

Smooth-Muscle-Cell-Specific Deletion of CD38 Protects Mice from AngII-Induced Abdominal Aortic Aneurysm through Inhibiting Vascular Remodeling
 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
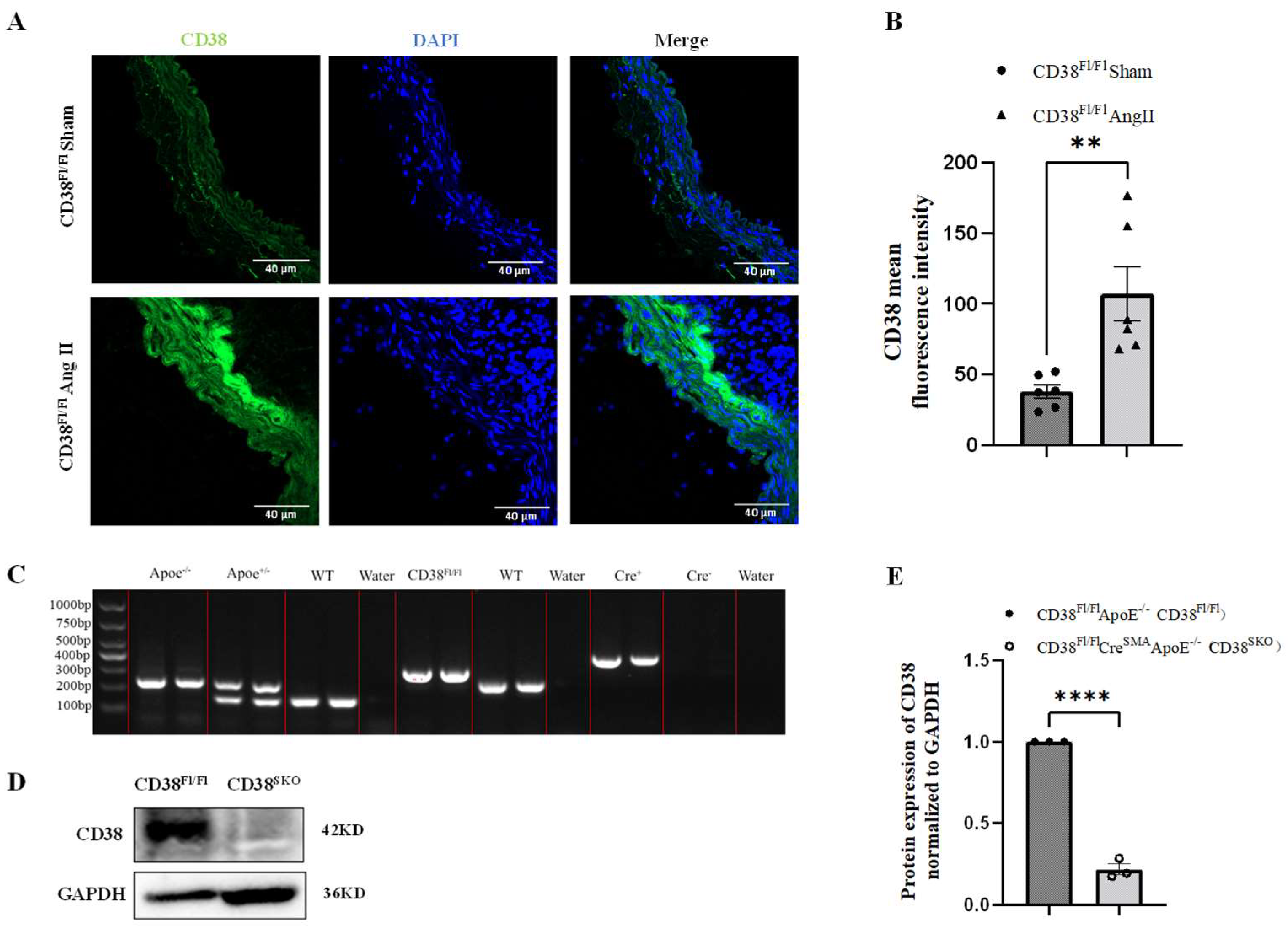
2.1. Generation and Identification of Smooth-Muscle-Cell-Specific CD38-Knockout Mice
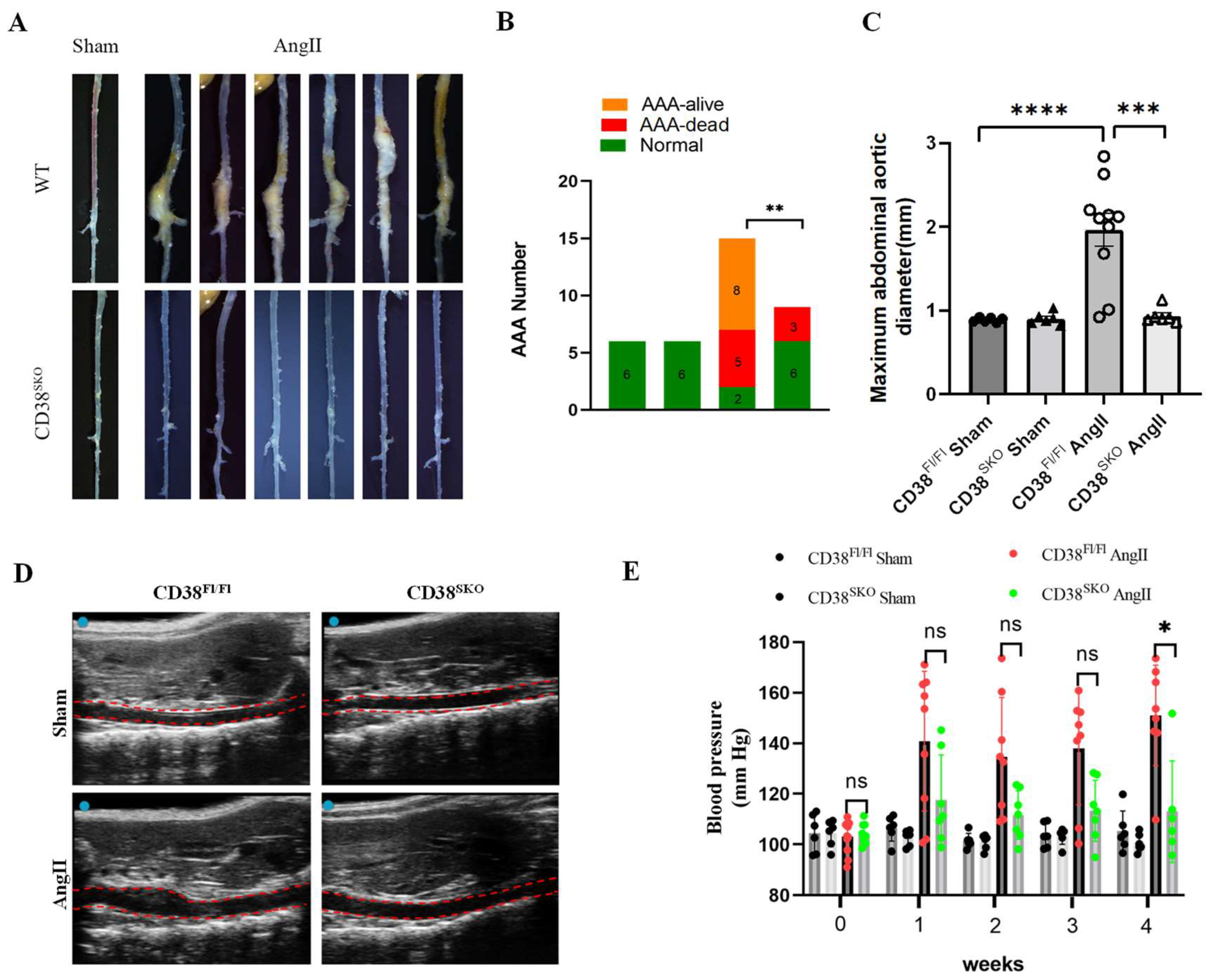
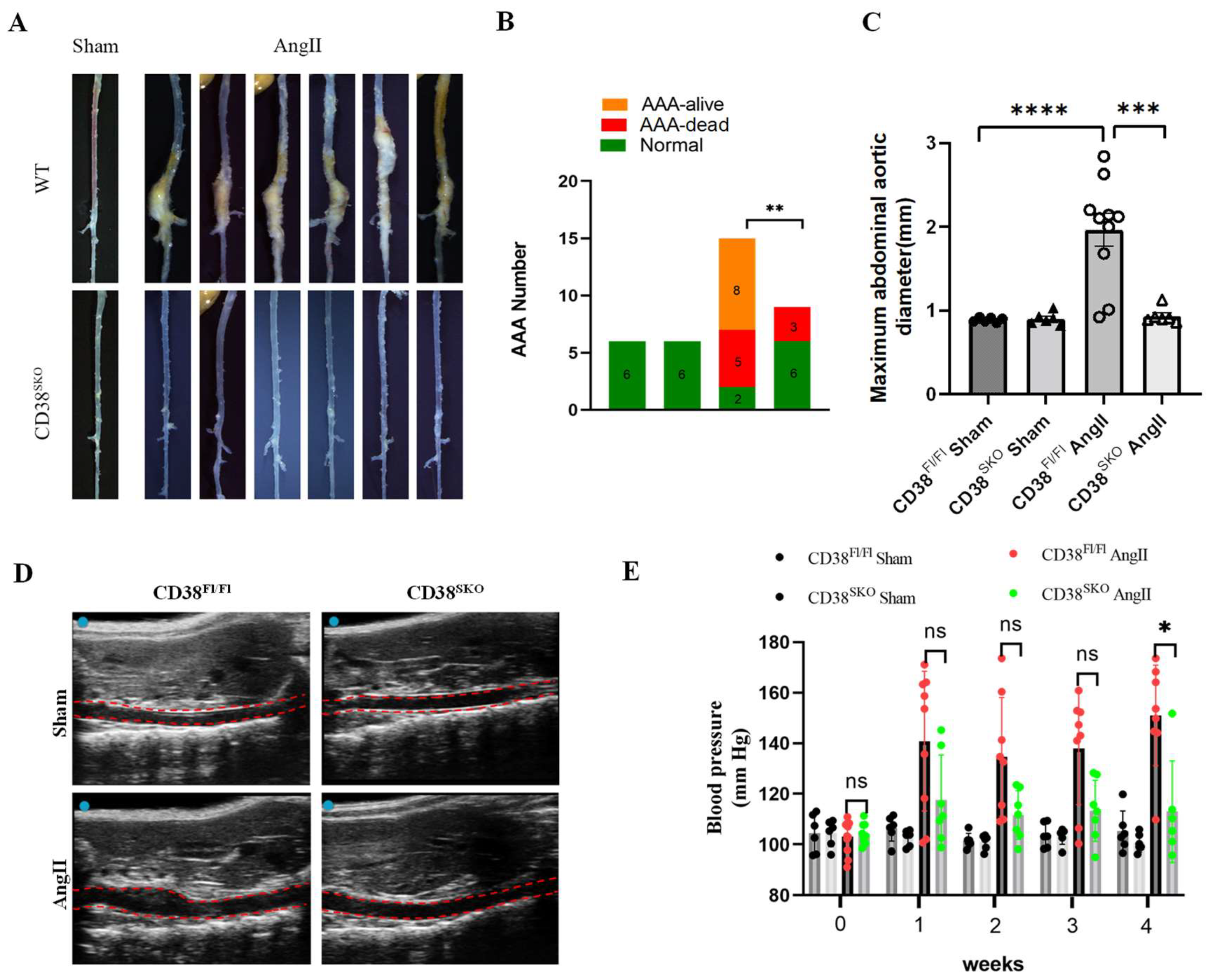
2.2. CD38 Deficiency in Smooth Muscle Cells (CD38SKO) Protected Mice from AngII-Induced AAA Formation
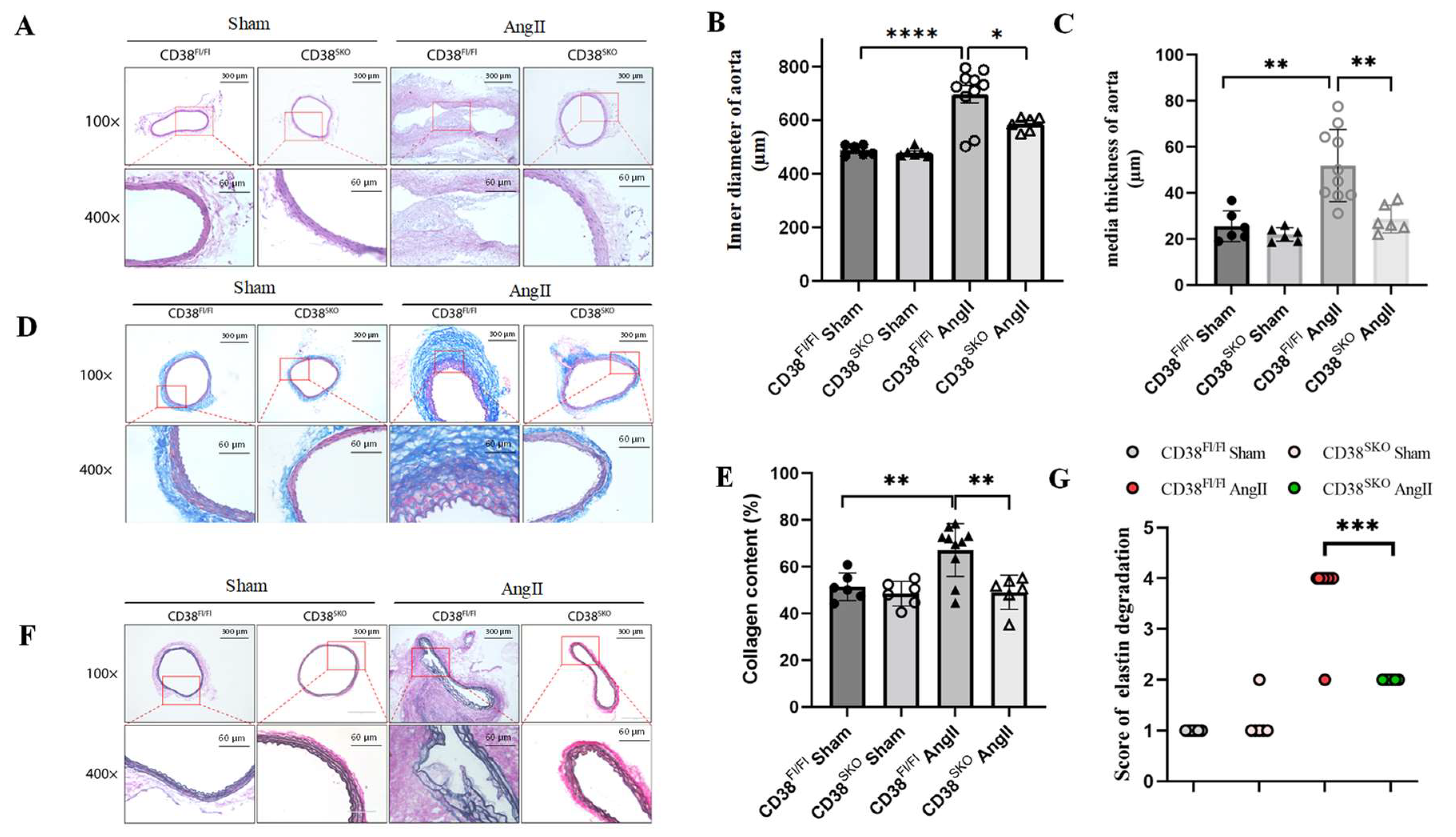
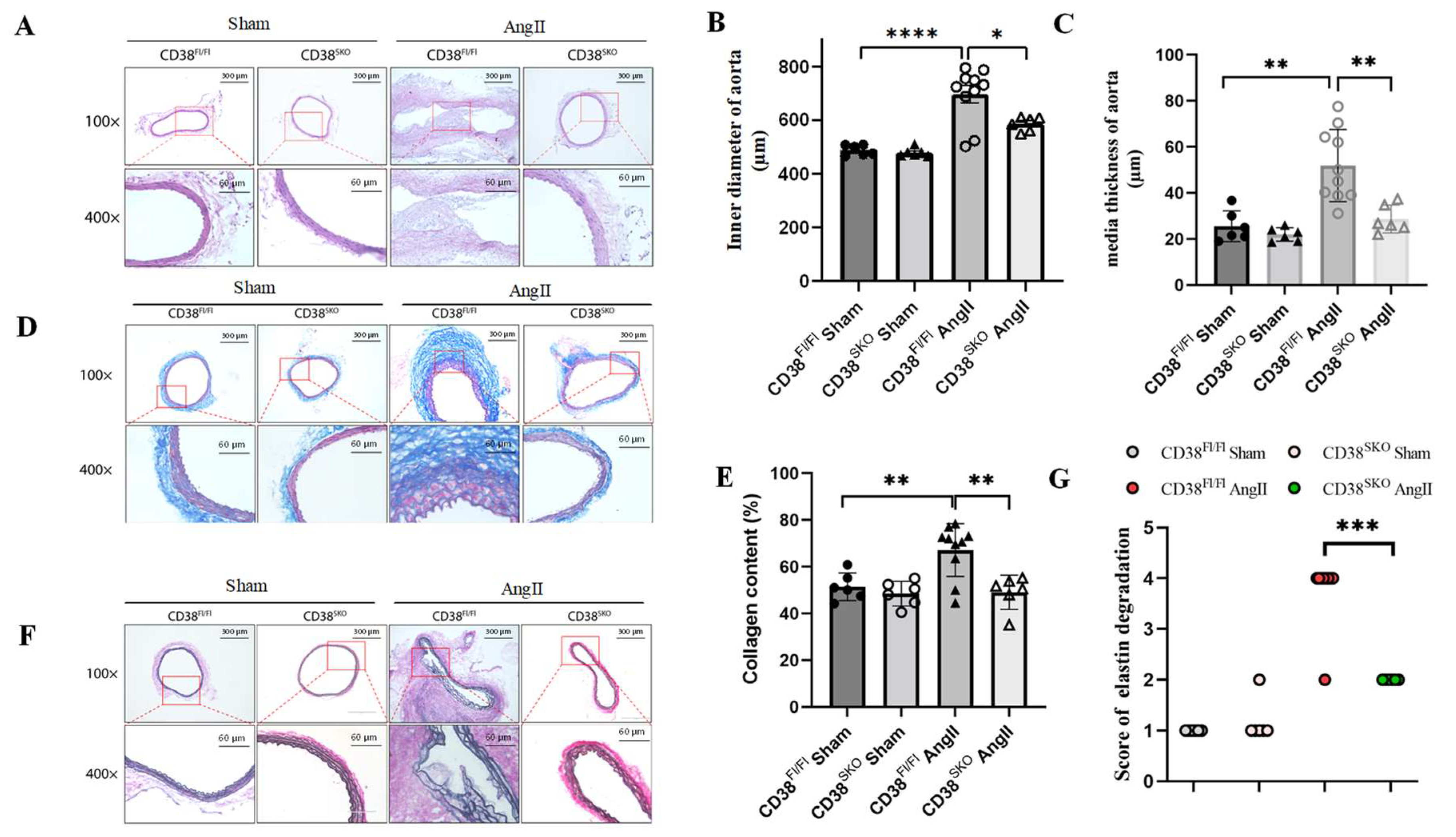
2.3. CD38SKO Mitigated Vascular Remodeling in Mouse AAA Models
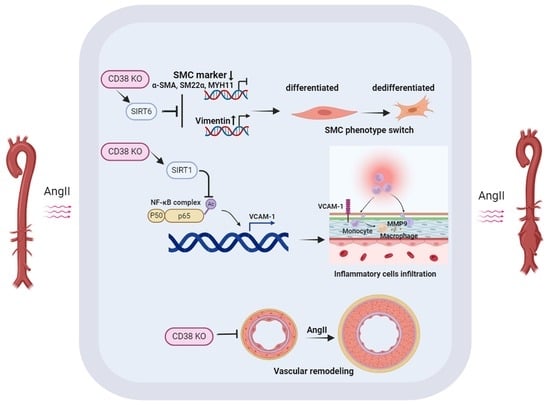
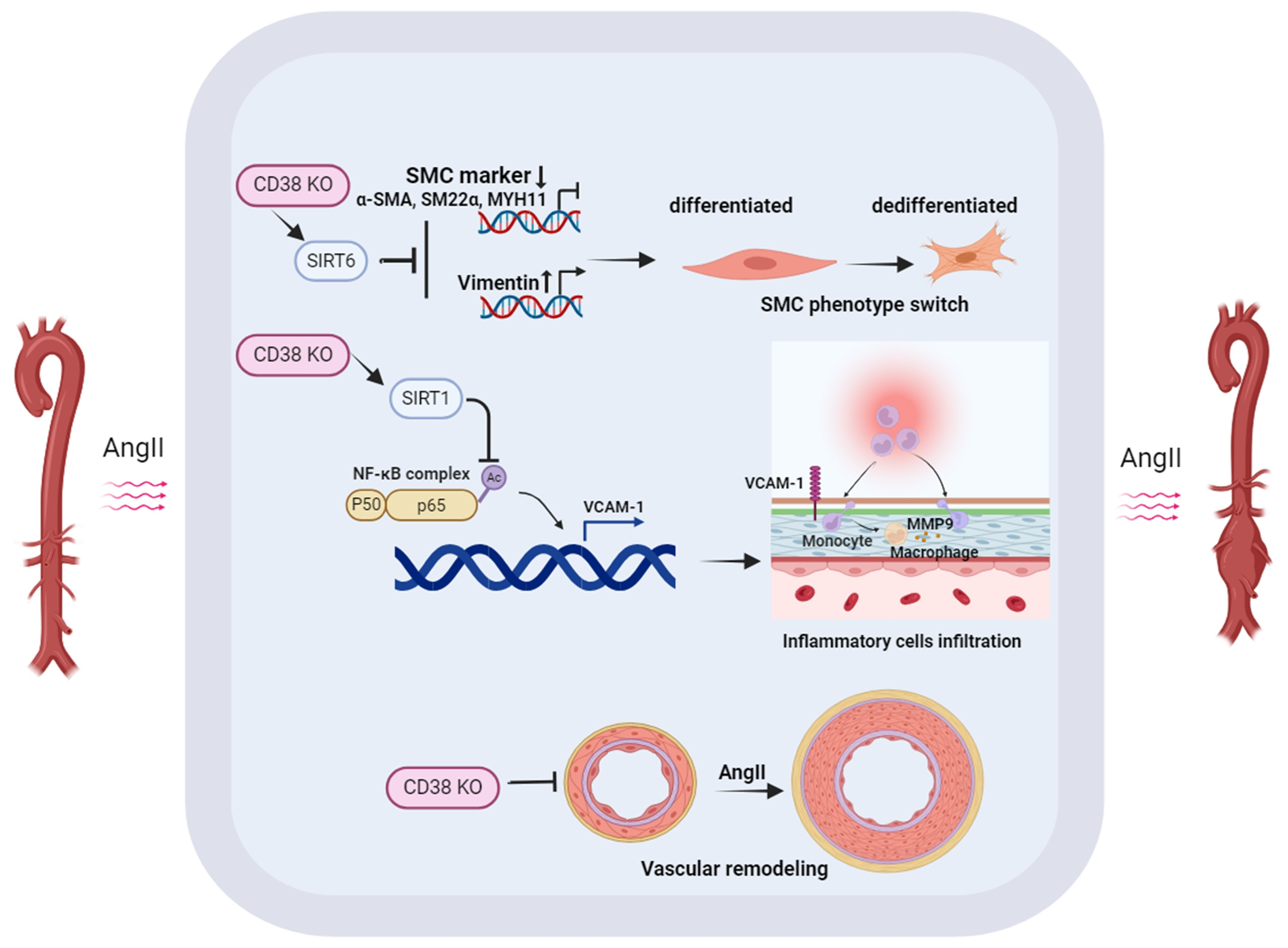
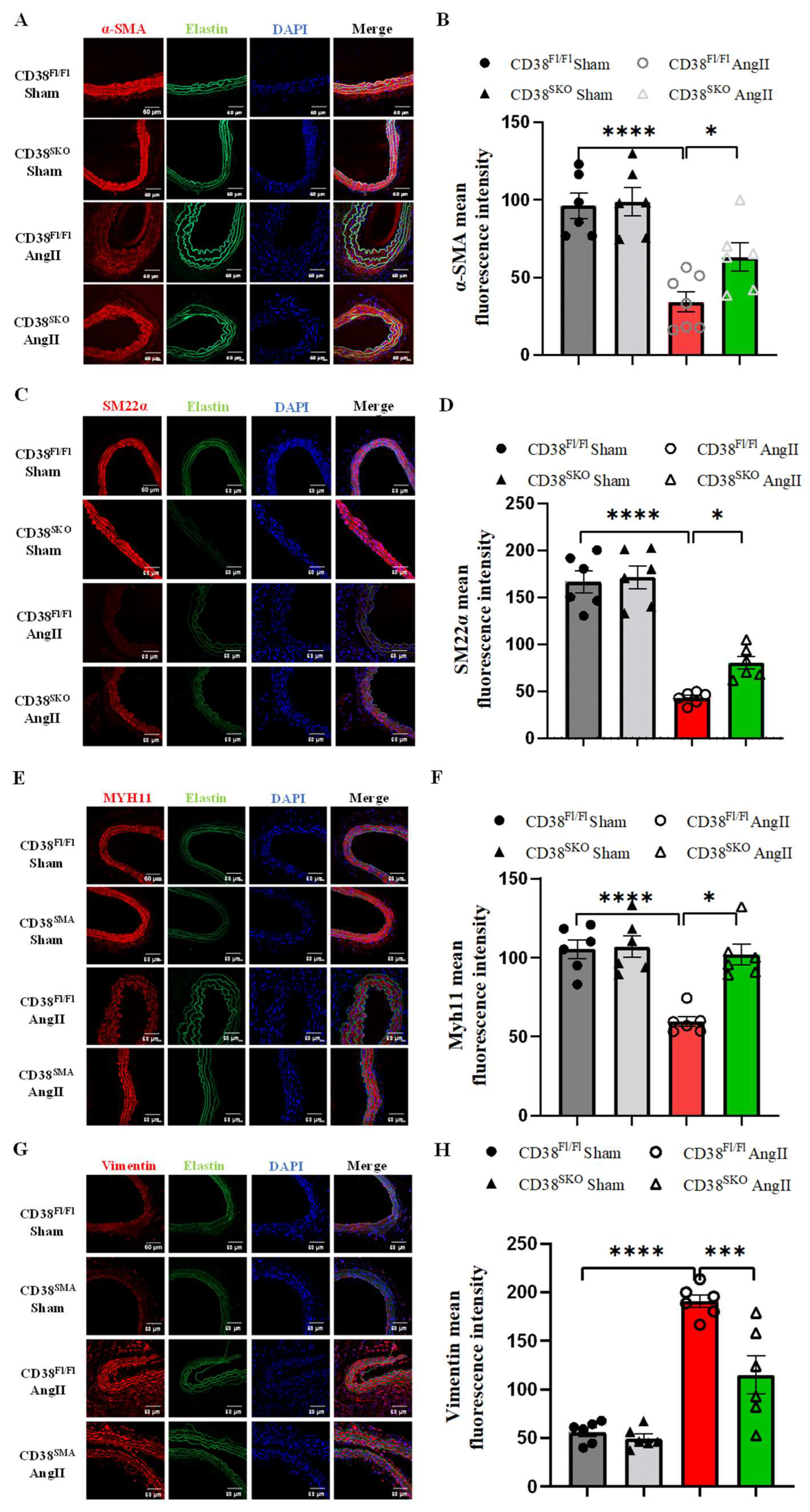
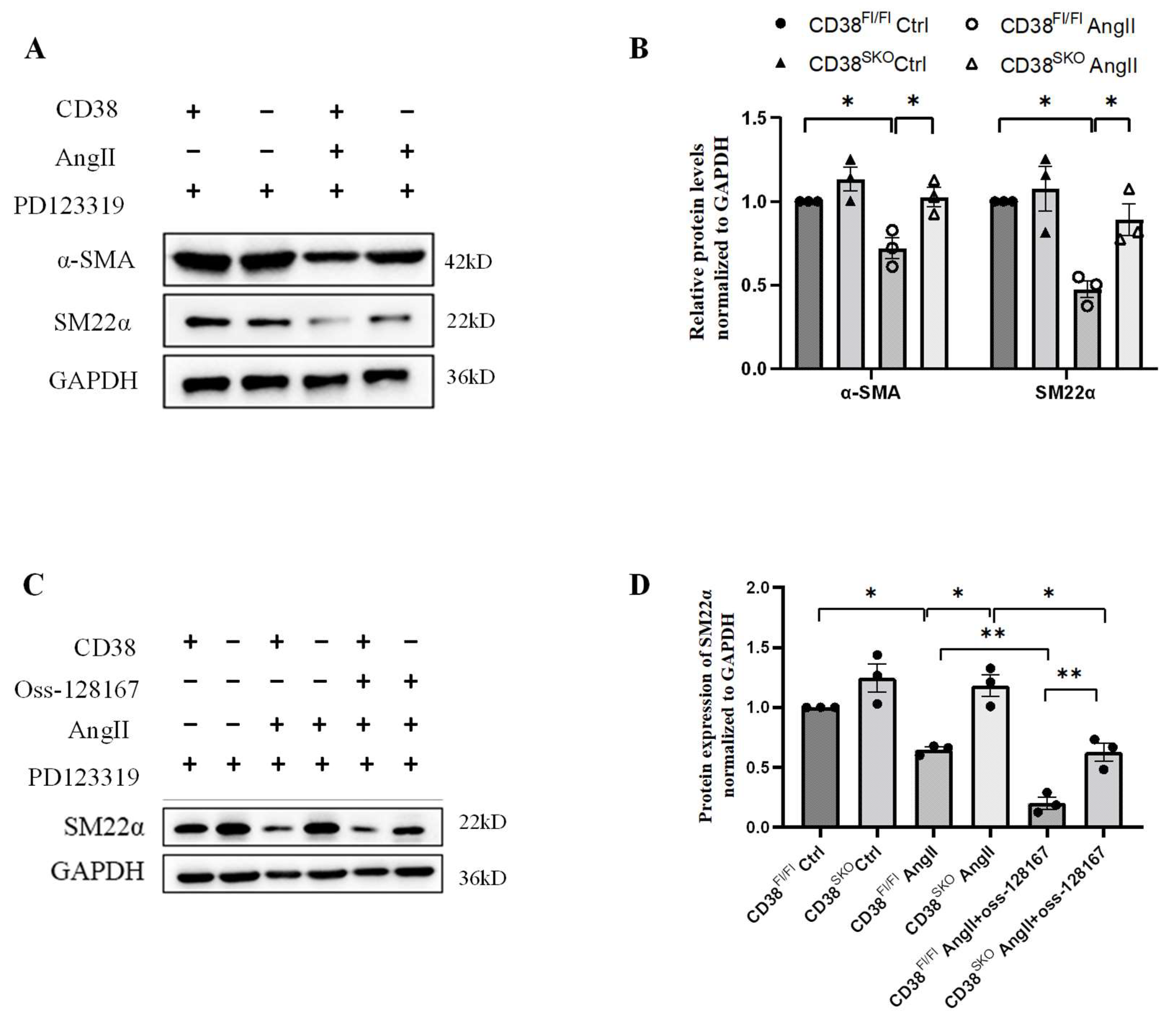
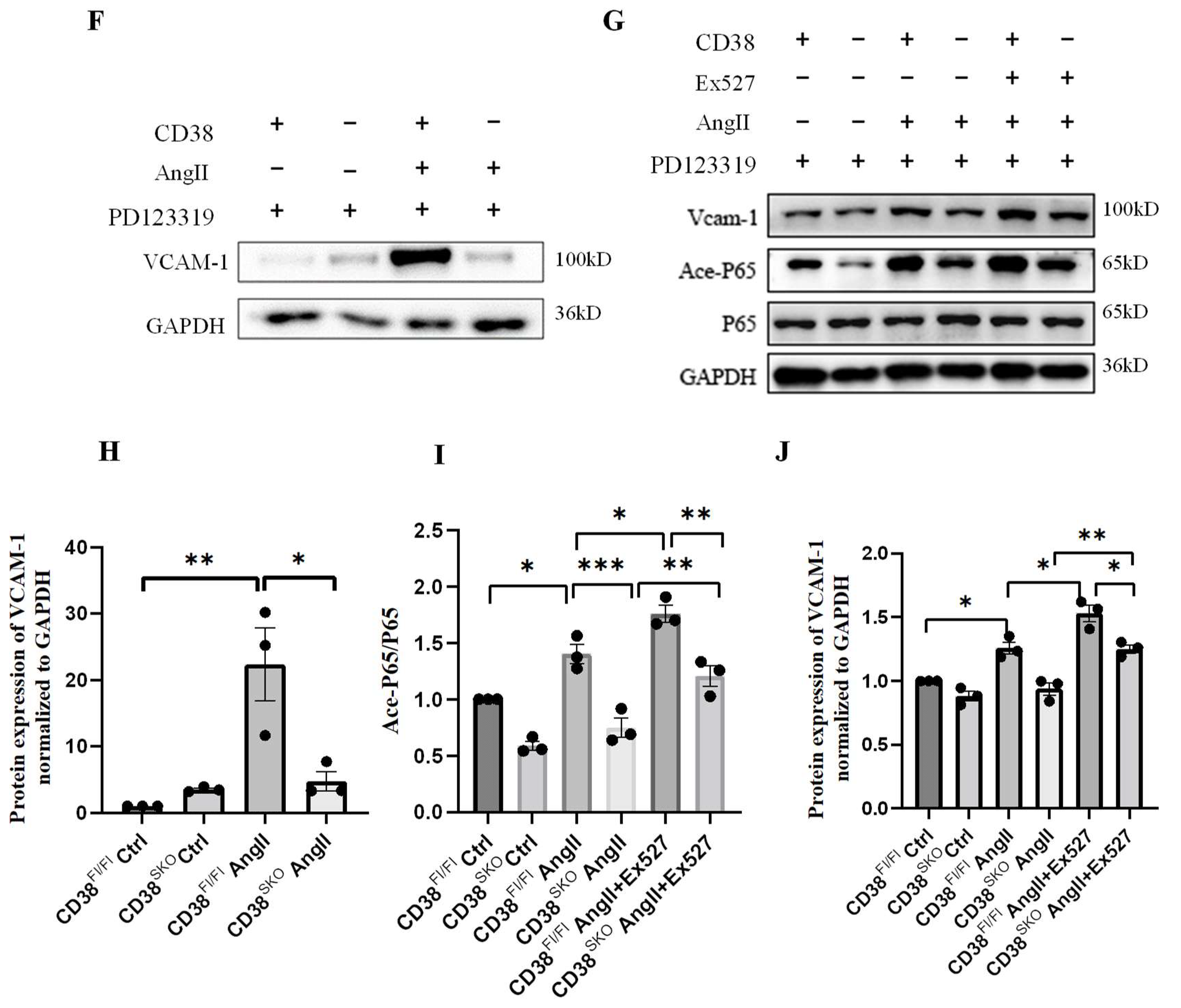
2.4. CD38SKO Attenuated the AngII-Induced Phenotype Switch of SMCs
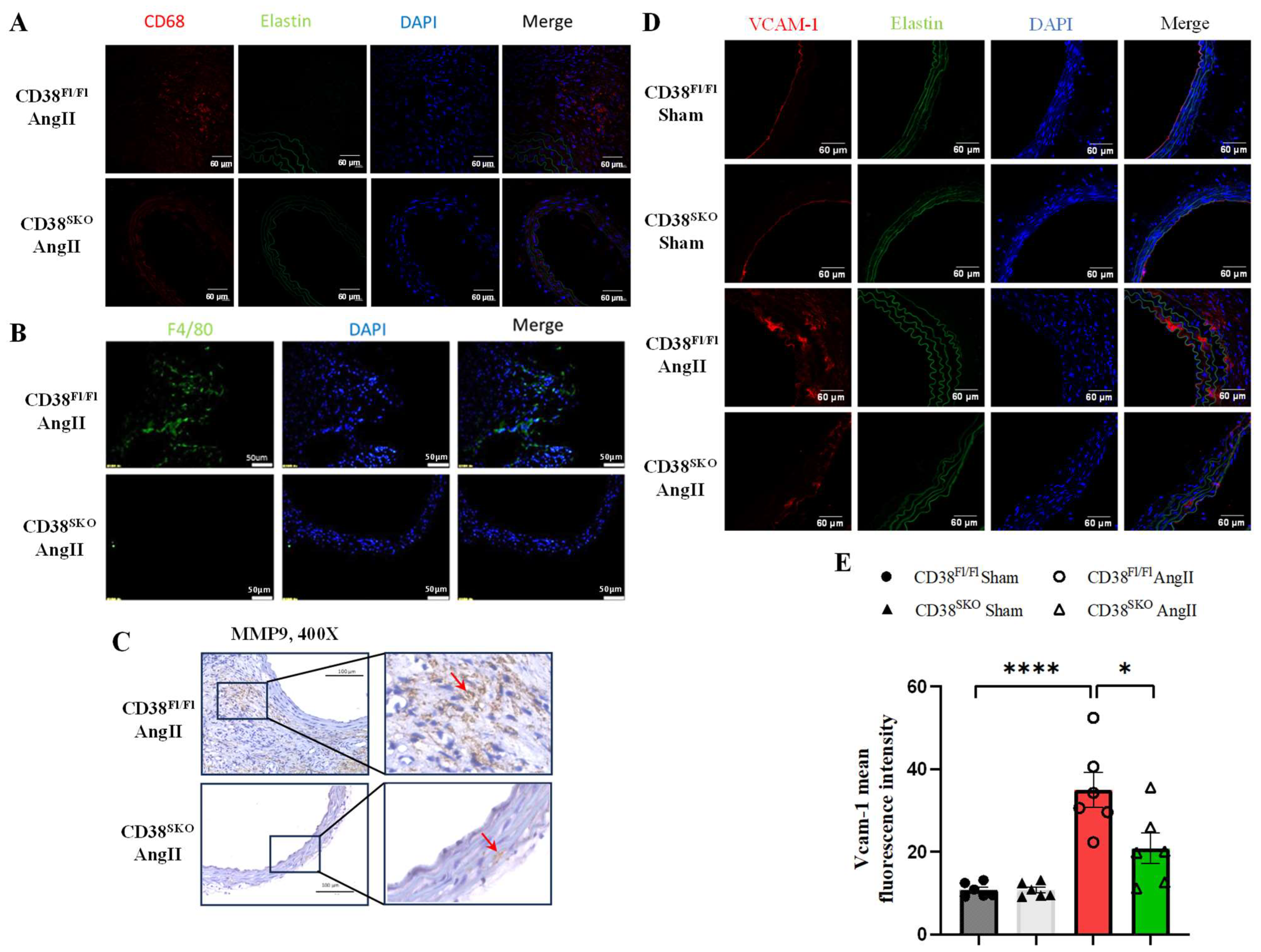
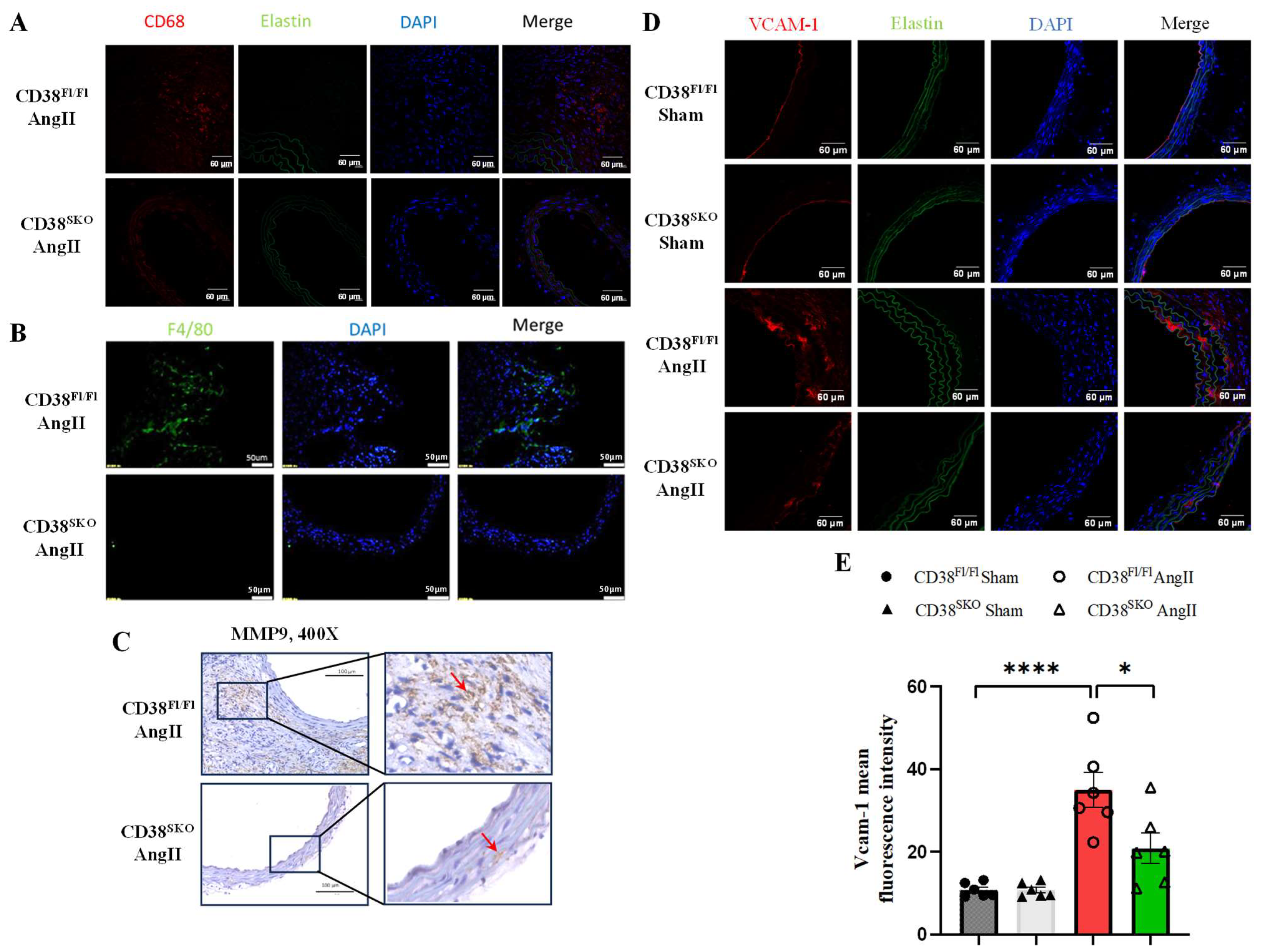
2.5. CD38SKO Reduced AngII-Induced Macrophage Infiltration and Inflammation in Aortas
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Cell Culture
4.3. AngII-Induced AAA Model
4.4. Blood Pressure Detection and Aortic Aneurysm Ultrasonography
4.5. Morphological and Histological Examination
4.6. Immunohistochemistry and Immunofluorescence
4.7. Western Blot
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahimtoola, S.H. Diagnosis and monitoring of abdominal aortic aneurysm: Current status and future prospects. Foreword. Curr. Probl. Cardiol. 2010, 35, 509. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Norman, P.E. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis 2011, 217, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ailawadi, G.; Eliason, J.L.; Upchurch, G.R., Jr. Current concepts in the pathogenesis of abdominal aortic aneurysm. J. Vasc. Surg. 2003, 38, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Aoki, H. Recent advances in pharmacotherapy development for abdominal aortic aneurysm. Int. J. Vasc. Med. 2012, 2012, 648167. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kong, W. Cellular signaling in Abdominal Aortic Aneurysm. Cell. Signal. 2020, 70, 109575. [Google Scholar] [CrossRef] [PubMed]
- Chaikof, E.L.; Dalman, R.L.; Eskandari, M.K.; Jackson, B.M.; Lee, W.A.; Mansour, M.A.; Mastracci, T.M.; Mell, M.; Murad, M.H.; Nguyen, L.L.; et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J. Vasc. Surg. 2018, 67, 2–77.e2. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Guo, D.C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Zhong, L.; He, X.; Si, X.; Wang, H.; Li, B.; Hu, Y.; Li, M.; Chen, X.; Liao, W.; Liao, Y.; et al. SM22alpha (Smooth Muscle 22alpha) Prevents Aortic Aneurysm Formation by Inhibiting Smooth Muscle Cell Phenotypic Switching Through Suppressing Reactive Oxygen Species/NF-kappaB (Nuclear Factor-kappaB). Arterioscler. Thromb. Vasc. Biol. 2019, 39, e10–e25. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Levey, R.H.; Schlossman, S.F. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. USA 1980, 77, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Escande, C.; White, T.A.; Thompson, M.; Soares, S.; Benech, J.C.; Chini, E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006, 349, 353–359. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, D.; Liu, J.; Chen, E.; Chen, C.; Liu, L.; Hu, H.; Guan, X.; Ma, W.; Zhang, Y.; et al. CD38 deficiency alleviates Ang II-induced vascular remodeling by inhibiting small extracellular vesicle-mediated vascular smooth muscle cell senescence in mice. Signal Transduct. Target. Ther. 2021, 6, 223. [Google Scholar] [CrossRef]
- Guan, X.H.; Liu, X.H.; Hong, X.; Zhao, N.; Xiao, Y.F.; Wang, L.F.; Tang, L.; Jiang, K.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Protects the Heart from Ischemia/Reperfusion Injury through Activating SIRT1/FOXOs-Mediated Antioxidative Stress Pathway. Oxid. Med. Cell Longev. 2016, 2016, 7410257. [Google Scholar] [CrossRef]
- Wang, L.F.; Li, Q.; Wen, K.; Zhao, Q.H.; Zhang, Y.T.; Zhao, J.L.; Ding, Q.; Guan, X.H.; Xiao, Y.F.; Deng, K.Y.; et al. CD38 Deficiency Alleviates Diabetic Cardiomyopathy by Coordinately Inhibiting Pyroptosis and Apoptosis. Int. J. Mol. Sci. 2023, 24, 16008. [Google Scholar] [CrossRef]
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Sharma, N.; Dev, R.; Ruiz-Rosado, J.D.; Partida-Sanchez, S.; Guerau-de-Arellano, M.; Dhakal, P.; Kuivaniemi, H.; Hans, C.P. Pharmacological inhibition of Notch signaling regresses pre-established abdominal aortic aneurysm. Sci. Rep. 2019, 9, 13458. [Google Scholar] [CrossRef]
- Ding, Y.N.; Wang, T.T.; Lv, S.J.; Tang, X.; Wei, Z.Y.; Yao, F.; Xu, H.S.; Chen, Y.N.; Wang, X.M.; Wang, H.Y.; et al. SIRT6 is an epigenetic repressor of thoracic aortic aneurysms via inhibiting inflammation and senescence. Signal Transduct. Target. Ther. 2023, 8, 255. [Google Scholar] [CrossRef]
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- McNeill, E.; Iqbal, A.J.; White, G.E.; Patel, J.; Greaves, D.R.; Channon, K.M. Hydrodynamic Gene Delivery of CC Chemokine Binding Fc Fusion Proteins to Target Acute Vascular Inflammation In Vivo. Sci. Rep. 2015, 5, 17404. [Google Scholar] [CrossRef] [PubMed]
- Rateri, D.L.; Howatt, D.A.; Moorleghen, J.J.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Prolonged infusion of angiotensin II in apoE(-/-) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol. 2011, 179, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, C.; Wang, W.; Liu, L.; Zhang, Q.; Zhang, J.; Wang, B.; Wang, S.; Hou, L.; Gao, C.; et al. PRDX2 Protects Against Atherosclerosis by Regulating the Phenotype and Function of the Vascular Smooth Muscle Cell. Front. Cardiovasc. Med. 2021, 8, 624796. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zheng, F.; Wu, N.; Zhu, G.Q.; Li, X.Z. Extracellular vesicles in vascular remodeling. Acta Pharmacol. Sin. 2022, 43, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun. Signal 2022, 20, 180. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef]
- Wang, S.; Jia, C. TRPV1 inhibits smooth muscle cell phenotype switching in a mouse model of abdominal aortic aneurysm. Channels 2020, 14, 59–68. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, K.; Liu, Z.; Xu, Q.; You, B.; Li, C.; Cao, J.; Zhou, H.; Li, X.; Chen, J.; et al. VPO1 Modulates Vascular Smooth Muscle Cell Phenotypic Switch by Activating Extracellular Signal-regulated Kinase 1/2 (ERK 1/2) in Abdominal Aortic Aneurysms. J. Am. Heart Assoc. 2018, 7, e010069. [Google Scholar] [CrossRef]
- Tummala, P.E.; Chen, X.L.; Sundell, C.L.; Laursen, J.B.; Hammes, C.P.; Alexander, R.W.; Harrison, D.G.; Medford, R.M. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation 1999, 100, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; An, Q.; Zhao, W.; Song, Y.; Tang, X.; Wang, J.; Chang, C.C.; Zhao, G.; Hsiai, T.; Fan, G.; et al. Distinct patterns of responses in endothelial cells and smooth muscle cells following vascular injury. JCI Insight 2022, 7, e153769. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Kaschina, E.; Namsolleck, P.; Unger, T. AT2 receptors in cardiovascular and renal diseases. Pharmacol. Res. 2017, 125 Pt A, 39–47. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Nong, Z.; Yin, H.; O’Neil, C.; Fox, S.; Balint, B.; Guo, L.; Leo, O.; Chu, M.W.A.; Gros, R.; et al. Nicotinamide Phosphoribosyltransferase in Smooth Muscle Cells Maintains Genome Integrity, Resists Aortic Medial Degeneration, and Is Suppressed in Human Thoracic Aortic Aneurysm Disease. Circ. Res. 2017, 120, 1889–1902. [Google Scholar] [CrossRef]
- Horimatsu, T.; Blomkalns, A.L.; Ogbi, M.; Moses, M.; Kim, D.; Patel, S.; Gilreath, N.; Reid, L.; Benson, T.W.; Pye, J.; et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: Role of NAD+/nicotinamide. Cardiovasc. Res. 2020, 116, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tu, Y.; Gao, Y.; Chen, H.; Liu, J.; Zheng, J. Smooth Muscle Sirtuin 1 Blocks Thoracic Aortic Aneurysm/Dissection Development in Mice. Cardiovasc. Drugs Ther. 2020, 34, 641–650. [Google Scholar] [CrossRef]
- Katz, D.J.; Stanley, J.C.; Zelenock, G.B. Gender differences in abdominal aortic aneurysm prevalence, treatment, and outcome. J. Vasc. Surg. 1997, 25, 561–568. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.-P.; Wang, Y.-K.; Wang, X.-Y.; Gong, L.-N.; Tan, H.-L.; Jiang, M.-X.; Wang, L.-F.; Yu, G.-H.; Deng, K.-Y.; Xin, H.-B. Smooth-Muscle-Cell-Specific Deletion of CD38 Protects Mice from AngII-Induced Abdominal Aortic Aneurysm through Inhibiting Vascular Remodeling. Int. J. Mol. Sci. 2024, 25, 4356. https://doi.org/10.3390/ijms25084356
Yu Z-P, Wang Y-K, Wang X-Y, Gong L-N, Tan H-L, Jiang M-X, Wang L-F, Yu G-H, Deng K-Y, Xin H-B. Smooth-Muscle-Cell-Specific Deletion of CD38 Protects Mice from AngII-Induced Abdominal Aortic Aneurysm through Inhibiting Vascular Remodeling. International Journal of Molecular Sciences. 2024; 25(8):4356. https://doi.org/10.3390/ijms25084356
Chicago/Turabian StyleYu, Zhen-Ping, Yi-Kai Wang, Xiao-Yu Wang, Li-Na Gong, Hui-Lan Tan, Mei-Xiu Jiang, Ling-Fang Wang, Guan-Hui Yu, Ke-Yu Deng, and Hong-Bo Xin. 2024. "Smooth-Muscle-Cell-Specific Deletion of CD38 Protects Mice from AngII-Induced Abdominal Aortic Aneurysm through Inhibiting Vascular Remodeling" International Journal of Molecular Sciences 25, no. 8: 4356. https://doi.org/10.3390/ijms25084356
APA StyleYu, Z.-P., Wang, Y.-K., Wang, X.-Y., Gong, L.-N., Tan, H.-L., Jiang, M.-X., Wang, L.-F., Yu, G.-H., Deng, K.-Y., & Xin, H.-B. (2024). Smooth-Muscle-Cell-Specific Deletion of CD38 Protects Mice from AngII-Induced Abdominal Aortic Aneurysm through Inhibiting Vascular Remodeling. International Journal of Molecular Sciences, 25(8), 4356. https://doi.org/10.3390/ijms25084356