
Whole-Transcriptome Sequencing of Knee Joint Cartilage from Kashin–Beck Disease and Osteoarthritis Patients
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
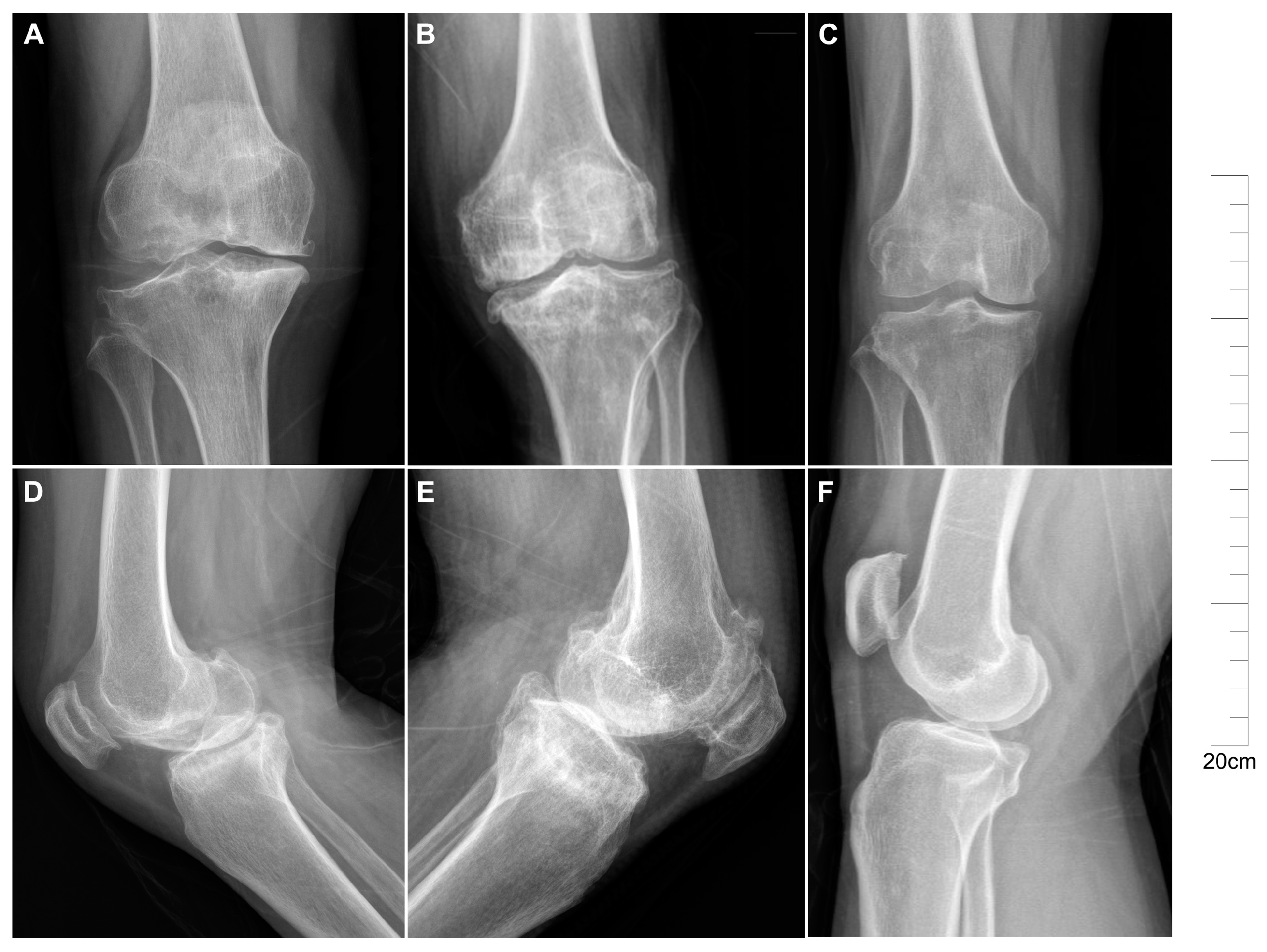
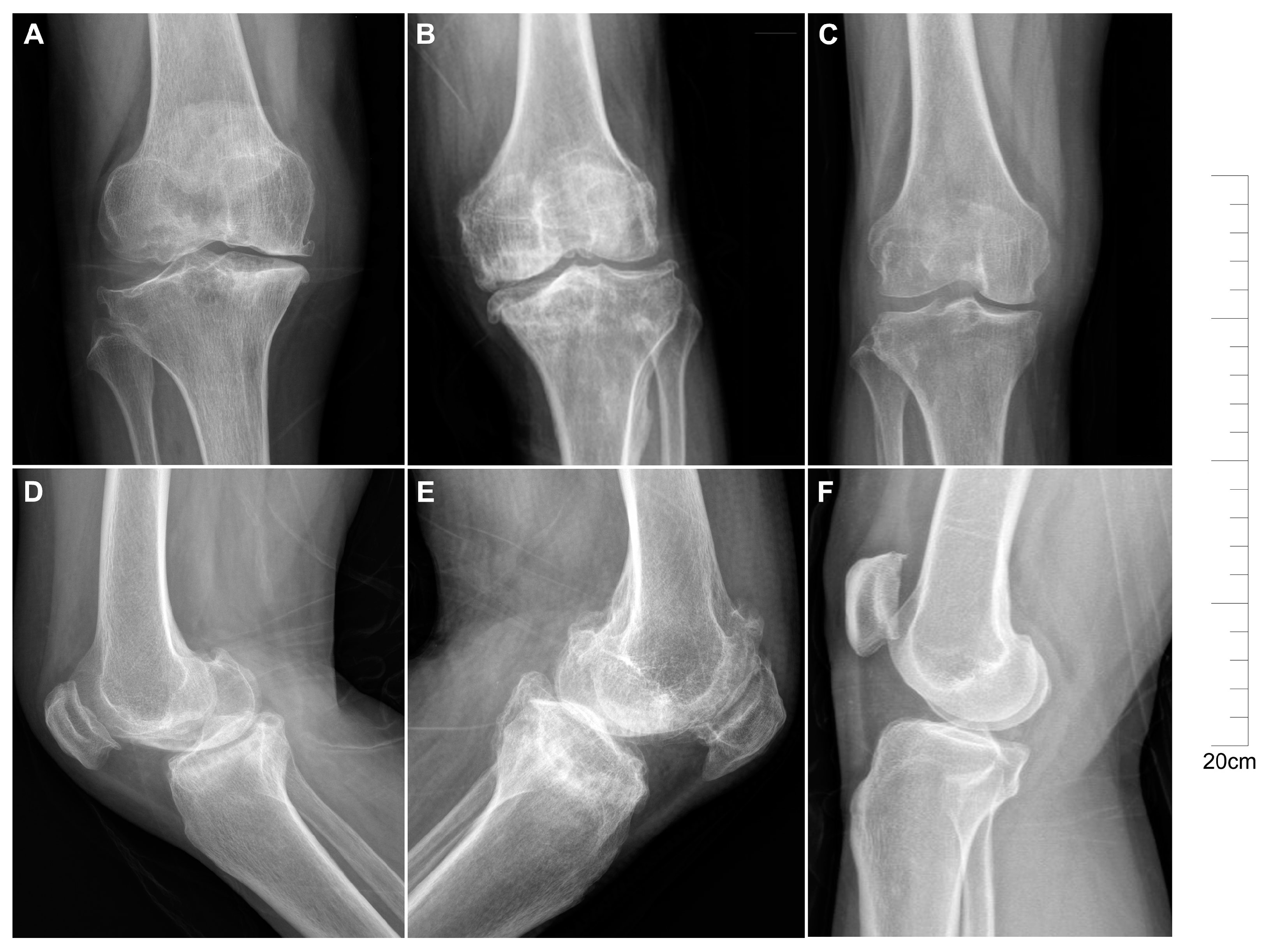
2.1. Descriptive Characteristics of Patients with KBD and OA
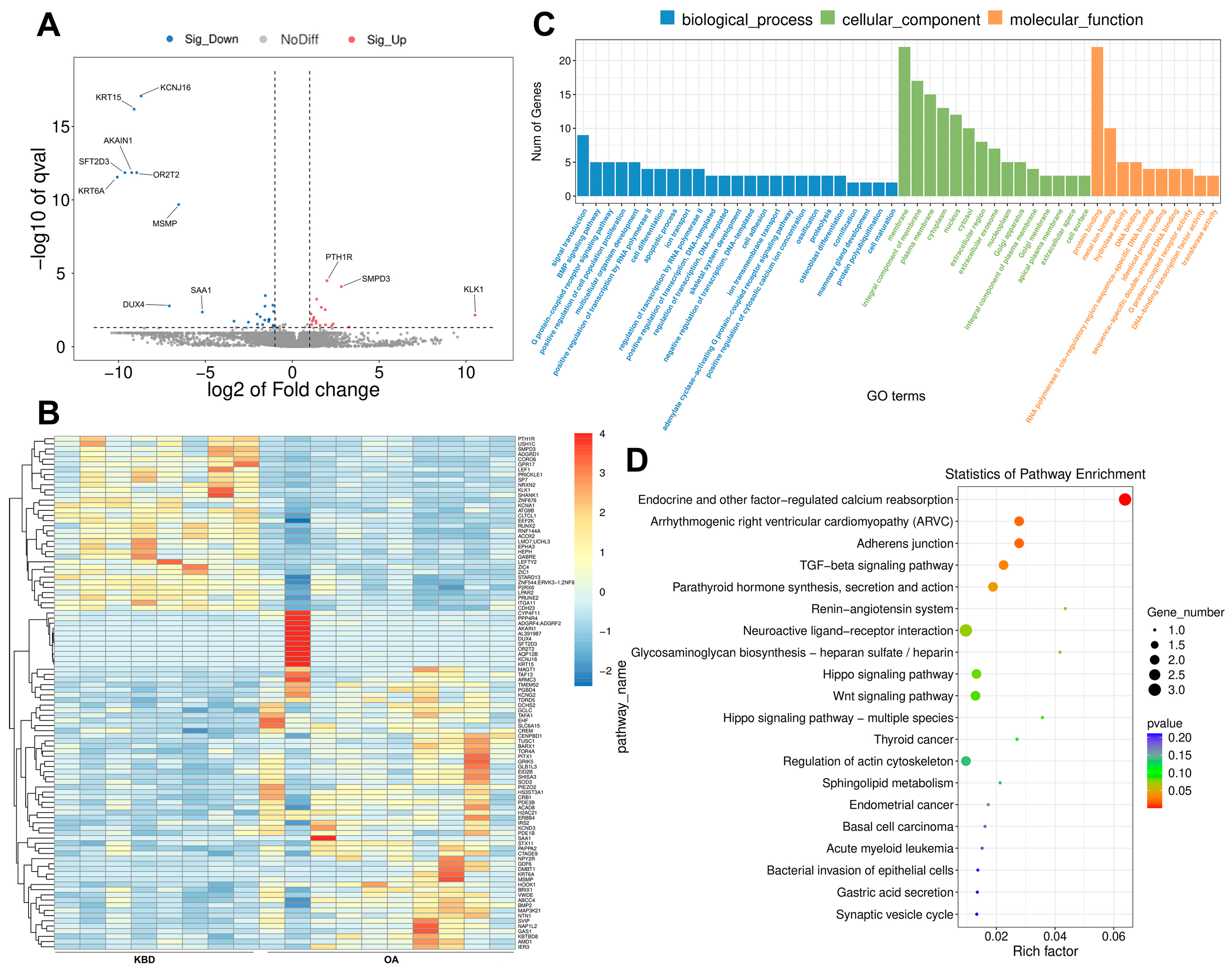
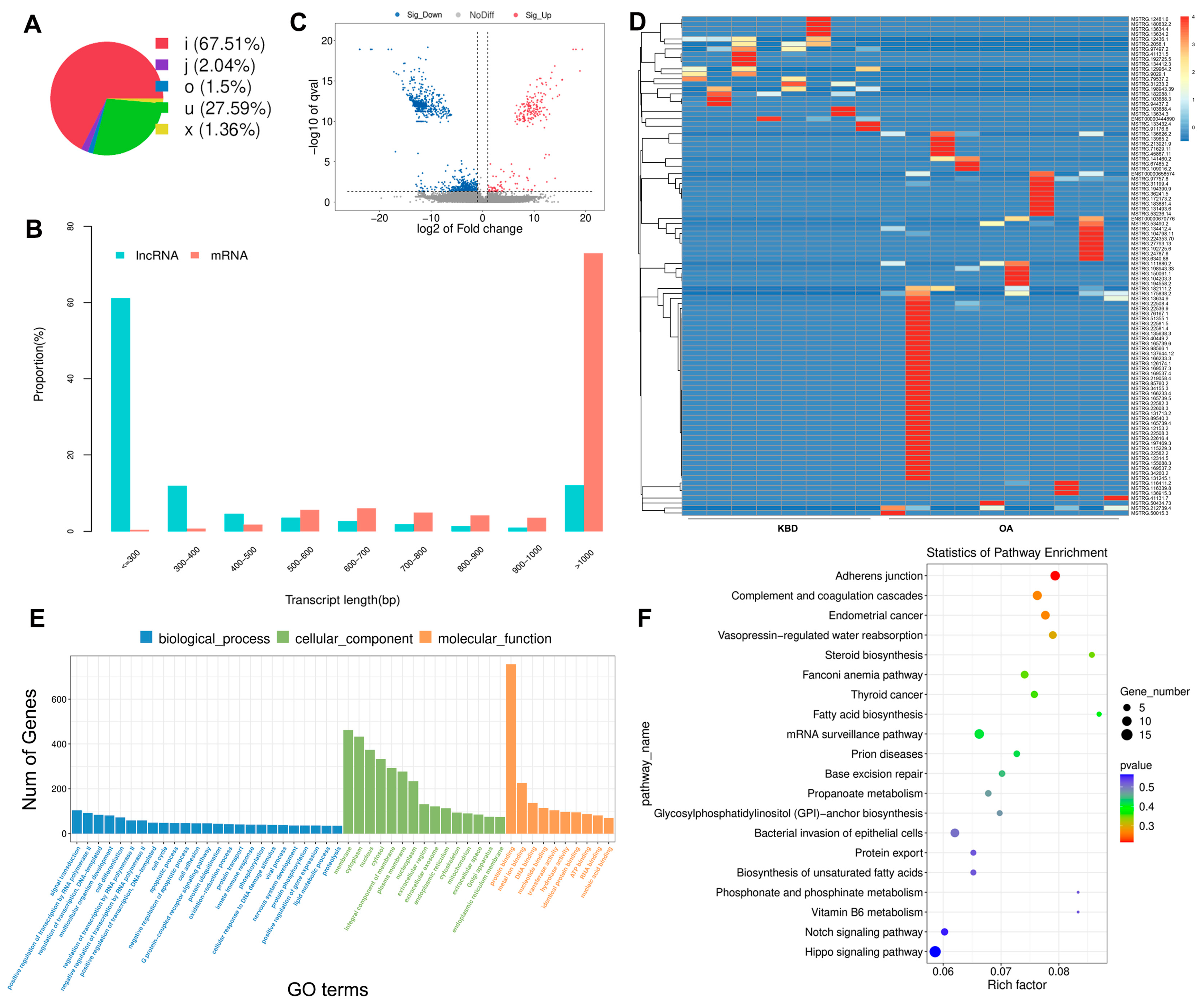
2.2. Identification of DE mRNAs between KBD and OA
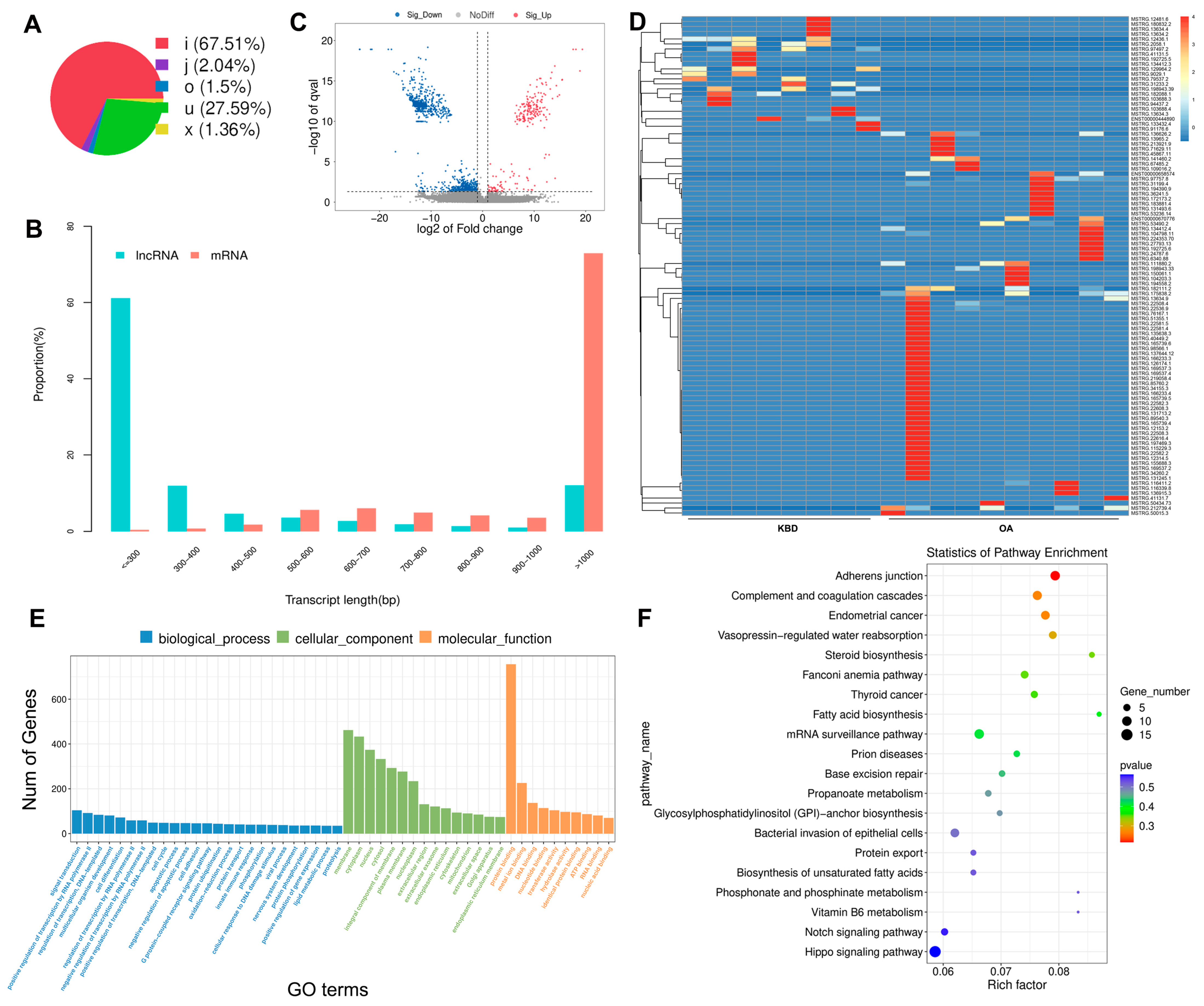
2.3. Prediction and Function Analysis of the lncRNA Target Genes
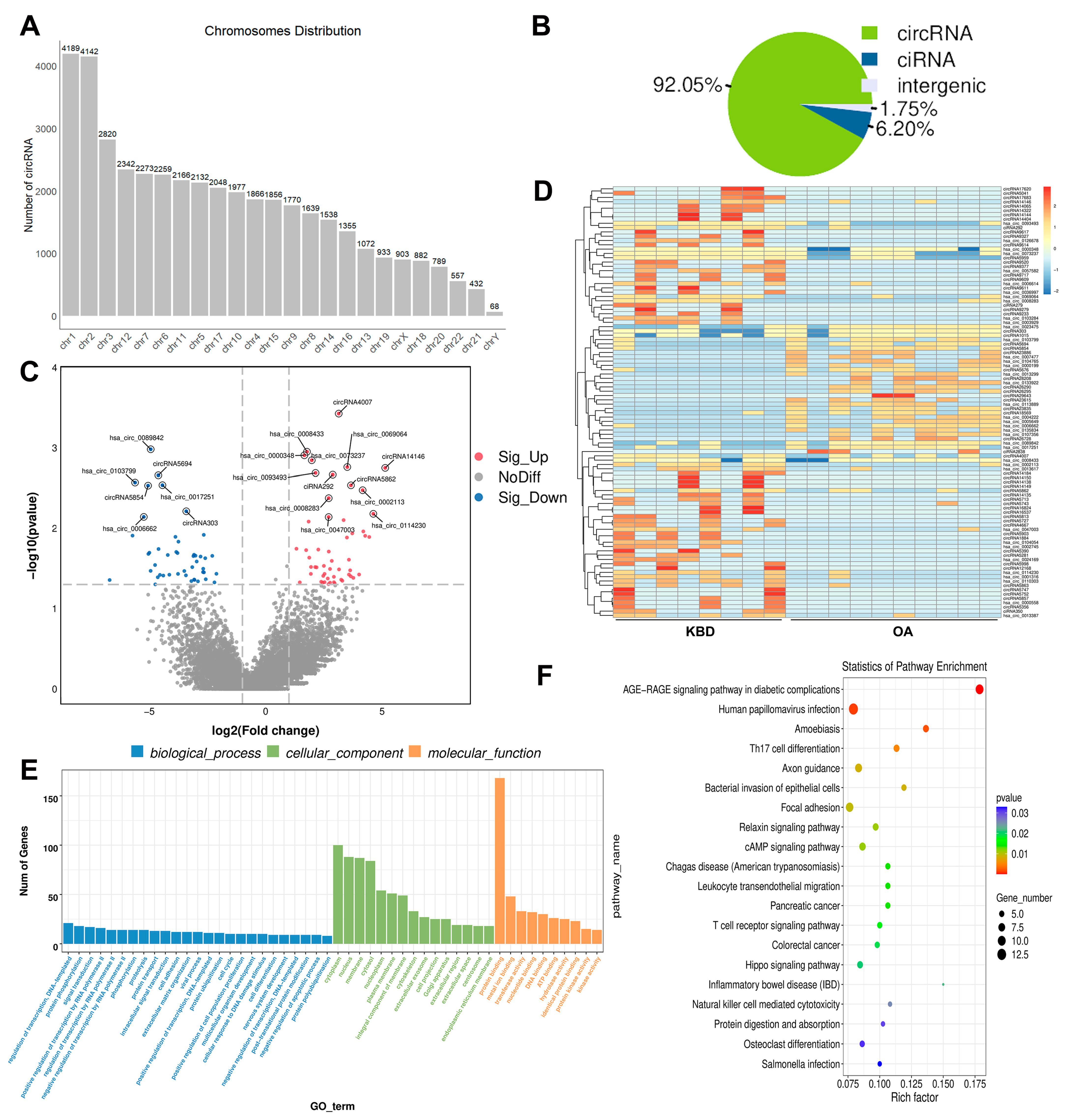
2.4. Overview of the circRNA Sequencing Data
2.5. Construction of the Potential lncRNA-mRNA and circRNA-miRNA Interactions
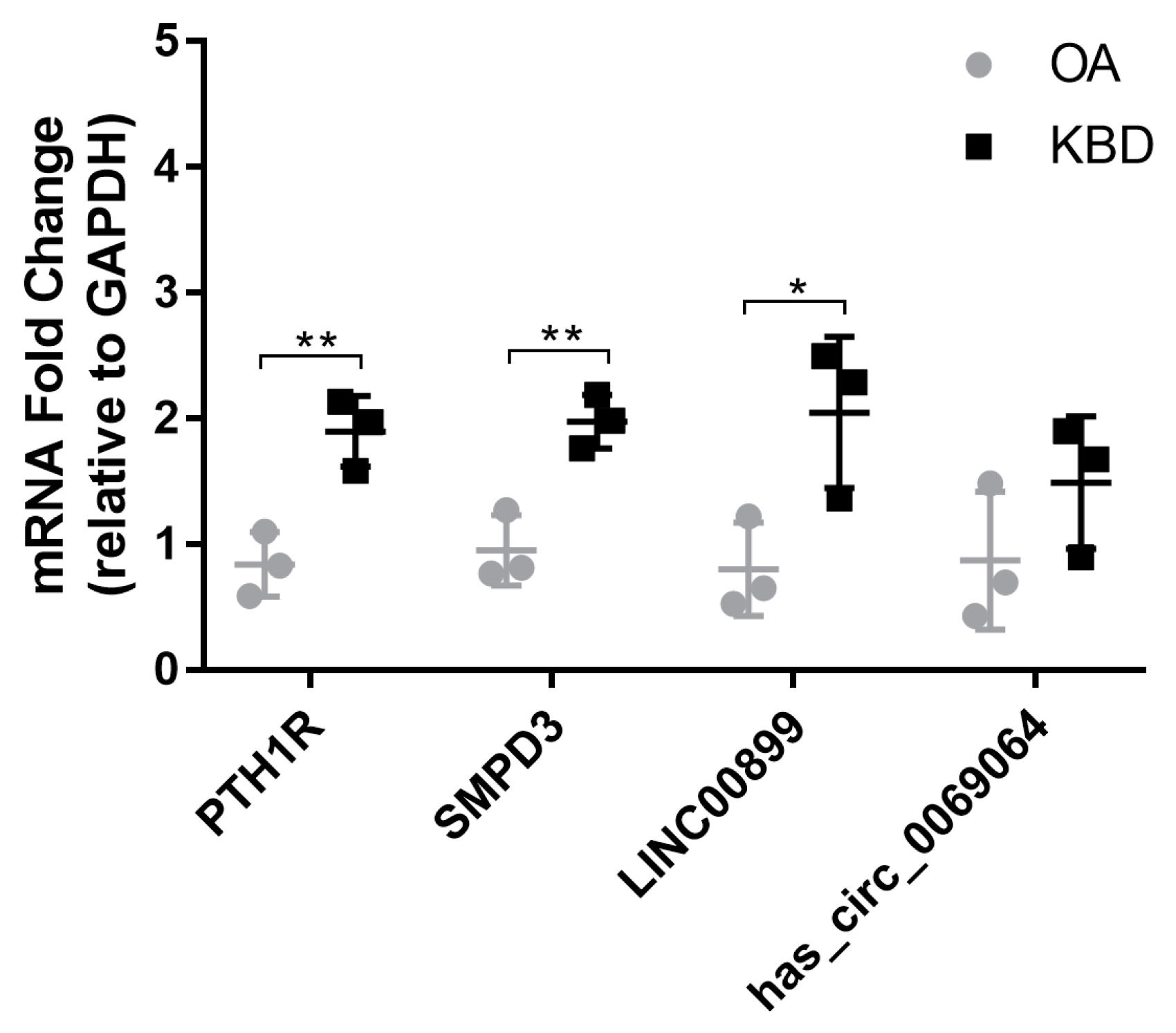
2.6. Validation of Differentially Expressed mRNAs and ncRNAs in Human
3. Discussion
4. Materials and Methods
4.1. Ethic Statement
4.2. Patients and Samples
4.3. RNA Library Construction and Sequencing
4.4. Preprocessing for Whole RNA Sequencing Data
4.5. Differential Expression Analysis of mRNAs and lncRNAs
4.6. Potential Target Gene Prediction and Functional Analysis of lncRNAs
4.7. Prediction, Annotation, and miRNA Interaction of Novel circRNAs
4.8. Functional Enrichment Analysis
4.9. Validation of the Differential Expression of mRNAs, lncRNAs, and circRNAs
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allander, E. Kashin-Beck disease. An analysis of research and public health activities based on a bibliography 1849–1992. Scand. J. Rheumatol. Suppl. 1994, 99, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, S.; Shi, Z.; Yue, Y.; Sun, J.; Chen, J.; Fu, Q.; Hughes, C.; Caterson, B. Articular cartilage metabolism in patients with Kashin–Beck Disease: An endemic osteoarthropathy in China. Osteoarthr. Cartil. 2008, 16, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, H.H.; Sun, Z.G.; Tang, H.B.; Min, J.K. Whole-transcriptome sequencing of knee joint cartilage from osteoarthritis patients. Bone Jt. Res. 2019, 8, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Guo, X.; Zhang, X.; Yu, H.; Yan, H.; Gao, Y.; Ma, W.; Gao, Z.; Xu, P.; Lammi, M. Comparative analysis of gene expression profiles between primary knee osteoarthritis and an osteoarthritis endemic to Northwestern China, Kashin-Beck disease. Arthritis Rheum. 2010, 62, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H.; et al. ENCODE whole-genome data in the UCSC Genome Browser: Update 2012. Nucleic Acids Res. 2011, 40, D912–D917. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle dif-ferentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Jiang, J.; Xu, C.; Kang, J.; Xiao, L.; Wu, M.; Xiong, J.; Guo, X.; Liu, H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 2013, 25, 69–80. [Google Scholar] [CrossRef]
- Dai, Y.; Jian, C.; Wang, X.; Dai, X. Comprehensive expression profiles of mRNAs, lncRNAs and miRNAs in Kashin-Beck disease identified by RNA-sequencing. Mol. Omics 2022, 18, 154–166. [Google Scholar] [CrossRef]
- He, A.; Ning, Y.; Wen, Y.; Cai, Y.; Xu, K.; Han, J.; Liu, L.; Du, Y.; Liang, X.; Li, P.; et al. Use of integrative epigenetic and mRNA expression analyses to identify significantly changed genes and functional pathways in osteoarthritic cartilage. Bone Jt. Res. 2018, 7, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, X.; Duan, C.; Ma, W.; Xu, P.; Gao, Z.; Wang, Z.; Yan, H.; Zhang, Y.; Yu, Y.; et al. Comparative analysis of gene expression profiles between the normal human cartilage and the one with endemic osteoarthritis. Osteoarthr. Cartil. 2009, 17, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sun, J.; Zhang, Y.; Guo, X.; Zhao, G. Comprehensive comparative analysis of histopathology and gene expression in subchondral bone between kashin-beck disease and primary osteoarthritis. Front. Genet. 2022, 13, 942326. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, A.; Wen, Y.; Xiao, X.; Hao, J.; Zhang, F.; Guo, X. Comparison of microRNA expression profiles of Kashin-Beck disease, osteoarthritis and rheumatoid arthritis. Sci. Rep. 2017, 7, 540. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, H.; Zhang, F.; Shao, W.; Yang, L.; Ning, Y.; Wang, S.; Zhao, G.; Lee, B.J.; Lammi, M.; et al. Long noncoding RNA expression profile reveals lncRNAs signature associated with extracellular matrix degradation in kashin-beck disease. Sci. Rep. 2017, 7, 17553. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, C.; Zhang, Y.; Yang, Y.; Ren, Z.; Lammi, M.J.; Guo, X. Screening for differentially expressed circRNA between Kashin–Beck disease and osteoarthritis patients based on circRNA chips. Clin. Chim. Acta 2020, 501, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ning, Y.; Zhang, P.; Poulet, B.; Huang, R.; Gong, Y.; Hu, M.; Li, C.; Zhou, R.; Lammi, M.J.; et al. Comparison of the major cell populations among osteoarthritis, Kashin-Beck disease and healthy chondrocytes by single-cell RNA-seq analysis. Cell Death Dis. 2021, 12, 551. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Ding, D.; Wang, Z.; Zhang, J.; Bai, C. The research in selenium and Kashin-Beck relationship for 20 years. Prophyl. Treat. Chin. Endemiol. 1997, 12, 18–21. [Google Scholar]
- Zhang, B.; Yang, L.; Wang, W.; Li, Y.; Li, H. Environmental selenium in the Kaschin-Beck disease area, Tibetan Plateau, China. Environ. Geochem. Health 2011, 33, 495–501. [Google Scholar] [CrossRef]
- Wu, H.; Ding, S.; Zhang, Q.; Ding, Q. Analysis of natural decreased regularity and corrected factor about Kashin-beck disease in Banma Country. Chin. J. Endemic. 2004, 23, 558–559. [Google Scholar]
- Becher, C.; Szuwart, T.; Ronstedt, P.; Ostermeier, S.; Skwara, A.; Fuchs-Winkelmann, S.; O Tibesku, C. Decrease in the expression of the type 1 PTH/PTHrP receptor (PTH1R) on chondrocytes in animals with osteoarthritis. J. Orthop. Surg. Res. 2010, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Tatarczuch, L.; Mirams, M. The skeleton: A multi-functional complex organ: The growth plate chondrocyte and en-dochondral ossification. J. Endocrinol. 2011, 211, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of Rate of Cartilage Differentiation by Indian Hedgehog and PTH-Related Protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Guo, X.; Zhang, R.; Wang, S.; Zuo, H.; Zhang, Z.; Geng, D.; Yu, Y.; Su, M. Effects of selenium and iodine deficiency on bone, cartilage growth plate and chondrocyte differentiation in two generations of rats. Osteoarthr. Cartil. 2007, 15, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Xian, L.; Crane, J.; Wen, C.; Hilton, M.; Lu, W.; Newman, P.; Cao, X. PTH receptor signaling in osteoblasts regulates endochondral vascu-larization in maintenance of postnatal growth plate. J. Bone Miner. Res. 2015, 30, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, M.; Liu, Q.; Zhang, H.; Zhang, J.; Lu, L.; Xie, M.; Chen, D.; Wang, M. Inhibition of Ihh Reverses Temporomandibular Joint Osteoarthritis via a PTH1R Signaling Dependent Mechanism. Int. J. Mol. Sci. 2019, 20, 3797. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Park, Y.; Lee, J.; An, H.J. In Vitro Study of Licorice on IL-1β-Induced Chondrocytes and In Silico Approach for Osteoarthritis. Pharmaceuticals 2021, 14, 1337. [Google Scholar] [CrossRef]
- Klinedinst, N.J.; Huang, W.; Nelson, A.K.; Resnick, B.; Renn, C.; Kane, M.A.; Dorsey, S.G. Inflammatory and Immune Protein Pathways Possible Mechanisms for Pain Following Walking in Knee Osteoarthritis. Nurs. Res. 2022, 71, 328–335. [Google Scholar] [CrossRef]
- Liang, J.; Wei, X.; Liu, Z.; Cao, D.; Tang, Y.; Zou, Z.; Zhou, C.; Lu, Y. Long noncoding RNA CYTOR in cancer: A TCGA data review. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 483, 227–233. [Google Scholar] [CrossRef]
- Hu, P.; Sun, F.; Ran, J.; Wu, L. Identify CRNDE and LINC00152 as the key lncRNAs in age-related degeneration of articular cartilage through comprehensive and integrative analysis. PeerJ 2019, 7, e7024. [Google Scholar] [CrossRef]
- Li, X.; Liao, Z.; Deng, Z.; Chen, N.; Zhao, L. Combining bulk and single-cell RNA-sequencing data to reveal gene expression pattern of chondrocytes in the osteoarthritic knee. Bioengineered 2021, 12, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, L.; Shen, H.; Hao, Q.; Fu, S.; Liu, X. Up-regulation of long non-coding RNA CYTOR induced by icariin promotes the viability and inhibits the apoptosis of chondrocytes. BMC Complement. Med. Ther. 2021, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Taheri, M.; Shojaei, S. A review on the role of MEG8 lncRNA in human disorders. Cancer Cell Int. 2022, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Jiang, L.; Huang, X.; Shang, H.; Gao, M.; You, W.; Tan, J.; Yan, H.; Sun, W. lncRNA MEG8 is downregulated in osteoarthritis and regulates chondrocyte cell proliferation, apoptosis and inflammation. Exp. Ther. Med. 2021, 22, 1153. [Google Scholar] [CrossRef]
- Xie, W.; Jiang, L.; Huang, X.; You, W.; Sun, W. Hsa_circ_00046621 accelerates the progression of osteoarthritis via the mi-croRNA-424-5p/VEGFA axis. Curr. Mol. Med. 2024, 24, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Duan, C.; Zhang, X.; Yao, D.; Si, G.; Gao, Y.; Gao, Z.; Umer, F.; Guo, X. RNA-seq analysis reveals different gene ontologies and pathways in rheumatoid arthritis and Kashin–Beck disease. Int. J. Rheum. Dis. 2018, 21, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Amhare, A.F.; Wang, L.; Lv, Y.; Deng, H.; Gao, H.; Guo, X.; Han, J.; Lammi, M.J. Proteomic analysis of knee cartilage reveals potential signaling pathways in pathological mechanism of Kashin-Beck disease compared with osteoarthritis. Sci. Rep. 2020, 10, 6824. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef]
- Feng, G.; Zhou, Y.; Yan, J.; Wang, Z.; Yang, Y.; Zhao, W.; Wang, N.; Lu, Z.; Chen, Y.; Jin, Q. Proteomic and N-glycoproteomic analyses of total subchondral bone protein in patients with primary knee osteoarthritis. J. Proteom. 2023, 280, 104896. [Google Scholar] [CrossRef]
- Huang, M.; Zhong, Z.; Lv, M.; Shu, J.; Tian, Q.; Chen, J. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget 2016, 7, 47186–47200. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Peffers, M.J.; Ormseth, M.J.; Jurisica, I.; Kapoor, M. The non-coding RNA interactome in joint health and disease. Nat. Rev. Rheumatol. 2021, 17, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Ren, G.; Day, C.R.; Zhao, K.; Chow, C.C.; Larson, D.R. Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity. Cell 2019, 176, 213–226.e218. [Google Scholar] [CrossRef]
- Maroni, P.; Gomarasca, M.; Lombardi, G. Long non-coding RNAs in bone metastasis: Progresses and perspectives as potential diagnostic and prognostic biomarkers. Front. Endocrinol. 2023, 14, 1156494. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Heilmeier, U.; Weilner, S.; Grillari, J. Circulating microRNAs as novel biomarkers for bone diseases-Complex sig-natures for multifactorial diseases? Mol. Cell. Endocrinol. 2016, 432, 83–95. [Google Scholar] [CrossRef] [PubMed]
- WS/T 207-2010; Diagnosis of Kashin-Beck Disease. National Health and Family Planning Commission of People’s Republic of China: Beijing, China, 2010.
- Yi, L. Determination of fusarium mycotoxins in corn and wheat from kaschin beck disease areas. Chin. J. Control Endem. Dis. 1992, 7, 71–75. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. 2011, 12, R72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-O.; Dong, R.; Zhang, Y.; Zhang, J.-L.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary Sequence-Mediated Exon Circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Decision letter: Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Zhang, Y.; Wen, Y.; Wang, W.; Wang, S.; Yang, T.; Shen, H.; Chen, X.; Tian, Q.; et al. Genome-wide copy number variation study and gene expression analysis identify ABI3BP as a susceptibility gene for Kashin–Beck disease. Hum. Genet. 2014, 133, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Liang, C.; Yang, X.; Li, P.; Liu, L.; Cheng, S.; Jia, Y.; Zhang, L.; Ma, M.; Qi, X.; et al. Genetic association scan of 32 osteoarthritis susceptibility genes identified TP63 associated with an endemic osteoarthritis, Kashin-Beck disease. Bone 2021, 150, 115997. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Haqqi, T.M. Interleukin-1β induced Stress Granules Sequester COX-2 mRNA and Regulates its Stability and Translation in Human OA Chondrocytes. Sci. Rep. 2016, 6, 27611. [Google Scholar] [CrossRef]
- Fleige, S.; Walf, V.; Huch, S.; Prgomet, C.; Sehm, J.; Pfaffl, M.W. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol. Lett. 2006, 28, 1601–1613. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, L.; Cheng, B.; Wei, W.; Liu, L.; Cheng, S.; Liu, H.; Jia, Y.; Wen, Y.; Zhang, F. Whole-Transcriptome Sequencing of Knee Joint Cartilage from Kashin–Beck Disease and Osteoarthritis Patients. Int. J. Mol. Sci. 2024, 25, 4348. https://doi.org/10.3390/ijms25084348
Han L, Cheng B, Wei W, Liu L, Cheng S, Liu H, Jia Y, Wen Y, Zhang F. Whole-Transcriptome Sequencing of Knee Joint Cartilage from Kashin–Beck Disease and Osteoarthritis Patients. International Journal of Molecular Sciences. 2024; 25(8):4348. https://doi.org/10.3390/ijms25084348
Chicago/Turabian StyleHan, Lixin, Bolun Cheng, Wenming Wei, Li Liu, Shiqiang Cheng, Huan Liu, Yumeng Jia, Yan Wen, and Feng Zhang. 2024. "Whole-Transcriptome Sequencing of Knee Joint Cartilage from Kashin–Beck Disease and Osteoarthritis Patients" International Journal of Molecular Sciences 25, no. 8: 4348. https://doi.org/10.3390/ijms25084348