.; Ito, J.; Sato, K. Virological Characteristics of the SARS-CoV-2 Omicron XBB.1.16 Variant. Lancet Infect Dis. 2023, 23, 655–656. [Google Scholar] [CrossRef]
- Tsujino, S.; Deguchi, S.; Nomai, T.; Padilla-Blanco, M.; Plianchaisuk, A.; Wang, L.; Begum, M.M.; Uriu, K.; Mizuma, K.; Nao, N.; et al. Virological characteristics of the SARS-CoV-2 Omicron EG.5.1 variant. BioRxiv 2023. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Zhang, R.M.; Ho, J.; Mohri, H.; Valdez, R.; Manthei, D.M.; Gordon, A.; Liu, L.; Ho, D.D. Antibody Neutralization of Emerging SARS-CoV-2 Subvariants: EG.5.1 and XBC.1.6. Lancet Infect Dis. 2023, 23, e397–e398. [Google Scholar] [CrossRef] [PubMed]
- Faraone, J.N.; Qu, P.; Goodarzi, N.; Zheng, Y.-M.; Carlin, C.; Saif, L.J.; Oltz, E.M.; Xu, K.; Jones, D.; Gumina, R.J.; et al. Immune Evasion and Membrane Fusion of SARS-CoV-2 XBB Subvariants EG.5.1 and XBB.2.3. Emerg. Microbes Infect. 2023, 12, 2270069. [Google Scholar] [CrossRef]
- Kosugi, Y.; Plianchaisuk, A.; Putri, O.; Uriu, K.; Kaku, Y.; Hinay, A.A., Jr.; Chen, L.; Kuramochi, J.; Sadamasu, K.; Yoshimura, K.; et al. Characteristics of the SARS-CoV-2 Omicron HK.3 Variant Harbouring the FLip Substitution. Lancet Microbe 2024, 5, E313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Bowen, A.; Mellis, I.A.; Valdez, R.; Gherasim, C.; Gordon, A.; Liu, L.; Ho, D.D. XBB.1.5 Monovalent mRNA Vaccine Booster Elicits Robust Neutralizing Antibodies against Emerging SARS-CoV-2 Variants. Cell Host Microbe 2023, 32, 25–34.e5. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Feng, L.; Yang, S.; Yu, Y.; Wang, L.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; et al. Convergent Evolution of SARS-CoV-2 XBB Lineages on Receptor-Binding Domain 455-456 Synergistically Enhances Antibody Evasion and ACE2 Binding. PLoS Pathog. 2023, 19, e1011868. [Google Scholar] [CrossRef] [PubMed]
- Roemer, C.; Sheward, D.J.; Hisner, R.; Gueli, F.; Sakaguchi, H.; Frohberg, N.; Schoenmakers, J.; Sato, K.; O’Toole, Á.; Rambaut, A.; et al. SARS-CoV-2 Evolution in the Omicron Era. Nat. Microbiol. 2023, 8, 1952–1959. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Liu, L.; Schwanz, L.T.; Li, Z.; Nair, M.S.; Ho, J.; Zhang, R.M.; Iketani, S.; Yu, J.; et al. Antigenicity and Receptor Affinity of SARS-CoV-2 BA.2.86 Spike. Nature 2023, 624, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; Wang, J.; Yu, L.; et al. Antigenicity and Infectivity Characterization of SARS-CoV-2 BA.2.86. Lancet Infect Dis. 2023, 23, e457–e459. [Google Scholar] [CrossRef]
- Lasrado, N.; Collier, A.Y.; Hachmann, N.P.; Miller, J.; Rowe, M.; Schonberg, E.D.; Rodrigues, S.L.; LaPiana, A.; Patio, R.C.; Anand, T.; et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariant BA.2.86. Vaccine 2023, 41, 6904–6909. [Google Scholar] [CrossRef]
- Tamura, T.; Mizuma, K.; Nasser, H.; Deguchi, S.; Padilla-Blanco, M.; Oda, Y.; Uriu, K.; Tolentino, J.E.M.; Tsujino, S.; Suzuki, R.; et al. Virological Characteristics of the SARS-CoV-2 BA.2.86 Variant. Cell Host Microbe 2024, 32, 170–180.e12. [Google Scholar] [CrossRef]
- Qu, P.; Xu, K.; Faraone, J.N.; Goodarzi, N.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; Saif, L.J.; et al. Immune Evasion, Infectivity, and Fusogenicity of SARS-CoV-2 BA.2.86 and FLip Variants. Cell 2024, 187, 585–595.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast Evolution of SARS-CoV-2 BA.2.86 to JN.1 under Heavy Immune Pressure. Lancet Infect Dis. 2024, 24, e70–e72. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Stewart, C.M.; Walls, A.C.; Hannon, W.W.; Veesler, D.; Bloom, J.D. Deep Mutational Scans for ACE2 Binding, RBD Expression, and Antibody Escape in the SARS-CoV-2 Omicron BA.1 and BA.2 Receptor-Binding Domains. PLoS Pathog. 2022, 18, e1010951. [Google Scholar] [CrossRef]
- Dadonaite, B.; Crawford, K.H.D.; Radford, C.E.; Farrell, A.G.; Yu, T.C.; Hannon, W.W.; Zhou, P.; Andrabi, R.; Burton, D.R.; Liu, L.; et al. A Pseudovirus System Enables Deep Mutational Scanning of the Full SARS-CoV-2 Spike. Cell 2023, 186, 1263–1278.e20. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R.; Ferri, E.; Farrell, A.G.; Dadonaite, B.; McCallum, M.; et al. Shifting Mutational Constraints in the SARS-CoV-2 Receptor-Binding Domain during Viral Evolution. Science 2022, 377, 420–424. [Google Scholar] [CrossRef]
- Moulana, A.; Dupic, T.; Phillips, A.M.; Chang, J.; Nieves, S.; Roffler, A.A.; Greaney, A.J.; Starr, T.N.; Bloom, J.D.; Desai, M.M. Compensatory Epistasis Maintains ACE2 Affinity in SARS-CoV-2 Omicron BA.1. Nat. Commun. 2022, 13, 7011. [Google Scholar] [CrossRef] [PubMed]
- Witte, L.; Baharani, V.A.; Schmidt, F.; Wang, Z.; Cho, A.; Raspe, R.; Guzman-Cardozo, C.; Muecksch, F.; Canis, M.; Park, D.J.; et al. Epistasis Lowers the Genetic Barrier to SARS-CoV-2 Neutralizing Antibody Escape. Nat. Commun. 2023, 14, 302. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Brown, J.; McMahon, T.E.; Farrell, A.G.; Asarnow, D.; Stewart, C.; Logue, J.; Murrell, B.; Chu, H.Y.; Veesler, D.; et al. Full-Spike Deep Mutational Scanning Helps Predict the Evolutionary Success of SARS-CoV-2 Clades. bioRxiv 2023. Preprint. [Google Scholar] [CrossRef]
- Yisimayi, A.; Song, W.; Wang, J.; Jian, F.; Yu, Y.; Chen, X.; Xu, Y.; Yang, S.; Niu, X.; Xiao, T.; et al. Repeated Omicron Exposures Override Ancestral SARS-CoV-2 Immune Imprinting. Nature 2023, 625, 148–156. [Google Scholar] [CrossRef]
- Taylor, A.L.; Starr, T.N. Deep Mutational Scans of XBB.1.5 and BQ.1.1 Reveal Ongoing Epistatic Drift during SARS-CoV-2 Evolution. PLoS Pathog. 2023, 19, e1011901. [Google Scholar] [CrossRef]
- Neverov, A.D.; Fedonin, G.; Popova, A.; Bykova, D.; Bazykin, G. Coordinated Evolution at Amino Acid Sites of SARS-CoV-2 Spike. Elife 2023, 12, e82516. [Google Scholar] [CrossRef]
- Bazykin, G.A.; Kondrashov, F.A.; Ogurtsov, A.Y.; Sunyaev, S.; Kondrashov, A.S. Positive Selection at Sites of Multiple Amino Acid Replacements since Rat–Mouse Divergence. Nature 2004, 429, 558–562. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond shielding: The roles of glycans in the SARS-CoV-2 spike potein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls the opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 Spike Opening Dynamics and Energetics Reveal the Individual Roles of Glycans and Their Collective Impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Dokainish, H.M.; Re, S.; Mori, T.; Kobayashi, C.; Jung, J.; Sugita, Y. The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein. Elife 2022, 11, e75720. [Google Scholar] [CrossRef]
- Singh, J.; Vashishtha, S.; Kundu, B. Spike Protein Mutation-Induced Changes in the Kinetic and Thermodynamic Behavior of Its Receptor Binding Domains Explain Their Higher Propensity to Attain Open States in SARS-CoV-2 Variants of Concern. ACS Cent. Sci. 2023, 9, 1894–1904. [Google Scholar] [CrossRef]
- Cao, W.; Dong, C.; Kim, S.; Hou, D.; Tai, W.; Du, L.; Im, W.; Zhang, X.F. Biomechanical characterization of SARS-CoV-2 spike RBD and human ACE2 protein-protein interaction. Biophys. J. 2021, 120, 1011–1019. [Google Scholar] [CrossRef]
- Oliva, R.; Shaikh, A.R.; Petta, A.; Vangone, A.; Cavallo, L. D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect. Molecules 2021, 26, 2622. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G. Balancing Functional Tradeoffs between Protein Stability and ACE2 Binding in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB Lineages: Dynamics-Based Network Models Reveal Epistatic Effects Modulating Compensatory Dynamic and Energetic Changes. Viruses 2023, 15, 1143. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P. Probing Conformational Landscapes of Binding and Allostery in the SARS-CoV-2 Omicron Variant Complexes Using Microsecond Atomistic Simulations and Perturbation-Based Profiling Approaches: Hidden Role of Omicron Mutations as Modulators of Allosteric Signaling and Epistatic Relationships. Phys. Chem. Chem. Phys. 2023, 25, 21245–21266. [Google Scholar] [CrossRef]
- Xiao, S.; Alshahrani, M.; Gupta, G.; Tao, P.; Verkhivker, G. Markov State Models and Perturbation-Based Approaches Reveal Distinct Dynamic Signatures and Hidden Allosteric Pockets in the Emerging SARS-Cov-2 Spike Omicron Variant Complexes with the Host Receptor: The Interplay of Dynamics and Convergent Evolution Modulates Allostery and Functional Mechanisms. J. Chem. Inf. Model. 2023, 63, 5272–5296. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G. Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites. Viruses 2023, 15, 2009. [Google Scholar] [CrossRef]
- Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P.; Verkhivker, G. Comparative Analysis of Conformational Dynamics and Systematic Characterization of Cryptic Pockets in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB.1 Spike Complexes with the ACE2 Host Receptor: Confluence of Binding and Structural Plasticity in Mediating Networks of Conserved Allosteric Sites. Viruses 2023, 15, 2073. [Google Scholar] [CrossRef]
- Zuzic, L.; Samsudin, F.; Shivgan, A.T.; Raghuvamsi, P.V.; Marzinek, J.K.; Boags, A.; Pedebos, C.; Tulsian, N.K.; Warwicker, J.; MacAry, P.; et al. Uncovering Cryptic Pockets in the SARS-CoV-2 Spike Glycoprotein. Structure 2022, 30, 1062–1074.e4. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Benvenuto, D.; Borsetti, A.; Azzena, I.; Casu, M.; Fiori, P.L.; Giovanetti, M.; Maruotti, A.; Ceccarelli, G.; et al. Genetic and Structural Data on the SARS-CoV-2 Omicron BQ.1 Variant Reveal Its Low Potential for Epidemiological Expansion. Int. J. Mol. Sci. 2022, 23, 15264. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Evans, J.P.; Faraone, J.N.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Enhanced Neutralization Resistance of SARS-CoV-2 Omicron Subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 2023, 31, 9–17.e3. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Computer Simulations and Network-Based Profiling of Binding and Allosteric Interactions of SARS-CoV-2 Spike Variant Complexes and the Host Receptor: Dissecting the Mechanistic Effects of the Delta and Omicron Mutations. Int. J. Mol. Sci. 2022, 23, 4376. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets. Int. J. Mol. Sci. 2022, 23, 11542. [Google Scholar] [CrossRef]
- Zhu, X.; Mannar, D.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Saville, J.W.; Leopold, K.; Li, W.; Dimitrov, D.S.; Tuttle, K.S.; et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021, 19, e3001237. [Google Scholar] [CrossRef]
- Ni, D.; Turelli, P.; Beckert, B.; Nazarov, S.; Uchikawa, E.; Myasnikov, A.; Pojer, F.; Trono, D.; Stahlberg, H.; Lau, K. Cryo-EM Structures and Binding of Mouse and Human ACE2 to SARS-CoV-2 Variants of Concern Indicate That Mutations Enabling Immune Escape Could Expand Host Range. PLoS Pathog. 2023, 19, e1011206. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Ito, J.; Suzuki, R.; Uriu, K.; Itakura, Y.; Zahradnik, J.; Kimura, K.T.; Deguchi, S.; Wang, L.; Lytras, S.; Tamura, T.; et al. Convergent Evolution of SARS-CoV-2 Omicron Subvariants Leading to the Emergence of BQ.1.1 Variant. Nat. Commun. 2023, 14, 2671. [Google Scholar] [CrossRef]
- Ren, W.; Lan, J.; Ju, X.; Gong, M.; Long, Q.; Zhu, Z.; Yu, Y.; Wu, J.; Zhong, J.; Zhang, R.; et al. Mutation Y453F in the Spike Protein of SARS-CoV-2 Enhances Interaction with the Mink ACE2 Receptor for Host Adaption. PLoS Pathog. 2021, 17, e1010053. [Google Scholar] [CrossRef]
- Philip, A.M.; Ahmed, W.S.; Biswas, K.H. Reversal of the Unique Q493R Mutation Increases the Affinity of Omicron S1-RBD for ACE2. Comput. Struct. Biotechnol. J. 2023, 21, 1966–1977. [Google Scholar] [CrossRef]
- Starr, T.N.; Thornton, J.W. Epistasis in Protein Evolution. Prot. Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Metzger, B.P.H.; Thornton, J.W. Epistatic Drift Causes Gradual Decay of Predictability in Protein Evolution. Science 2022, 376, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; De Marco, A.; Starr, T.N.; Liu, Z.; Pinto, D.; Walls, A.C.; Zatta, F.; Zepeda, S.K.; Bowen, J.E.; Sprouse, K.R.; et al. Antibody-Mediated Broad Sarbecovirus Neutralization through ACE2 Molecular Mimicry. Science 2022, 375, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent Cross-Reactive Antibodies Following Omicron Breakthrough in Vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Fu, H.; Jian, F.; Cao, Y.; Li, M. Immune Evasion and ACE2 Binding Affinity Contribute to SARS-CoV-2 Evolution. Nat. Ecol. Evol. 2023, 7, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Arnold, F.H. In the Light of Directed Evolution: Pathways of Adaptive Protein Evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 9995–10000. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.I.; Suchard, M.A.; Bloom, J.D. Stability-mediated epistasis constrains the evolution of an influenza protein. Elife 2013, 2, e00631. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent Protein Structure Refinement by Optimizing Hydrogen Bonding Network and Atomic-Level Energy Minimization. Proteins 2013, 81, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An Interactive Web Server for Efficient Protein Structure Refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store-A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J Comput Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, C.; Zhou, H.Y.; Zhou, Y.Q. A physical reference state unifies the structure-derived potential of mean force for protein folding and binding. Proteins 2004, 56, 93–101. [Google Scholar] [CrossRef]
- Gonzalez, T.R.; Martin, K.P.; Barnes, J.E.; Patel, J.S.; Ytreberg, F.M. Assessment of Software Methods for Estimating Protein-Protein Relative Binding Affinities. PLoS ONE 2020, 15, e0240573. [Google Scholar] [CrossRef]
- Pucci, F.; Rooman, M. Prediction and Evolution of the Molecular Fitness of SARS-CoV-2 Variants: Introducing SpikePro. Viruses 2021, 13, 935. [Google Scholar] [CrossRef]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: A community-hopping model of allosteric regulation and communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef]
- Newman, M.E. Modularity and Community Structure in Networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G. Coarse-Grained Molecular Simulations and Ensemble-Based Mutational Profiling of Protein Stability in the Different Functional Forms of the SARS-CoV-2 Spike Trimers: Balancing Stability and Adaptability in BA.1, BA.2 and BA.2.75 Variants. Int. J. Mol. Sci. 2023, 24, 6642. [Google Scholar] [CrossRef]
- Palla, G.; Derényi, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar] [CrossRef]
- Adamcsek, B.; Palla, G.; Farkas, I.J.; Derényi, I.; Vicsek, T. CFinder: Locating cliques and overlapping modules in biological networks. Bioinformatics 2006, 22, 1021–1023. [Google Scholar] [CrossRef]
- Ming, D.; Chen, R.; Huang, H. Amino-Acid Network Clique Analysis of Protein Mutation Non-Additive Effects: A Case Study of Lysozme. Int. J. Mol. Sci. 2018, 19, 1427. [Google Scholar] [CrossRef]
- Bridgham, J.T.; Ortlund, E.A.; Thornton, J.W. An Epistatic Ratchet Constrains the Direction of Glucocorticoid Receptor Evolution. Nature 2009, 461, 515–519. [Google Scholar] [CrossRef]
- Buda, K.; Miton, C.M.; Tokuriki, N. Pervasive Epistasis Exposes Intramolecular Networks in Adaptive Enzyme Evolution. Nat. Commun. 2023, 14, 8508. [Google Scholar] [CrossRef]
- Wagner, A. Evolvability-Enhancing Mutations in the Fitness Landscapes of an RNA and a Protein. Nat. Commun. 2023, 14, 624. [Google Scholar] [CrossRef] [PubMed]
- Ogbunugafor, C.B. Mutations That Enhance Evolvability May Open Doors to Faster Adaptation. Nat. Commun. 2023, 14, 6310. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omicron Variant | Mutational Landscape |
|---|---|
| BA.1 | A67, T95I, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F |
| BA.2 | T19I, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| BA.4 | T19I, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| BA.5 | T19I, LPPA24-27S, Del 69-70, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| BQ.1.1 | T19I, LPPA24-27S, H69del, V70del, V213G, G142D, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, K444T, L452R, N460K, S477N, T478K, E484A, F486V, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| XBB.1 | T19I, V83A, G142D, Del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486S, F490S, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| XBB.1.5 | T19I, V83A, G142D, Del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486P, F490S, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| XBB.1.5 + F456L | T19I, V83A, G142D, Del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, F456L, N460K, S477N, T478K, E484A, F486P, F490S, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
| XBB.1.5 + L455F/F456L | T19I, V83A, G142D, Del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, L455F, F456L, N460K, S477N, T478K, E484A, F486P, F490S, R493Q reversal, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raisinghani, N.; Alshahrani, M.; Gupta, G.; Verkhivker, G. Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape. Int. J. Mol. Sci. 2024, 25, 4281. https://doi.org/10.3390/ijms25084281
Raisinghani N, Alshahrani M, Gupta G, Verkhivker G. Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape. International Journal of Molecular Sciences. 2024; 25(8):4281. https://doi.org/10.3390/ijms25084281
Chicago/Turabian StyleRaisinghani, Nishank, Mohammed Alshahrani, Grace Gupta, and Gennady Verkhivker. 2024. "Ensemble-Based Mutational Profiling and Network Analysis of the SARS-CoV-2 Spike Omicron XBB Lineages for Interactions with the ACE2 Receptor and Antibodies: Cooperation of Binding Hotspots in Mediating Epistatic Couplings Underlies Binding Mechanism and Immune Escape" International Journal of Molecular Sciences 25, no. 8: 4281. https://doi.org/10.3390/ijms25084281