1. Introduction
Receptor tyrosine kinases (RTKs) are a crucial family of proteins that play a key role in the regulation of various cell functions, including cell proliferation, differentiation, and survival [
1]. These transmembrane receptors are integral components of cellular signaling pathways that modulate a wide range of physiological processes [
1]. Dysregulation of RTKs, often caused by gain-of-function mutations, genomic amplifications, chromosomal rearrangements, or autocrine signaling mechanisms, has been implicated in the pathogenesis of numerous human cancers [
2,
3,
4]. This abnormal activation disrupts normal cell regulation, leading to uncontrolled cell growth and disease progression [
1,
5]. Aberrant signaling of RTKs has been identified in more than fifty human RTKs in twenty subfamilies, contributing to the complexity and diversity of their pathological roles [
1]. These dysfunctions are closely associated with the initiation and progression of various oncogenic and developmental disorders [
6,
7,
8,
9,
10,
11]. The dysregulation of RTK extends beyond oncology to neurodegeneration, psychiatric conditions such as depression and addiction, and genetic and developmental disorders [
5].
Misregulation of RTKs through mechanisms such as overexpression, mutations, and cell-autonomous ligand–receptor interactions after loss of cell polarity underscores the critical need for targeted therapeutic interventions [
5]. Pathological activation of RTKs is a key driver of tumorigenesis and metastasis, making RTKs a focal point for the development of targeted cancer therapies [
12,
13]. The emergence of small-molecule inhibitors and the approval of several of such agents for clinical use underscore the growing emphasis on RTK-targeted interventions in oncology [
1,
13]. RTKs are additionally involved in a variety of genetic and developmental disorders, including brain overgrowth syndromes that cause severe neurological impairments [
5]. The disruption of the RTK signaling pathways is also associated with defects in the assembly or function of the primary cilia, resulting in a group of conditions known as ciliopathies [
5]. Exploring RTKs in these contexts not only provides insight into their fundamental biological functions but also opens avenues for novel therapeutic approaches to address a wide range of diseases driven by RTK dysregulation [
2].
The human FGFR2 protein (Uniprot P21802 · FGFR2_HUMAN), a member of the fibroblast growth factor receptor (FGFR) family, plays a crucial role in the RTK signaling network [
14]. Structurally, FGFR2 shares the characteristic features of RTKs, including three immunoglobulin-like domains (Ig domains) in its extracellular region, a single-pass transmembrane domain, and an intracellular tyrosine kinase domain. Functionally, FGFR2 is involved in mediating cellular responses to fibroblast growth factors, influencing cell growth, survival, and differentiation. While mutations in human FGFR2, predominantly missense, occur across various domains of this protein, a significant portion of those with clinical implications are found in the FGFR2 tyrosine kinase domain, as this domain is central to the intracellular signaling cascade initiated upon binding of fibroblast growth factors to the extracellular domain of FGFR2. This dysregulation is associated with various cancers and developmental disorders, highlighting the importance of FGFR2 in the broader context of RTK-mediated diseases. The focus of this study is the FGFR2 tyrosine kinase domain. In the interest of convenience, we will refer to it from here on as FGFR2-TKD.
Mutations in FGFR2-TKD have been associated with craniosynostosis syndromes, such as Crouzon syndrome and Apert syndrome, highlighting the involvement of this receptor in skeletal disorders [
15,
16]. Mutations have been additionally implicated in promoting breast tumorigenicity by maintaining breast tumor-initiating cells [
17] as well as in gastric, lung, endometrial, colorectal, and pancreatic cancers and in the development of drug resistance highlighting the importance of FGFR2-TKD in malignant neoplasms [
14,
17,
18,
19,
20,
21,
22]. Elucidating the biological and clinical implications of aberrations/mutations in the broader FGFR family in paediatric and young adult cancers (and other disorders) is central for effective therapeutic treatment but currently challenging [
23], in part because identifying patients most likely to benefit from FGFR inhibition currently rests on identifying activating FGFR mutations.
In FGFR2, the functional dynamics is key to clinical implications and consists of two primary states of FGFR2-TKD: an inactive, unphosphorylated state and an active, phosphorylated state crucial for signaling. Evidence of such states can be found in experimentally resolved tertiary structures of the FGFR2-TKD in wildtype and variant forms. Such structures, deposited in the Protein Data Bank (PDB) [
24], show the receptor in its inactive and active structural states. These states are shown in
Figure 1; the main structural alteration visually observed is that in the cytoplasmic region of FGFR2 (the activation loop colored in yellow).
Understanding the structural dynamics of FGFR2 is believed to be essential to the elucidating role of mutations in the prognosis of the disease, as well as in the development of targeted therapeutic interventions [
25]. Hence, this paper formulates and seeks to address key questions on (i) how and to what extent do mutations in FGFR2-TKD impact the structural dynamics, (ii) how does the dynamics relate to functional dynamics, and (iii) how does mutation-impacted structural dynamics percolate to functional dynamics and then to potentially distinct disorders?
Contemporary predictive models of functional dynamics have traditionally focused on amino acid sequences alone, overlooking the critical role of the tertiary structure and therefore structural dynamics [
26]. In this paper we take an integrative approach to connecting sequence variations to functional dynamics and disorders through structural dynamics. In particular, we utilize all known structures (deposited in the PDB) of FGFR2-TKD to capture and characterize the structural dynamics. Based on the principle of conformation selection [
27,
28], which in summary states that mutations alter the population probabilities of particular structures but do not remove from or add to the structure space, and a body of work in the Shehu laboratory capturing and characterizing the structure space of mutation-rich receptors [
29,
30,
31,
32,
33,
34,
35,
36], we seek to understand the biological and clinical implications of FGFR2-TKD mutations through a characterization of structural dynamics. In this paper we summarize the structure space documented in the PDB for FGFR2-TKD (over wildtype and variants) through machine learning methodologies that can capture both linear and nonlinear dynamics. The overlay of such dynamics over the sequence space allows us to identify regions that connect structure to function and provide insight on the functional implications of variant forms of FGFR2. In addition, connecting particular variants to diseases in which certain mutations are implicated allows us to identify disorder-specific signatures of dynamics, thus connecting structural and functional dynamics with disease.
Figure 1.
Structures of inactive and active forms of FGFR2-TKD. Panel (
a) displays inactive FGFR2-TKD, highlighting the structural readiness of the kinase for activation, including the kinase activation loop and key unphosphorylated tyrosine residues. Color coding identifies the various known functional regions (see
Table 1: nucleotide binding loop in gray, Alpha C helix in pink, kinase hinge in purple, kinase insert in blue, catalytic pocket in black, and activation loop in yellow). Panel (
b) shows the active and phosphorylated FGFR2-TKD. The active structure is shown to accompany phosphorylation, enhancing substrate recognition and catalytic activity. The activation loop is distinct in the inactive and active structures, colored in yellow. The transition from an inactive to an active state is fundamental in various cellular processes, including growth and tissue repair.
Figure 1.
Structures of inactive and active forms of FGFR2-TKD. Panel (
a) displays inactive FGFR2-TKD, highlighting the structural readiness of the kinase for activation, including the kinase activation loop and key unphosphorylated tyrosine residues. Color coding identifies the various known functional regions (see
Table 1: nucleotide binding loop in gray, Alpha C helix in pink, kinase hinge in purple, kinase insert in blue, catalytic pocket in black, and activation loop in yellow). Panel (
b) shows the active and phosphorylated FGFR2-TKD. The active structure is shown to accompany phosphorylation, enhancing substrate recognition and catalytic activity. The activation loop is distinct in the inactive and active structures, colored in yellow. The transition from an inactive to an active state is fundamental in various cellular processes, including growth and tissue repair.
Table 1.
Distilling literature in [
37,
38,
39], we identify the following functional regions: the nucleotide-binding loop, the Alpha C helix at the kinase N-lobe, the gate keeper, the kinase hinge (and its vicinity), the kinase insert, the catalytic pocket, the Alpha-C tether, and the activation-loop region. Among these, the activation-loop region is reported as the most critical for activation, especially the residues around position 659. In our collection of tertiary structure data from the PDB, we have removed chains missing any of these important regions. For more details, refer to
Table 2.
Table 1.
Distilling literature in [
37,
38,
39], we identify the following functional regions: the nucleotide-binding loop, the Alpha C helix at the kinase N-lobe, the gate keeper, the kinase hinge (and its vicinity), the kinase insert, the catalytic pocket, the Alpha-C tether, and the activation-loop region. Among these, the activation-loop region is reported as the most critical for activation, especially the residues around position 659. In our collection of tertiary structure data from the PDB, we have removed chains missing any of these important regions. For more details, refer to
Table 2.
| Symbol | Region Name | Region Range |
|---|
| B | Nucleotide-binding loop | 480–490 |
| N | Alpha C helix at the kinase N-lobe | 525–539 |
| G | Gate keeper | 564 |
| H | Kinase hinge and its vicinity | 566–571, 549, 565 |
| K | Kinase insert | 579–599 |
| C | Catalytic pocket | 620–630 |
| T | Alpha-C tether | 650 |
| A | Activation loop | 643–649, 651–664 |
| O | Others | - |
Table 2.
Categorization of FGFR2-TKD Segment Structures by Activation State. The table provides a comprehensive categorization of 29 distinct tertiary structures of the cytoplasmic segment of FGFR2-TKD kinase sourced from the Protein Data Bank (PDB). These structures are classified into four primary activation states: active, inactive, intermediate, and unknown. The “unknown” category refers to structures whose activation status remains ambiguous because of interactions with inhibitory compounds, reflecting ongoing debates in the literature.
Table 2.
Categorization of FGFR2-TKD Segment Structures by Activation State. The table provides a comprehensive categorization of 29 distinct tertiary structures of the cytoplasmic segment of FGFR2-TKD kinase sourced from the Protein Data Bank (PDB). These structures are classified into four primary activation states: active, inactive, intermediate, and unknown. The “unknown” category refers to structures whose activation status remains ambiguous because of interactions with inhibitory compounds, reflecting ongoing debates in the literature.
| PDB ID (No. of Residue) | Status | Chains Removed |
|---|
| 1GJO (316) | Inactive/WT | - |
| 1OEC (316) | Inactive/WT | - |
| 2PSQ (370) | Inactive/WT | - |
| 2PVF (334) | Active/WT | - |
| 3RI1 (313) | Inactive/WT | - |
| 2PVY (324) | K659N/Active/A | removed, Missing residue 659 |
| 2PWL (324) | N549H/Active/K | - |
| 2PY3 (324) | E565G/Active/K | - |
| 2PZ5 (324) | N549T/Active/K | - |
| 2PZP (324) | K526E/Active/N | - |
| 2PZR (324) | K641R/Active/K | - |
| 2Q0B (324) | E565A/Active/K | - |
| 3B2T (311) | A628T/Active/C | - |
| 3CLY (334) | C491A/Unknown/O | - |
| 4J95 (324) | K659N/Active/A | removed, Missing residue 654, 659 |
| 4J96 (324) | K659M/Active/A | - |
| 4J97 (324) | K659E/Active/A | - |
| 4J98 (324) | K659Q/Active/A | - |
| 4J99 (324) | K659T/Active/A | removed, Missing residue 653 |
| 5EG3 (334) | Multiple/Unknown/O | - |
| 5UGL (324) | D650V/Active/T | removed, Missing residue 659 |
| 5UGX (324) | E565A/D650V/Active/HT | removed, Missing residue 659 |
| 5UHN (324) | E565A/N549H/Active/H | removed, Missing residue 659 |
| 5UI0 (324) | E565A/K659M/Active/HA | - |
| 6LVK (313) | Unknown/WT | - |
| 6LVL (313) | Unknown/WT | - |
| 6V6Q (411) | Multiple/Intermediate/O | removed, Missing residue 659 |
| 7KIA (308) | V564F/Unknown/G | - |
| 7KIE (308) | V564F/Unknown/G | - |
| 7OZY | 2cMissing .PDB file | |
| 8E1X | The reference paper information does not match with PDB structure. |
Another important contribution of this work is the enrichment of the analysis with computationally predicted tertiary structures; several FGFR2-TKD variants lack PDB-deposited tertiary structures. Utilizing and evaluating several methodologies, including AlphaFold2 and AlphaMissense, allows us to include in our analysis predicted tertiary structures. Connecting the structural and functional dynamics to disease then additionally allows us to elucidate potential functional and clinical implications of novel variants with no experimentally resolved structures.
The rest of this paper is organized as follows. In
Section 4, we describe the methodological approach summarized above to connect FGFR2-TKD mutations to structural and functional dynamics and disease.
Section 2 presents our findings. The paper concludes in
Section 3 with a discussion of the major findings and future avenues of inquiry.
2. Results
We now embark on a comprehensive analysis, with a specific focus on the cytoplasmic region of FGFR2. This effort is rooted in the pioneering work of Chen et al., who provided valuable information on the molecular architecture of FGFR2 [
37,
38,
39]. Our distillation of this literature reveals seven critical functional regions listed in
Table 1, each playing a unique role in FGFR2 activity and regulation. These regions include the nucleotide binding loop (amino-acid residues 480–490), essential for ATP binding and energy transfer; the Alpha C helix (residues 525–539) in the kinase N lobe, which contributes to structural stability; the gate keeper residue at position 564, crucial for substrate specificity; the kinase hinge region (566–571) and its vicinity, which form a pivotal junction for domain movement; the kinase insert region, a segment known for its variability among kinases; the catalytic pocket (620–630), the site of enzymatic activity; the Alpha-C tether at position 650, a key element in maintaining the inactive state; and the activation loop (643–649, 651–664), which controls the transition between the active and inactive states. It is worth noting that the molecular brake is a key autoinhibitory mechanism predominantly located in the kinase hinge region of receptor tyrosine kinases (RTKs) [
38]. It regulates kinase activity through a network of hydrogen bonds among three crucial amino acids, mediating autoinhibition when engaged and increasing kinase activity when disrupted by pathogenic mutations. This mechanism is directly linked with the development of various cancers and genetic disorders. Key functional regions affected by the molecular brake include: the kinase hinge region, where residues like E565, N549, and K641 form the core components of the brake; the Alpha C helix in the kinase’s N-lobe, which indirectly influences the molecular brake’s function, exemplified by the K526E mutation that creates new hydrogen bonds affecting the hinge area; and the activation loop, which contains multiple phosphorylation sites and whose conformation is directly impacted by the state of the molecular brake, indirectly regulating kinase activity through the stabilization of the A loop.
As detailed in
Table 2, the PDB-deposited structures encompass a wide range of known states, thus offering a valuable dataset for our investigation. Our dataset not only incorporates these core structures but also includes an array of single-point mutations. These are key to understanding the alterations in the structural and functional status of FGFR2. The dataset is further enhanced by including structures with multiple point mutations, broadening our scope to explore a wide range of structural modifications. This is instrumental in examining how mutations affects the functional characteristics of the FGFR2 kinase.
In addition to linking structural dynamics to function, we also aim to obtain clinical implications of mutation-impacted dynamics in disease. We incorporate data from several databases, such as the Online Mendelian Inheritance in Man (OMIM), the Catalog of Somatic Mutations in Cancer (COSMIC), and the Human Gene Mutation Database (HGMD). Building on these databases, we have curated known disease categories and specific disorders under each category for FGFR2 variants with known and unknown structures, as listed in
Table 3. Using these categories and diseases as labels, we analyze organizations of known (and predicted) tertiary structures of FGFR2-TKD wildtype and variants in a few dimensions that permit visualization of the structure space and so relate structural dynamics with disease.
2.1. From Structure to Function to Disease Linking Structure to Function to Disease for FGFR2 Variants with Known Structural Characterization
2.1.1. Extracting Structural Dynamics
As described in
Section 4, what we obtain from PCA are vectors/axes of a new space that that has been oriented to maximally capture the structural variation in a few dimensions. These vectors are the PCs. As also described in
Section 4, the eigenvalue obtained with each PC from PCA measures the amount of data variation along that PC, and this variation can be employed to understand whether a few dimensions (a few PCs) are all that is needed to capture most of the variation in the data, that is, whether an intrinsic organization/dynamics emerges from the data in a few dimensions (as opposed to the close to 800 dimensions in the Cartesian space of CA traces of collected FGFR2-TKD tertiary structures).
Figure 2 relates the cumulative variance (measured as related in
Section 4) as one includes PCs in the order of variation that they capture and shows the following: the first PC captures
of the structural variation, an indication that the structural variations are dominated by few concerted motions (in the PC space); with two PCs, one gets to
of the variation, and three PCs capture
of the variation. These results indicate that the top three PCs are meaningful and that focusing on these three alone misses less than
of the variation.
Figure 2 also shows that only six PCs are needed to capture
of the structural variation; this level of efficiency in variance interpretation, particularly for an atomic distance matrix comprised of close to 800 dimensions (
coordinates for the CA atoms), underscores the robustness and practicality of PCA.
We can further evaluate the effectiveness of PCA in exposing the intrinsic structural dynamics of FGFR2-TKD by leveraging its high interpretability. As related in
Section 4, we can “reconstruct” traces in the Cartesian coordinate space from projections on
i PCs, where
i can vary from 1 to higher values. Given the high variance captured by the top six PCs, we only focus on these in this exercise. In particular, we carry out the following exercise: Given a CA trace corresponding to a tertiary structure in our collected structures for FGFR2-TKD, we can obtain its projections over the top six PCs. So, each trace corresponds to a six-dimensional point in the space of the six PCs. If we zero out all but the first coordinate (the projection over PC1), each trace is now a one-dimensional point in the PC space. Per the equation in
Section 4, inverting the projection and adding the average trace, we can obtain a corresponding (reconstructed) CA trace with each one-dimensional point. Measuring the RMSD between this reconstructed trace and the original one we can understand the reconstruction error resulting from ignoring the other five PCs. We can repeat this process now zeroing out all but the first two PCs, then all but the first three PCs, and so on.
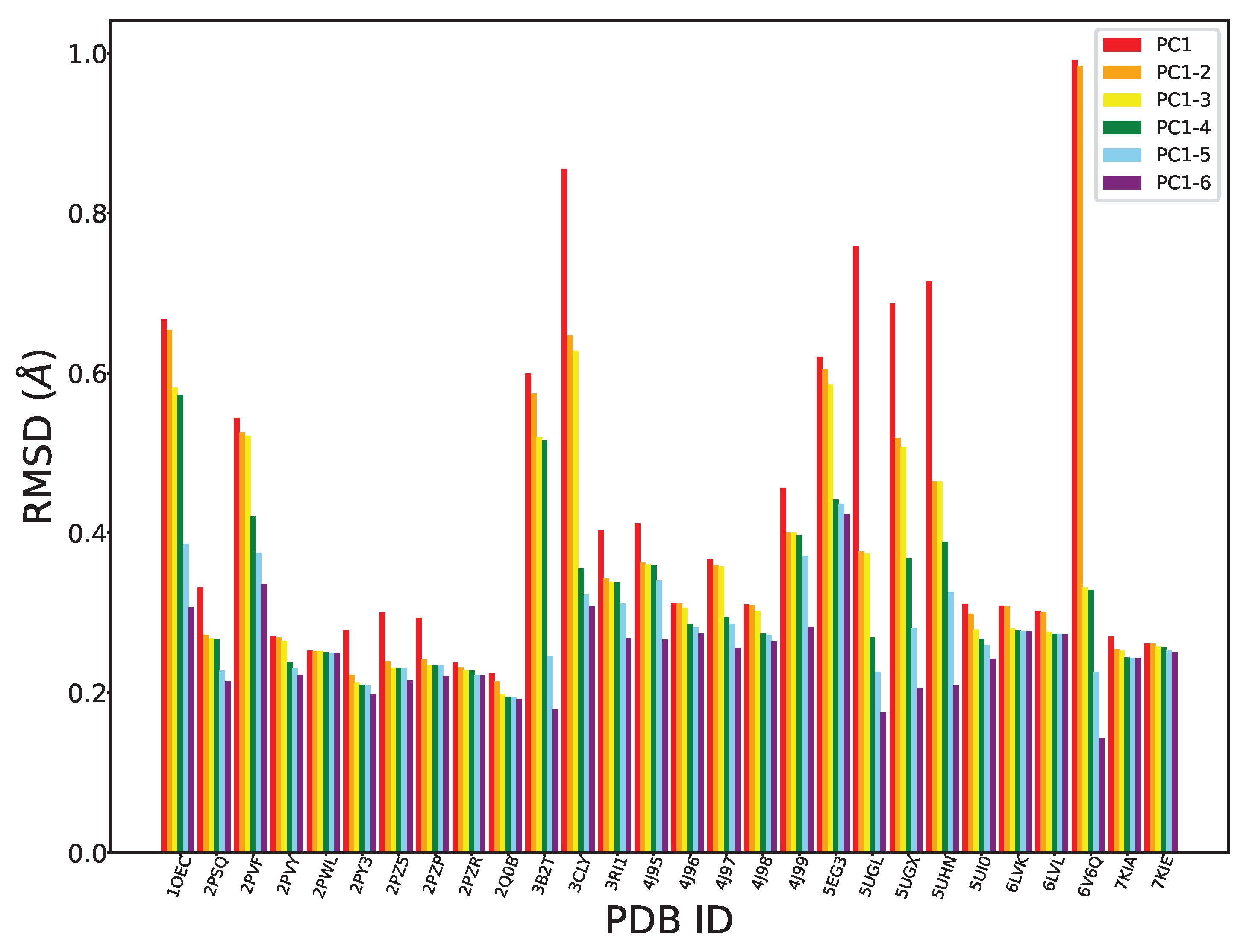
Figure 3 shows the reconstruction error (measured through the RMSD between the reconstructed and the original trace) as we include more PCs.
As expected, given that the top six PCs already capture more than 90% of the variance, the range for the RMSD-based reconstruction error is in the Å. Also as expected, the RMSD decreases with an increasing number of PCs. Moreover, our findings reveal a significant decrease when considering the first three PCs, further suggesting that these three top components capture most of the structural variability in our dataset. It is worth noting that some tertiary structures (PDB ID 3CLY and PDB ID 6V6Q) show significant deviation (higher RMSDs) from this trend, representing outliers. The RMSD values for the structures with PDB IDs 3B2T and 6V6Q are elevated in the context of the first and second PCs when contrasted with other structures. This discrepancy can be attributed to the presence of additional structural elements or motifs within 3B2T and 6V6Q, potentially encompassing unknown functional regions or variations. These elements are not adequately captured by PCA and so are not reflected in the variation captured by the first two PCs.
The above quantitative results suggest four main findings: (i) there is an intrinsic structural dynamics of FGFR2-TKD, and it occurs in three major modes/dimensions; (ii) PCA is an effective methodology for exposing this dynamics; (iii) inspection of each of these three PCs and the CA atoms that they impact ought to now map this dynamics to the functional regions; and (iv) projection of the traces onto a few PCs ought to be informative and, when visualized, may provide useful information on potential co-localization of structures based on known functional and/or clinical implications.
2.1.2. Linking Structural to Functional Variation
As related in
Section 4, PCA is highly interpretable. For instance, we can visualize the magnitude of atomic displacements for each PC (we restrict here our attention to the top three PCs) over the amino-acid sequence, which allows us to understand how the dynamics is “distributed” over the functional regions.
As shown in
Figure 4, the first three PCs greatly reflect the activation loop. This structural element is of paramount importance as it plays a decisive role in regulating the functional status of FGFR2. This loop plays an outsize role in the function of FGFR2, and our analysis captures this well. The structural variation present in the wildtype and variant forms of FGFR2-TKD is distilled well through PCA, and the top three modes of the intrinsic dynamics highlight the importance of this loop, connecting structure variation to function.
Furthermore, we observe that other functional regions, such as the kinase hinge and the nucleotide binding loop, although not predominantly featured in the first PC (PC1), are effectively captured in the second and third PCs. This pattern suggests that PC1 primarily encapsulates the most influential or critical functional regions of FGFR2. The absence of the kinase hinge and nucleotide binding loop in the primary component underscores the nuanced nature of protein dynamics, where linear methods like PCA may not fully capture the intricacies of non-linear structural changes. Moreover, this observation underlines the importance of examining multiple components to gain a comprehensive understanding of all functional dynamics within the protein. The selective prominence of these regions in PCs beyond the first also speaks to the need for broad analytical approaches that consider the complex interplay of structural elements across the protein. Another intriguing aspect of our findings is the prominence of residues in the 700 to 750 range within the PCA analysis despite these not being associated with a specific functional region. This could be attributed to the fact that these residues form a coil structure within the protein, which is inherently more unstable and thus exhibits more significant variations across different structures. Such fluctuations make this segment particularly distinct in our analysis, highlighting its unique role in the protein’s overall structure and behavior. This detailed analysis of intrinsic dynamics in FGFR2 not only corroborates the utility of PCA in protein structure analysis but also provides nuanced insights into the complex interplay of structural elements and their functional implications. The results not only validate established knowledge about critical regions such as the activation loop but also bring to light the dynamics of less prominent regions, offering a more holistic view of FGFR2-TKD structural dynamics.
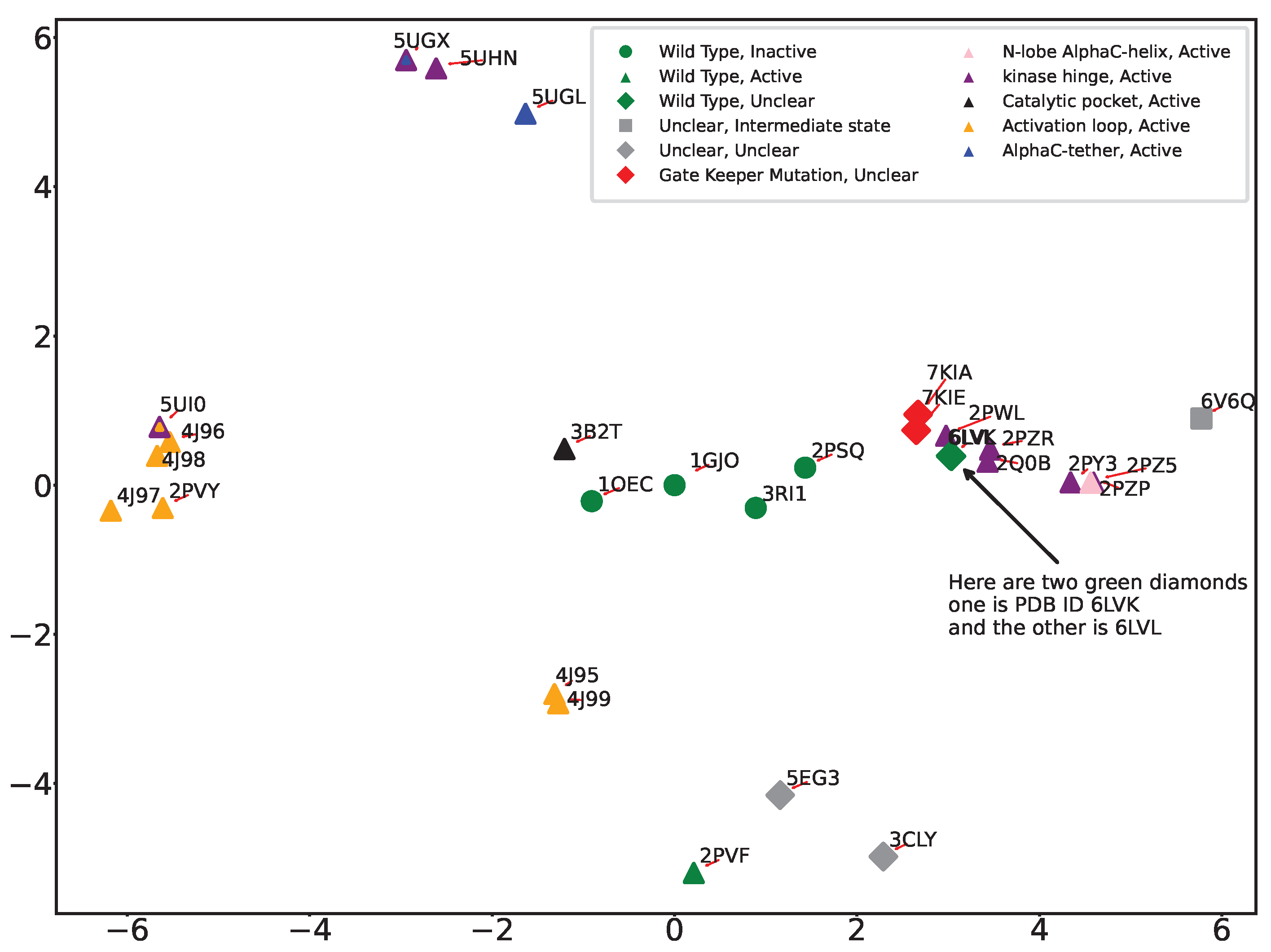
The PC-based summarization of structural dynamics provides us with yet another approach to linking structural dynamics to functional variation; we can visualize the structural variation of wildtype and variant FGFR2 by projecting all structures onto the top two PCs. Color-coding each projection with the functional designation of their corresponding tertiary structure allows us then to potentially better infer functional implications of mutations.
Figure 5 shows the 2D projection/embedding (over PC1 and PC2) of tertiary structures of wildtype and variant FGFR2-TKD. We observe that the PC1-PC2 projections effectively categorize mutations originating from the activation loop and kinase hinge regions, as we observe strong co-localization in the structure space. In addition to color-coded markers for different functional regions, we also introduce markers based on the activation status of the structure using different shapes. Circular markers represent inactive, triangular markers indicate active, rhombus markers signify uncertain status, and square markers represent an intermediate state, a status between active and inactive.
Incorporation of color and shape markers uncovers various clustering patterns. All active forms are well co-localized in various clusters in the 2D embedding. The PDB IDs 3CLY and 6V6Q remain distinctly separate from the others, aligned with our earlier observations of these structures as outliers. PDB ID 6V6Q is isolated, in agreement with the designation of this tertiary structure as in the intermediate state and unclear in functional implication. PDB ID 3CLY is also designated as unclear in both activity and functional region. 3B2T, the tertiary structure with a mutation in the catalytic pocket region, is distinct from the other variant forms and instead is co-localized with the wildtype inactive forms (PDB ID 1GJO and PDB ID 1OEC). This is due to the relatively conserved nature of the catalytic pocket region, located between beta sheets 6 and 8, and suggests that mutation of a catalytic pocket residue has minimal impact on the conformation of the activation loop. We also observe that 2PSQ, though labeled as ’inactive and unphosphorylated,’ lies away from 1GJO in the embedding. A detailed look reveals intriguing insights; PDB ID 2PSQ displays an activation loop region similar to the ’phosphorylated’ structure 2PVF. Despite its classification with inactive structures, 2PSQ’s spatial positioning in our analysis sets it apart from 1GJO and other unphosphorylated structures, hinting at a structural identity closer to the active state. This is consistent with previously published observations [
37] where authors also notice that the structure is similar to inactive ones. It is worth noting that no published evidence points to 3RI1 behaving as 2PSQ. PDB ID 5UI0, notable for mutations E565A and K659M, which significantly increase kinase activity, resembles the activity level of wildtype phosphorylated FGFR2-TKD with an activated A loop. This dual mutation affects the allosteric network at two crucial points, resulting in enhanced kinase activity. However, as revealed by the PC1-PC2 embedding, 5UI0’s distinct placement sets it apart from other structures starting with 5’, such as 5UGX and 5UHN. Generally, structures prefixed with ‘4’ and ‘5’ are categorized as ‘active’, displaying various degrees of kinase activity. We also highlight the co-localization in the PC1-PC2 embedding of PDB IDs 7KIA and 7KIE, the only two tertiary structures with the gatekeeper mutation V564F which affects the ATP binding pocket and the activation loop, influencing the binding of kinase inhibitors and kinase activity, and with bound inhibitors.
It is worth highlighting that the two main clusters of the active forms of FGFR2-TKD positioned on opposite sides of the first principal component (PC1), as depicted in
Figure 5, likely indicate substantial structural and functional diversity within these active states. This distribution suggests that PC1 captures the largest variance in the dataset, reflecting significant differences in activation mechanisms, such as variations in activation loop conformations, domain orientations, or Alpha C helix positioning. Additionally, these clusters may highlight functional diversities within active states, potentially influenced by ligand binding, post-translational modifications, or specific mutations that confer activation, each leading to unique structural alterations. The distinct clusters could also be a result of data-driven separation due to the conformational sampling present in the structural datasets used for PCA, with the protein adopting multiple active conformations throughout its functional cycle. The separation into clusters thus reflects the complex interplay of structural adaptations, functional requirements, and external modulatory factors that characterize the dynamic nature of protein activation and regulation.
As related in
Section 4, though not as interpretable as PCA, we also employ Isomap to extract the intrinsic dynamics of FGFR2-TKD. While Isomap does not reveal the amount of variation over amino acids per its components, projections of tertiary structures over the top two components can nonetheless be obtained and labeled as above, shown in
Figure 6.
Figure 6 reveals that Isomap is closely aligned with PCA in classifying the major functional regions. Both techniques successfully distinguish between active activation loop structures and wildtype inactive structures and isolate uniquely dissimilar structures, such as 3CLY and 6V6Q. Compared to PCA, Isomap better separates mutations in the gatekeeper region (in red) and the kinase hinge region (in purple). However, Isomap is not as effective as PCA in segregating activation loop mutations; Isomap places 2PSQ closer to 1GJO despite 2PSQ’s activation region being more akin to a phosphorylated state.
2.1.3. Linking Structural Variation to Disease Implication
Table 4 recapitulates the functional status of each structure and adds the disease classification where relevant. It is worth noting that not all the mutations/combinations of mutations were observed clinically; that is, the mutations that were “ported” from FGFR3.
The 2D embeddings of tertiary structures, whether obtained with PCA or Isomap, can now be re-analyzed by labeling projections of structures with their disease classification.
Figure 7 shows this analysis for the PC1-PC2 embedding. A clear segregation is observed between most of the genetic disorders and cancers, underscoring the role of structural dynamics in disease differentiation. Notable exceptions are “unclassified Craniosynostosis Syndrome” and “LADD1 syndrome”, represented by red and pink, respectively. Interestingly, both the UCS structures have the same mutation (K659N) that was identified in a patient not initially recognized as having Cruzon-like features [
40]. 3B2T was previously recognized as similar to wildtype FGFR2 [
41], LADD syndrome is clinically distinct from Pfeiffer and Crouzon syndromes, and the A628T mutation, in the catalytic pocket, decreases kinase activity rather than activates it. It is worth noting that the ambiguity re UCS and LADD1 can also be attributed to the limited sample size available for these conditions, highlighting the challenges with understanding the molecular basis of rare disorders.
Figure 8 relates the analysis over the Isomap-obtained embedding. The Isomap embedding shown in
Figure 8 does not exhibit superior classification when compared to the PCA projection and does not separate well between developmental and oncogenic disorders.
The lack of experimentally available structures motivates us to enrich the analysis with computationally predicted ones, as we relate next.
2.2. Extending the Analysis with Computational Models of Variants with No Structural Characterization
As related in
Section 4, we pursue three distinct structure prediction methods. First, we evaluate the faithfulness of these methods in reproducing known tertiary structures of FGFR2-TKD; we limit this analysis to single-nucleotide variants, of which there are 13 in the list of 29 with known tertiary structures listed in
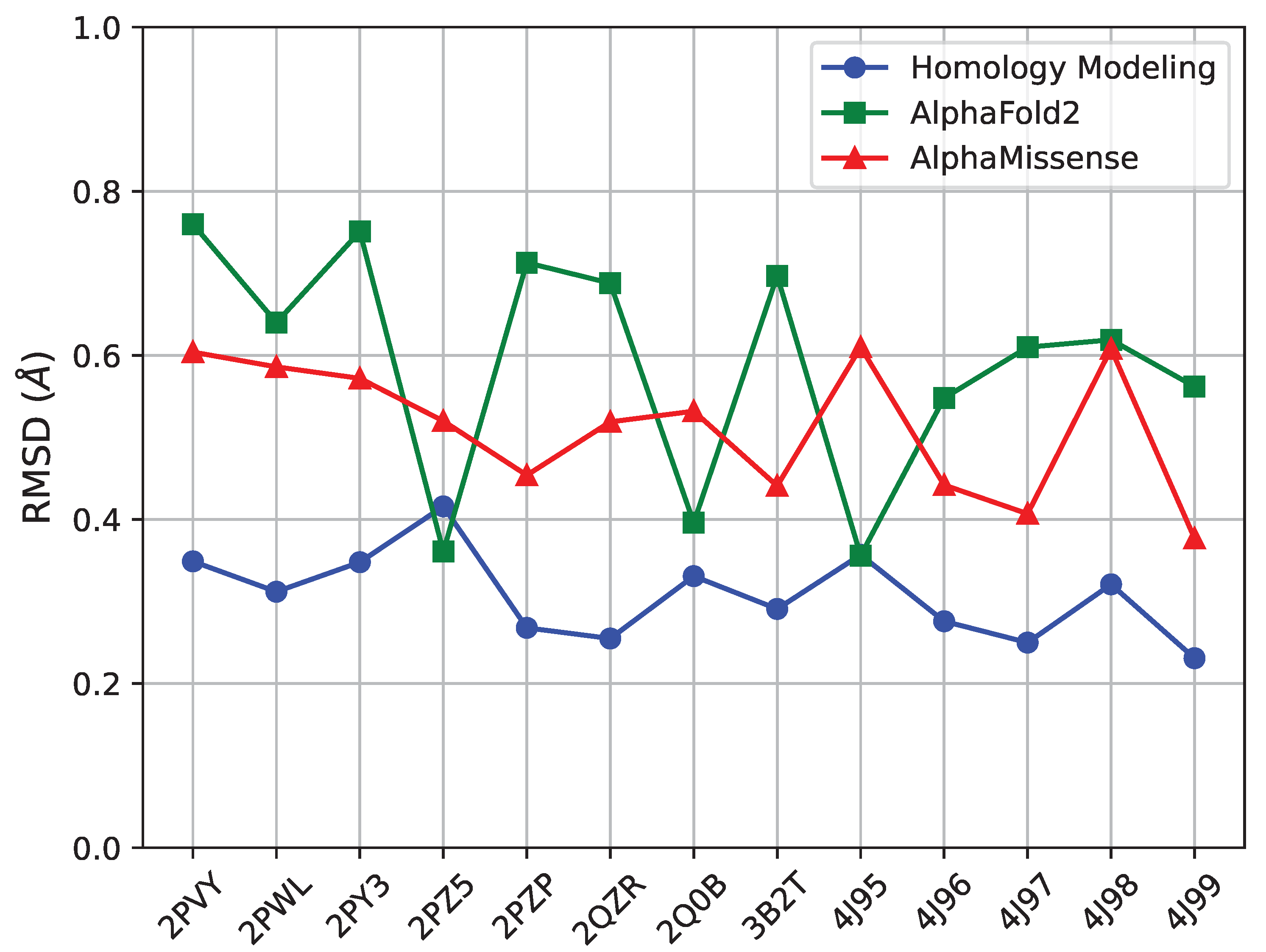
Table 2. For each, we measure the RMSD between the known and the predicted structure for each of the three methods (SWISS-MODEL, AlphaFold2, AlphaMissense) and relate it in
Figure 9. All three methods are effective at reproducing known structures, as their error is below
Å for each structure. However, the method with the lowest RMSD per structure is SWISS-MODEL, followed by AlphaMissense and then AlphaFold2.
The difference between a
Å RMSD (which is what SWISS-MODEL achieves on each structure) and a
–
Å RMSD (which is the range for AlphaFold2) is made evident in
Figure 10, which zooms in on the predicted and known structure for PDB ID 2PZ5.
Figure 10 offers several insights. AlphaFold2, despite its sophisticated algorithmic approach, faces challenges in coil regions. This issue could be attributed to potential biases within its training dataset, which might favor certain structures over others. In comparison, homology modeling through SWISS-MODEL displays a higher degree of adaptability to various structural states. This method seems to offer a more nuanced understanding of the protein structures, particularly in capturing the range of structural variations. It is also worth noting that both AlphaFold2 and AlphaMissense, while offering groundbreaking perspectives in protein modeling, show a noticeable decline in predictive accuracy in less structured regions like the activation loop in coil areas. Our findings underscore the need for a careful and critical approach when interpreting the results in these specific regions. The distinct methodologies, although robust in many aspects, require cautious application and interpretation, especially in the context of structurally complex and dynamically variable regions such as the activation loops within protein coils.
Taken altogether, the above analysis demonstrates the superiority of SWISS-MODEL in our case. The SWISS-MODEL-generated structures closely match the original PDB structures. The structures, further filled (with SCWRL4) and refined with NAMD2, exhibit a high degree of similarity to the actual structures in the PDB. Based on these observations, we decided to employ SWISS-MODEL homology modeling for all subsequent mutant modeling tasks.
Having established SWISS-MODEL to be a more faithful structure prediction method in our context, we apply it to variant sequences with no known structures. Followed by SCWRL4 and the energetic refinement described in
Section 4, we now enrich our prior embedding-based analysis with computed (and refined) structures.
Figure 11 shows the PC1-PC2 embedding of known and computed tertiary structures with colors and shapes utilized to indicate functional region and state designations. ’X’ is utilized to further indicate the computed structures.
Figure 11 suggests that R6127 and R678G are distinct, though possibly both in an active state, whereas A648T may represent an intermediate, unclear state in terms of activity.
Figure 12 now adds the computed structures to the PC1-PC2 embedding labeled with disease classifications. Some interesting observations emerge. Modeled structures with the mutations R678G (associated with Crouzon syndrome), A648T (associated with LADD1), G663E (associated with Pfeiffer syndrome), and R612T (associated with lung cancer) have been meticulously classified into specific disease-related categories. In particular, R678G and G663E align closely with mutations causing genetic/developmental disorders, showing a strong correlation with this group. On the contrary, A648T and R612T show a less precise alignment with their respective groups; A648T is somewhat close to the 3B2T marker, suggesting a less definitive association, while R612T aligns more loosely with cancer-causing mutations. It should also be noted that R612T is distinct in its position in the embedding, which is consistent with being the only mutant with a classification of lung cancer.
We note that the A628T (PDB entry 3B2T) mutation affects the intrinsic catalytic activity of FGFR2 kinase by compromising the catalytic activity of the kinase domain. The A628T mutation alters the configuration of key residues in the catalytic pocket, leading to decreased tyrosine phosphorylation of FGFR2 and recruitment of downstream signaling molecules. Similarly, A648T confers a loss of function demonstrated by decreased proliferation relative to wildtype FGFR2 in a competition assay and decreased transformation activity [
42], decreased protein kinase activity, and reduced downstream MAPK signaling pathway activation in cultured cells. Both mutations are reported with LADD syndrome, even though the mutation positions are not located in the same functional region.
3. Discussion
Elucidating the biological and clinical implications of mutations in proteins central to human health is challenging but key to effective, targeted, personalized therapeutics. In this paper, we have focused on FGFR2, a key protein in regulating cell proliferation, growth, and differentiation. Decades of computational and experimental research on the relationship between sequence, structure, and function have demonstrated the importance of structural dynamics in this relationship [
26]. In particular, a key realization is that mutations often impact the structural dynamics of a protein and through it they impact the ability of the protein to carry out its “wildtype” interactions with other molecules in the cell, percolating into disrupted chemical pathways that then demonstrate clinically in disorders and disease [
43].
We leverage this realization in this paper and aim to characterize and summarize the structural dynamics of FGFR2. By relying on the principle of conformation selection [
27,
28], we harness the structural diversity present among PDB-deposited tertiary structures of wildtype and variant forms of FGFR2 in various functional states. Through various machine learning methodologies we reveal intrinsic organizations of the structure space populated by the experimentally resolved structures that expose a few intrinsic modes of dynamics. The methodological approaches presented here provide a mesoscale global view of the structural states that highlight major modes of motion and parts of the structure with coordinated movements. It complements the atomic resolution results from molecular dynamics and X-ray crystallization studies that revealed changes in, for example, H bonding patterns of the molecular brake residues between the active and inactive states.
Through various structure prediction methods, we enrich this analysis with computationally modeled tertiary structures of FGFR2 variants currently with no structural characterization. Mapping the identified intrinsic modes of structural dynamics on the amino-acid sequence of FGFR2 and expanding this to wildtype and variant sequences reveals which functional regions are most implicated by the dynamics and in which variants and functional states, drawing a connection between sequence mutations, structural dynamics, and functional dynamics. Labeling the elucidated organization of the structure space with disorder information further completes the sequence–structure–function relationship by bringing into it clinical implications.
What have we learned about FGFR2 from this analysis? Several observations emerge. The dynamics captures well the prominent, outsize role of the activation loop in all the top intrinsic modes of dynamics. Other functional regions, such as the kinase hinge and nucleotide binding loop also feature prominently in the top modes. In particular, the detailed analysis of the intrinsic dynamics in FGFR2 in this paper not only corroborates the utility of PCA but also provides nuanced insights into the complex interplay of structural elements and their functional implications. For instance, the results presented earlier not only validate established knowledge about critical regions such as the activation loop but also expose the dynamics of less prominent regions, thus offering a more holistic view of FGFR2-TKD structural dynamics. In addition, the identified dynamics provides good co-localization of active forms of FGFR2 and even provides us with a better understanding of subtle structural indications in a tertiary structure documented as inactive in the PDB but with structural indications that correctly place it closer to active structures in our analysis.
The presented approach is able to distinguish the partially inactivated structure of 2PSQ from other structures in the autoinhibited state, a finding that had been overlooked by many previous studies. Future studies need to confirm our finding of 3RI1 behaving as 2PSQ. The results suggest that disease-associated mutations differ not only in their location in the protein sequence but also in their impact on the 3D structure. In addition to previously recognized differences in the activation loop for activating mutations, the models distinguished activating mutations in different regions and the inhibitory mutations associated with LADD syndrome, although they lie in different subregions of the protein. RTKs are a large family with many members of clinical importance and that share dysregulation of kinase activity as a disease mechanism. Future work will explore whether the approach applied here to FGFR2 can also contribute to the characterization of structure–function–disease relationships for other RTKs.
There are several limitations to our approach. First, it relies on crystal structures determined by different laboratories, using different methods, and in different space groups. Batch effects such as these could affect the 3D structures. We are unable to distinguish whether series of structures are more similar to each other because of their functional relationship or because of unaccounted for effects. Second, while the results suggest that there is some correlation between the functional impact of mutations and the associated disease, protein structural methods cannot account for other factors that contribute to the manifestation of the disorder, including clinical factors such as whether the mutation is germline or somatic. Indeed, some mutations could be associated with either germline or somatic diseases.
The results from using molecular models for mutations without existing protein structures shows promise for application to novel mutations with unclear mechanism of action. Previous studies have suggested that activating mutations function by changing the equilibrium between activated and inactivated forms. The method applied here captures snapshots of the structure space rather than the relative frequency of each structure but suggests that in addition to the balance of activated and inactivated forms of the protein, there may also be structural differences between different activated states or their intermediates.
Interesting additional observations emerge with regards to the impact of mutations on disease. We note that in addition to the analysis we have presented in the main paper, we relate here a complementary analysis on the ability of AlphaMissense to capture the pathogenicity or not of sequence mutations. While we have evaluated AlphaMisense in the main paper in its ability to reproduce known tertiary structures, AlphaMissense is a larger computational system that also predicts pathogenicities (or not) given a protein sequence. In
Table 5 we relate the scores and predictions from AlphaMissense on the potential pathogenicities of all FGFR2 variants in our dataset (the wildtype is correctly predicted as benign). Column 4 discretizes these scores into hits or not and shows that AlphaMissense correctly predicts that the FGFR2 variants we study are pathogenic in 14/16 cases (87.5% of the time).
We are not yet at the point where we can predict the actual pathogenicity from a given protein amino-acid sequence, but we are making progress, as the findings in this paper relate. We note among our findings that the identified structural dynamics for FGFR2-TKD separates well between developmental disorders and oncogenic disorders and reproduces the clinical implications of FGFR2 variants with no known structural characterization (for which we employ computationally modeled structures) from their positions in the PCA-reduced structure space. Further fine-grained differentiation suffers due to an imbalanced distribution of tertiary structures among disorders.
The availability of more tertiary structures in various forms and clinical settings for FGFR2 will further help the discriminative power of the methodology presented in this paper. As related in the detailed analysis, already we see its promise in providing a complete picture that brings together sequence, structure, dynamics, function, and clinical implications for a holistic understanding of the molecular basis of a disorder and so targeted therapeutic treatments.
Methodological Considerations
Understanding the structural basis of protein function and its alteration due to pathogenic mutations is crucial for the development of targeted therapeutic strategies. In our study, we primarily focused on Principal Component Analysis (PCA) and Isomap due to their broad applicability and robustness in capturing structural dynamics. However, we recognize the importance of discussing the strengths and limitations of these and other relevant methods in the context of pathogenicity analysis.
PCA is advantageous for its simplicity, interpretability, and efficiency in identifying the principal axes of variation within a dataset. Its linear nature allows for the straightforward projection of new data onto the derived principal components, facilitating the comparison of structural variants. However, PCA’s linear approach may not capture complex, non-linear relationships within the protein structures, potentially overlooking subtle but critical variations associated with pathogenicity.
Isomap, on the other hand, excels in mapping non-linear manifold structures, making it particularly useful for uncovering intricate structural dynamics that linear methods like PCA might miss. This capability enables a deeper understanding of the protein’s conformational space and its alteration by mutations. The main drawback of Isomap is its computational complexity and the difficulty of interpreting the resulting dimensions, which can limit its utility in large-scale or rapid analyses.
Other methods, such as t-Distributed Stochastic Neighbor Embedding (t-SNE) and Uniform Manifold Approximation and Projection (UMAP), have also been employed to explore structural diversity and pathogenicity. t-SNE is highly effective in visualizing clusters of high-dimensional data but is computationally intensive and sensitive to parameter settings, which can affect reproducibility. UMAP offers a balance between t-SNE’s detailed visualization capabilities and PCA’s scalability, providing high-quality embeddings with less computational demand. However, both t-SNE and UMAP can produce embeddings where the relative distances between points do not always represent meaningful biological relationships, potentially complicating the interpretation of pathogenic versus non-pathogenic structural variations.
In summary, each method has its unique strengths and limitations in the context of studying protein structural diversity and pathogenicity. Our choice of PCA and Isomap was guided by the objectives of our analysis, the nature of our dataset, and the need for a balance between computational efficiency and the ability to capture complex structural dynamics. We acknowledge the importance of considering a range of computational tools for a comprehensive understanding of the structural bases of protein pathogenicity and encourage future studies to explore the complementarity of these methods in greater detail.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










