
Loss of SAV1 in Kidney Proximal Tubule Induces Maladaptive Repair after Ischemia and Reperfusion Injury




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
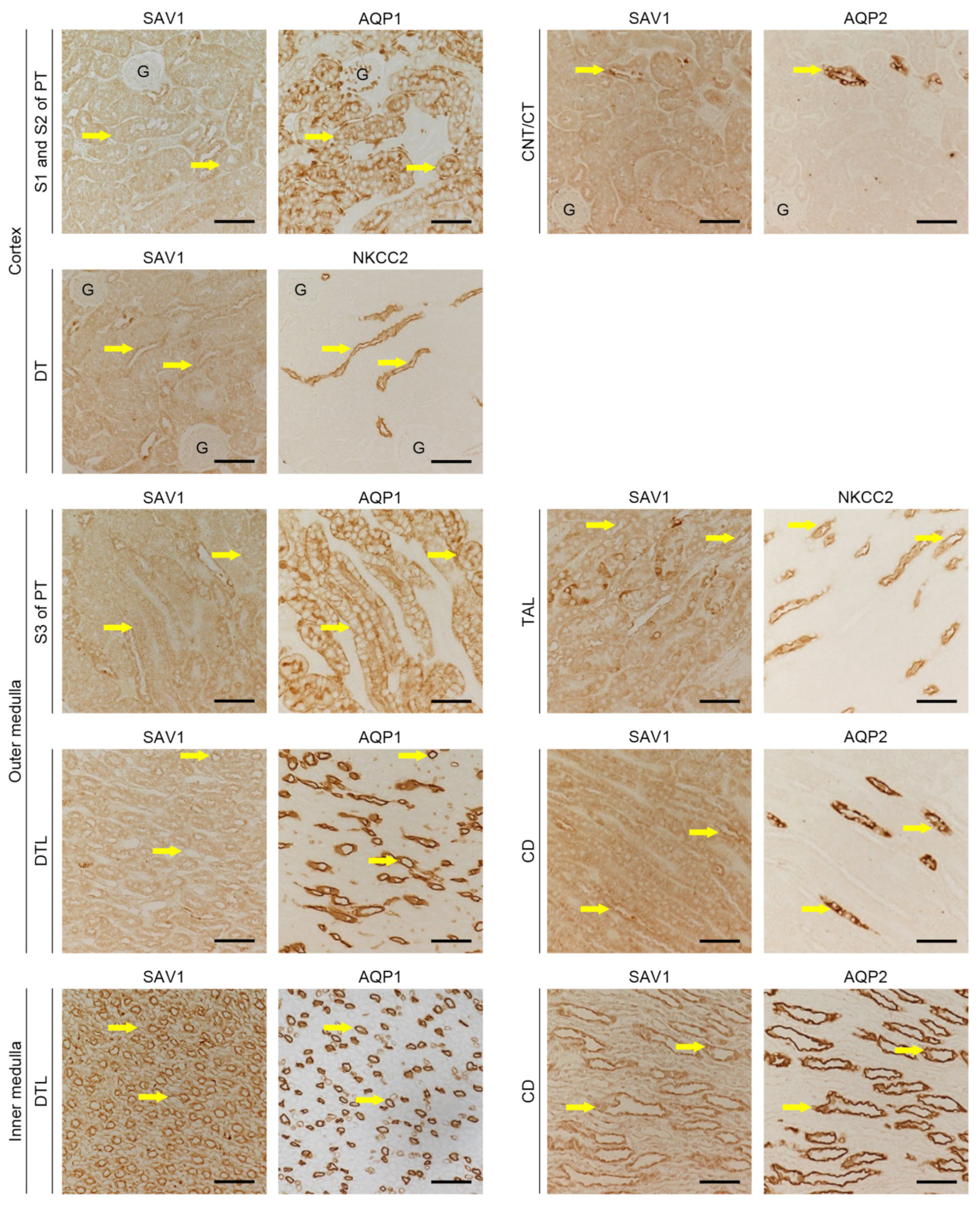
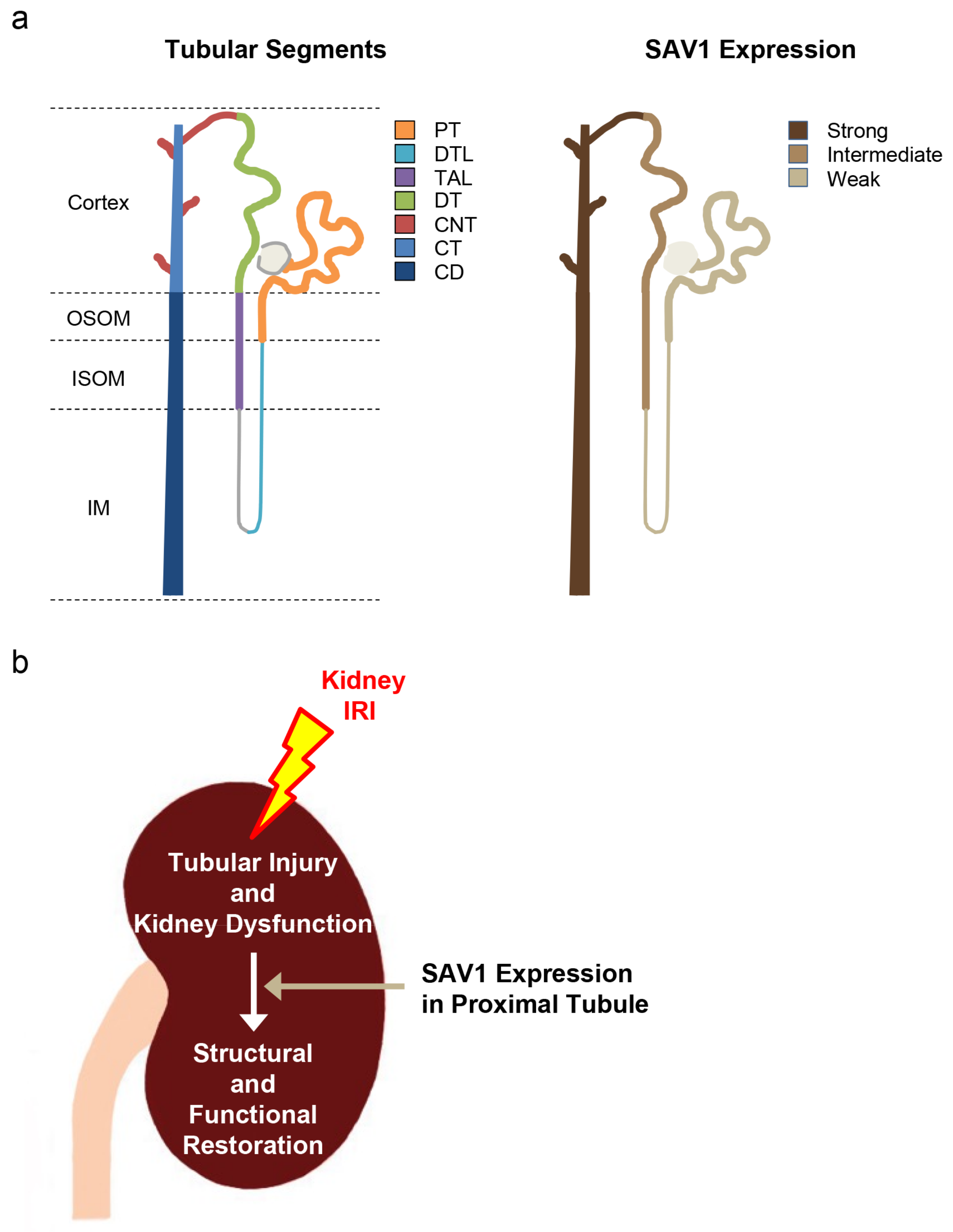
2.1. Distribution of SAV1 in Mouse Kidney Tuubles
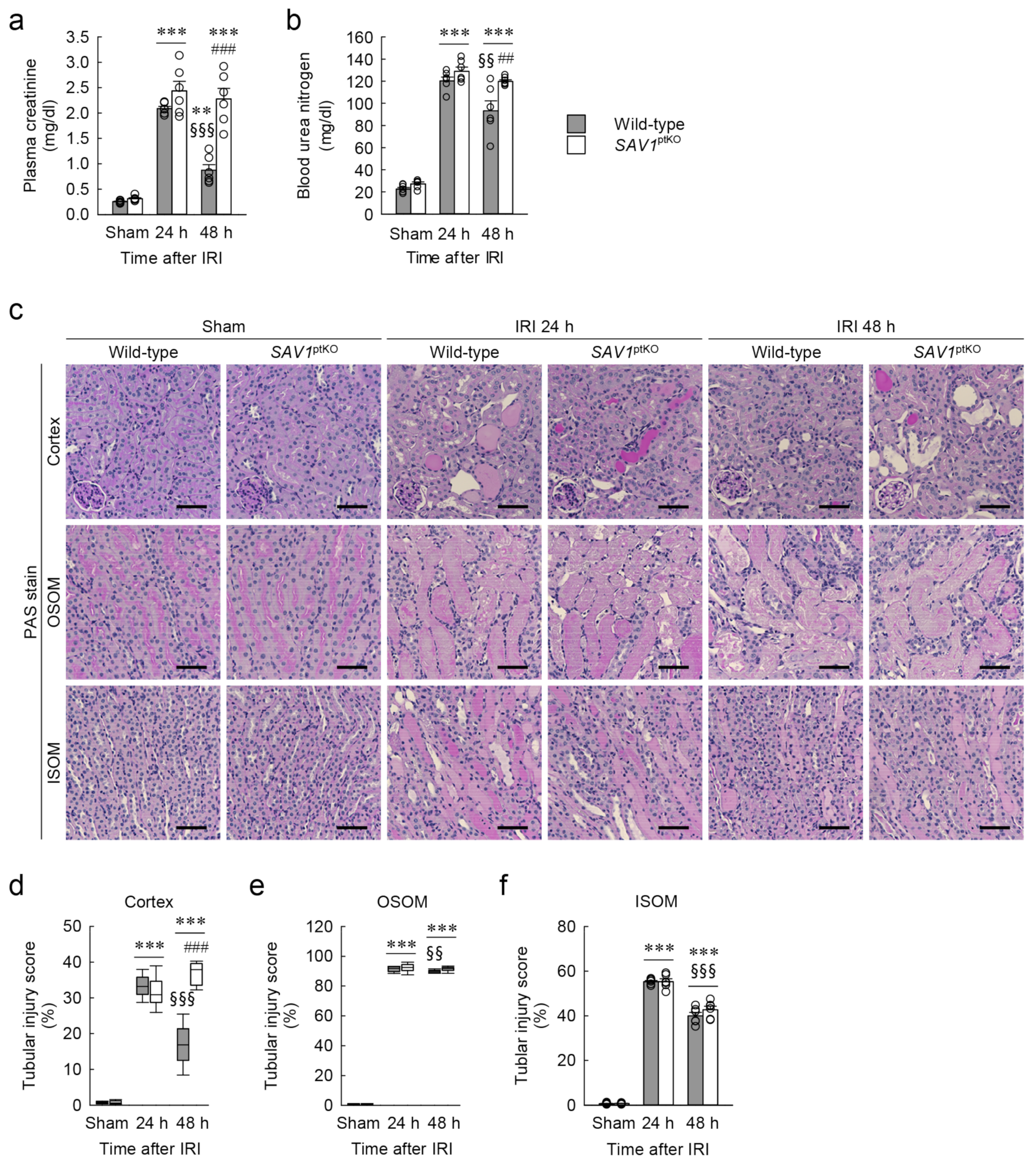
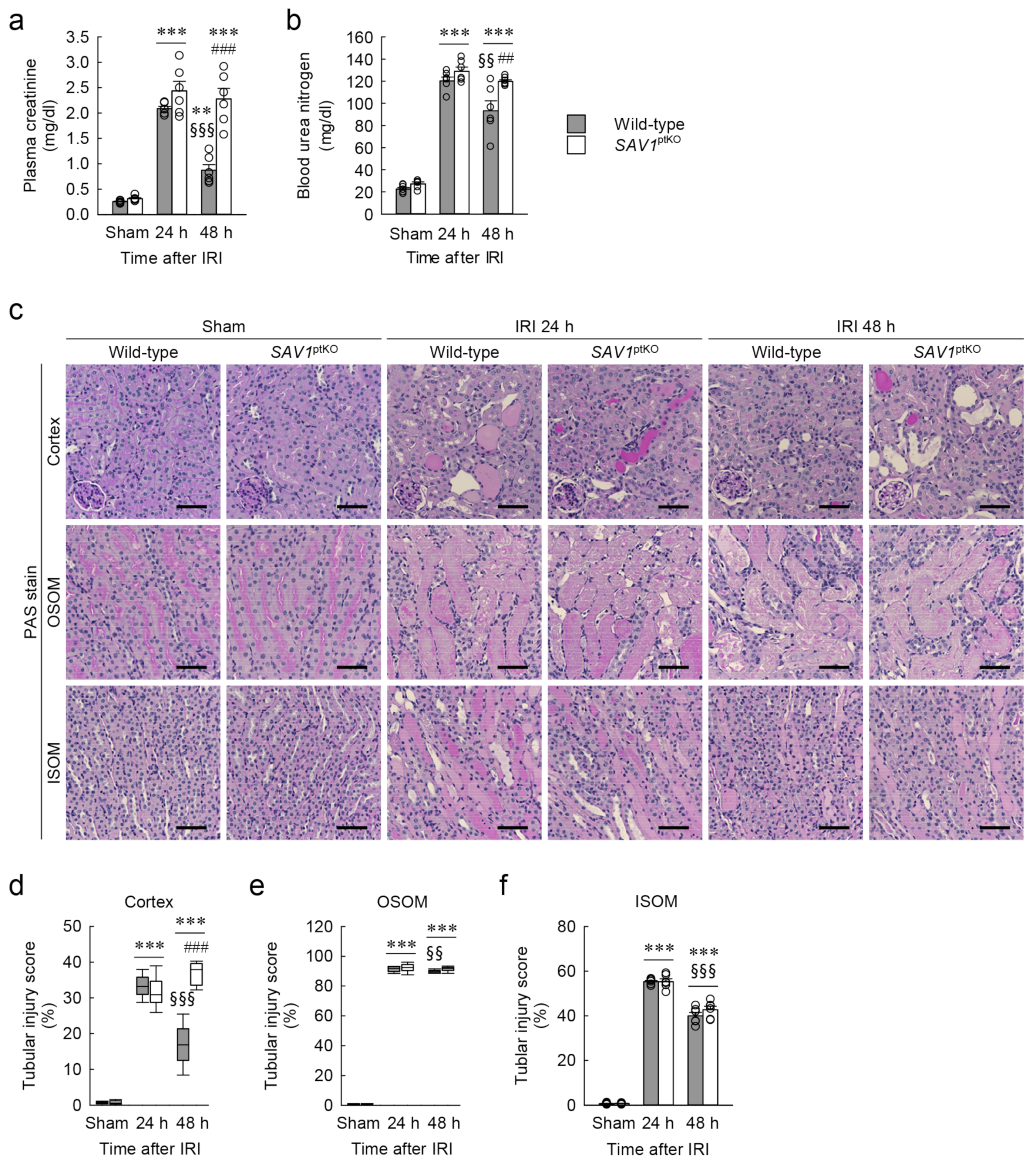
2.2. Proximal Tubule-Specific SAV1 Deficiency Induces Maladaptive Restoration of Kidney Function after IRI
2.3. Proximal Tubule-Specific SAV1 Deficiency Fails to Restore Cortical Tubular Structure after IRI
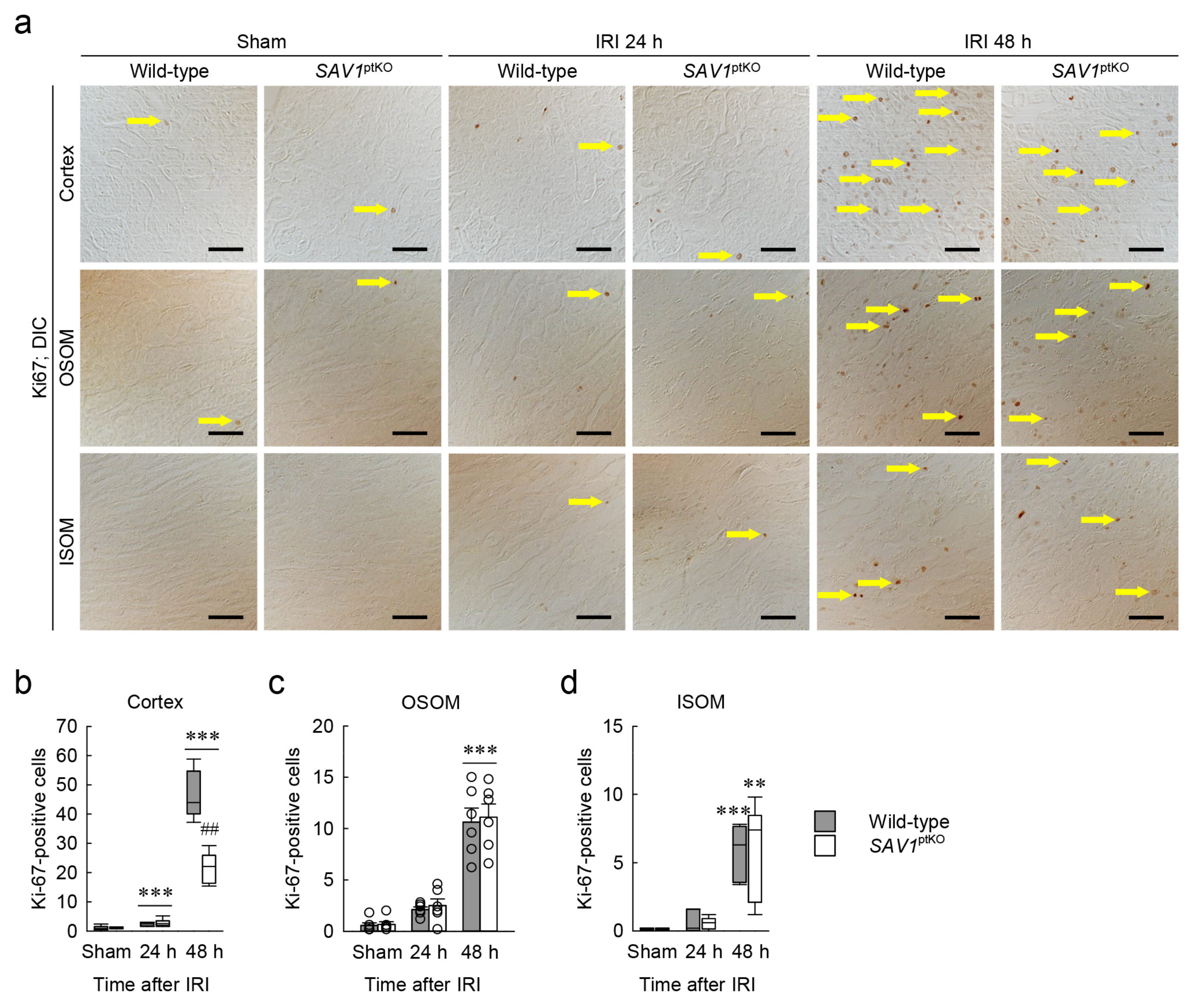
2.4. SAV1 Deficiency in Proximal Tubules Delays Cell Proliferation in the Cortex after IRI
2.5. SAV1 in Proximal Tubules Plays a Role beyond the Hippo Pathway in Kidneys Exposed to IRI
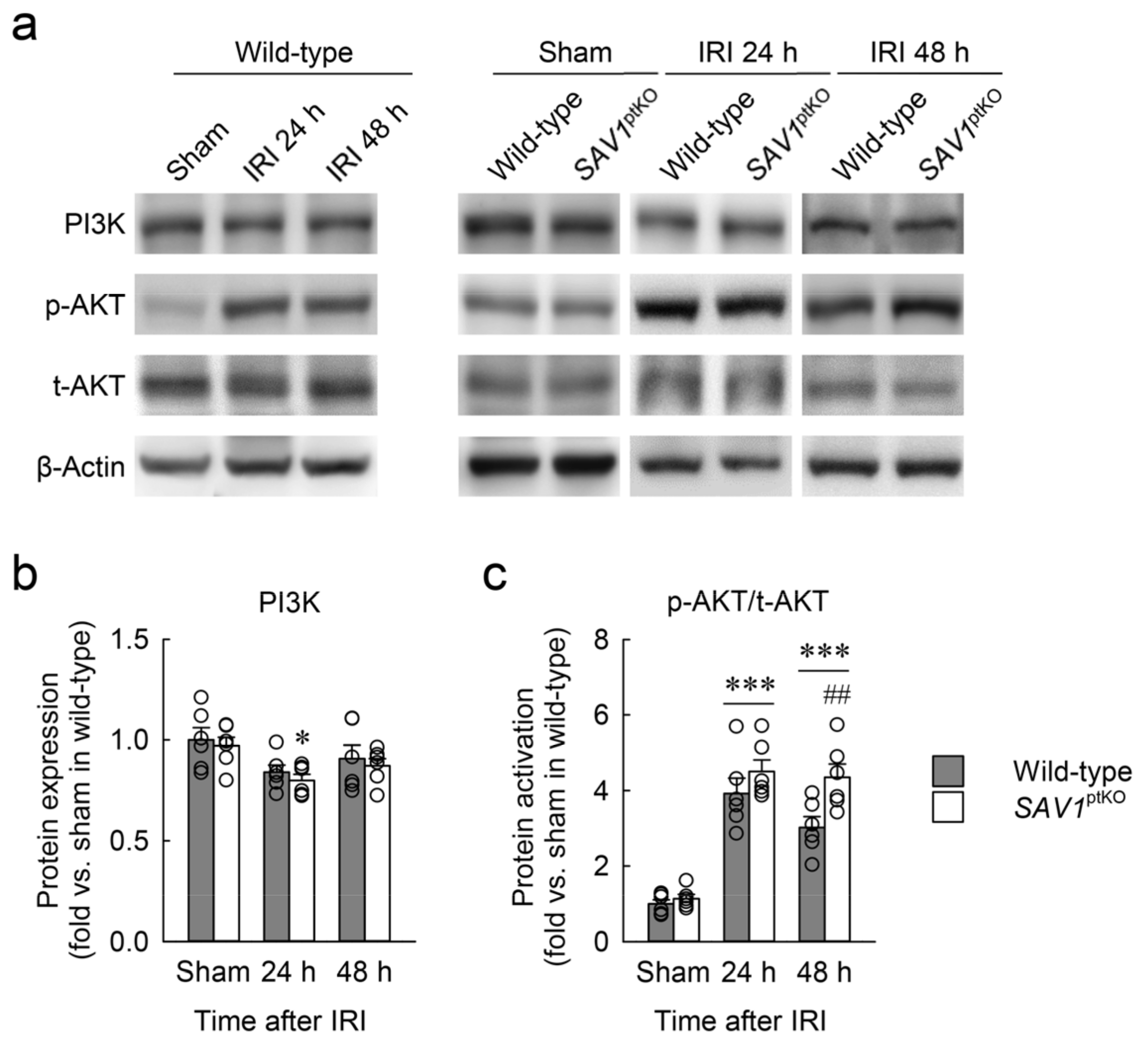
2.6. Loss of SAV1 in the Kidney Proximal Tubule Activates AKT after Kidney IRI
3. Discussion
4. Conclusions
5. Materials and Methods
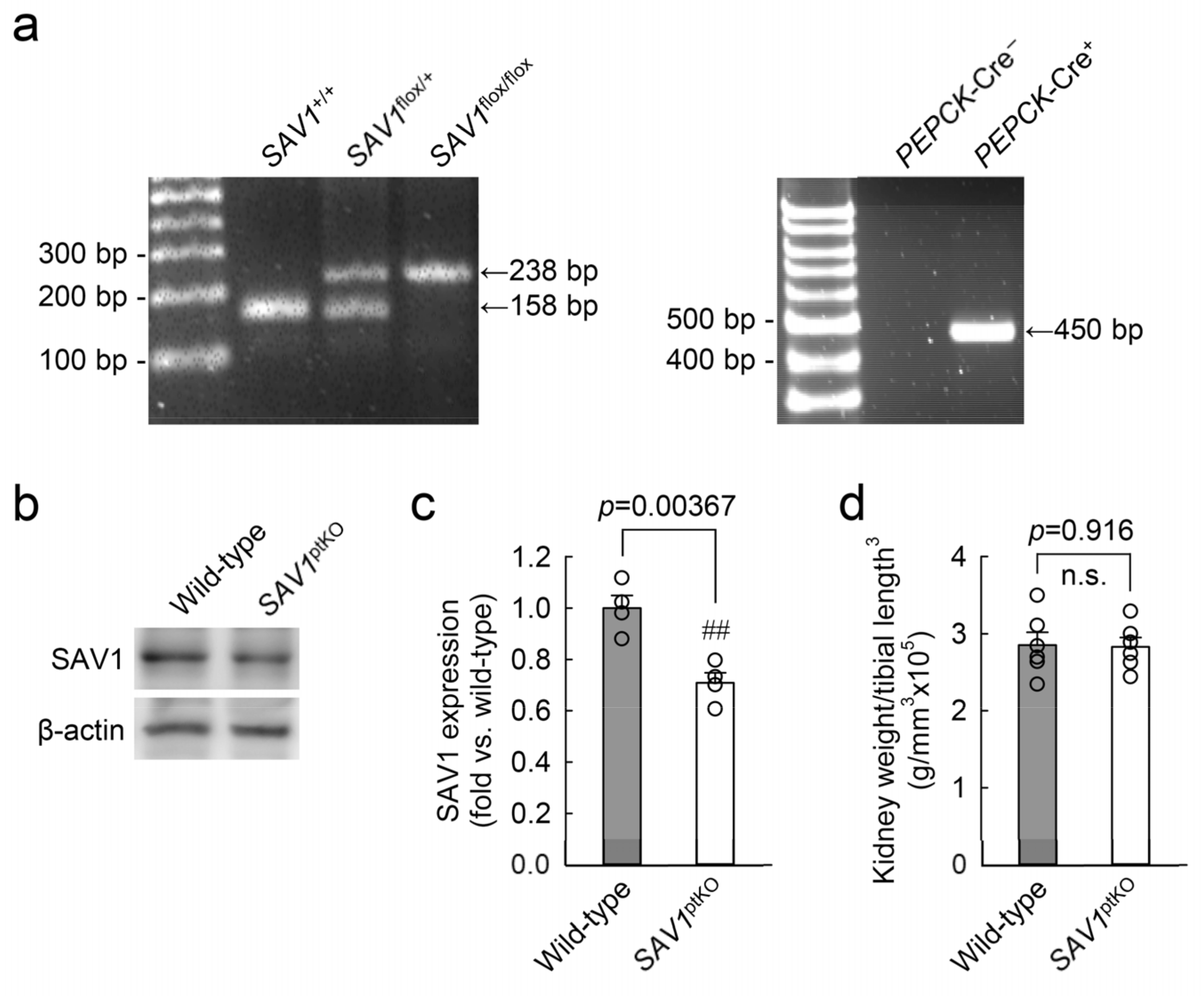
5.1. Animal Preparation
5.2. Surgery
5.3. Kidney Function
5.4. Immunohistochemistry
5.5. Tubular Injury and Cell Proliferation
5.6. Western Blot Analysis
5.7. Kidney Size
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shiva, N.; Sharma, N.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. Renal ischemia/reperfusion injury: An insight on in vitro and in vivo models. Life Sci. 2020, 256, 117860. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef]
- Stamellou, E.; Leuchtle, K.; Moeller, M.J. Regenerating tubular epithelial cells of the kidney. Nephrol. Dial. Transpl. 2021, 36, 1968–1975. [Google Scholar] [CrossRef]
- Kim, J. Spermidine rescues proximal tubular cells from oxidative stress and necrosis after ischemic acute kidney injury. Arch. Pharmacal Res. 2017, 40, 1197–1208. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.P.; Kim, J. Poly(ADP-ribose) polymerase 1 activation links ischemic acute kidney injury to interstitial fibrosis. J. Physiol. Sci. JPS 2015, 65, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wilflingseder, J.; Willi, M.; Lee, H.K.; Olauson, H.; Jankowski, J.; Ichimura, T.; Erben, R.; Valerius, M.T.; Hennighausen, L.; Bonventre, J.V. Enhancer and super-enhancer dynamics in repair after ischemic acute kidney injury. Nat. Commun. 2020, 11, 3383. [Google Scholar] [CrossRef]
- Daehn, I.S.; Duffield, J.S. The glomerular filtration barrier: A structural target for novel kidney therapies. Nat. Rev. Drug Discov. 2021, 20, 770–788. [Google Scholar] [CrossRef]
- Kishi, S.; Brooks, C.R.; Taguchi, K.; Ichimura, T.; Mori, Y.; Akinfolarin, A.; Gupta, N.; Galichon, P.; Elias, B.C.; Suzuki, T.; et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Investig. 2019, 129, 4797–4816. [Google Scholar] [CrossRef]
- Chevalier, R.L. Why is chronic kidney disease progressive? Evolutionary adaptations and maladaptations. Am. J. Physiol.-Ren. Physiol. 2023, 325, F595–F617. [Google Scholar] [CrossRef]
- Fu, Y.; Xiang, Y.; Li, H.L.; Chen, A.Q.; Dong, Z. Inflammation in kidney repair: Mechanism and therapeutic potential. Pharmacol. Ther. 2022, 237, 108240. [Google Scholar] [CrossRef]
- Kim, J.; Seok, Y.M.; Jung, K.J.; Park, K.M. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am. J. Physiol. Ren. Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.J. The unfolding of the Hippo signaling pathway. Dev. Biol. 2022, 487, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Habshi, T.; Shelke, V.; Kale, A.; Lech, M.; Gaikwad, A.B. Hippo signaling in acute kidney injury to chronic kidney disease transition: Current understandings and future targets. Drug Discov. Today 2023, 28, 103649. [Google Scholar] [CrossRef]
- Elster, D.; von Eyss, B. Hippo signaling in regeneration and aging. Mech. Ageing Dev. 2020, 189, 111280. [Google Scholar] [CrossRef]
- Wu, Z.; Guan, K.L. Hippo Signaling in Embryogenesis and Development. Trends Biochem. Sci. 2021, 46, 51–63. [Google Scholar] [CrossRef]
- Zhang, C.; Li, C.L.; Xu, K.X.; Zheng, Z.H.; Cheng, G.Z.; Wu, H.J.; Liu, J. The Hippo pathway and its correlation with acute kidney injury. Zool. Res. 2022, 43, 897–910. [Google Scholar] [CrossRef]
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37, e00565. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Kim, W.Y.; Hur, J.; Kim, H.; Nam, S.A.; Choi, A.; Kim, Y.M.; Park, S.H.; Chung, C.; Kim, J.; et al. The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Sci. Rep. 2016, 6, 31931. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; López-Novoa, J.M.; López-Hernández, F.J. Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin. Kidney J. 2015, 8, 548–559. [Google Scholar] [CrossRef]
- Lin, Z.; Xie, R.; Guan, K.; Zhang, M. A WW tandem-mediated dimerization mode of SAV1 essential for Hippo signaling. Cell Rep. 2020, 32, 108118. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xu, Y.; Feng, Y.; Shen, M.; Yuan, F.; Yuan, Y. YAP nuclear-cytoplasmic translocation is regulated by mechanical signaling, protein modification, and metabolism. Cell Biol. Int. 2020, 44, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Liu, H.; Han, S.; Fu, Z.R.; Wang, J.Y.; Chen, Y.; Wang, L.M. Melatonin pretreatment alleviates renal ischemia-reperfusion injury by promoting autophagic flux via TLR4/MyD88/MEK/ERK/mTORC1 signaling. FASEB J. 2020, 34, 12324–12337. [Google Scholar] [CrossRef] [PubMed]
- Mziaut, H.; Henniger, G.; Ganss, K.; Hempel, S.; Wolk, S.; McChord, J.; Chowdhury, K.; Ravassard, P.; Knoch, K.P.; Krautz, C.; et al. MiR-132 controls pancreatic beta cell proliferation and survival through Pten/Akt/Foxo3 signaling. Mol. Metab. 2020, 31, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Curtis, L.M. Sex and Gender Differences in AKI. Kidney360 2024, 5, 160–167. [Google Scholar] [CrossRef]
- Kim, J.; Kil, I.S.; Seok, Y.M.; Yang, E.S.; Kim, D.K.; Lim, D.G.; Park, J.W.; Bonventre, J.V.; Park, K.M. Orchiectomy attenuates post-ischemic oxidative stress and ischemia/reperfusion injury in mice—A role for manganese superoxide dismutase. J. Biol. Chem. 2006, 281, 20349–20356. [Google Scholar] [CrossRef]
- Hukriede, N.A.; Soranno, D.E.; Sander, V.; Perreau, T.; Starr, M.C.; Yuen, P.S.T.; Siskind, L.J.; Hutchens, M.P.; Davidson, A.J.; Burmeister, D.M.; et al. Experimental models of acute kidney injury for translational research. Nat. Rev. Nephrol. 2022, 18, 277–293. [Google Scholar] [CrossRef]
- He, L.Y.; Wei, Q.Q.; Liu, J.; Yi, M.X.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.M.; Liu, F.Y.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef]
- Ray, S.C.; Mason, J.; O’Connor, P.M. Ischemic renal injury: Can renal anatomy and associated vascular congestion explain why the medulla and not the cortex is where the trouble starts? Semin. Nephrol. 2019, 39, 520–529. [Google Scholar] [CrossRef]
- Lee, K.P.; Lee, J.H.; Kim, T.S.; Kim, T.H.; Park, H.D.; Byun, J.S.; Kim, M.C.; Jeong, W.I.; Calvisi, D.F.; Kim, J.M.; et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef]
- Kai, T.; Tsukamoto, Y.; Hijiya, N.; Tokunaga, A.; Nakada, C.; Uchida, T.; Daa, T.; Iha, H.; Takahashi, M.; Nomura, T.; et al. Kidney-specific knockout of in the mouse promotes hyperproliferation of renal tubular epithelium through suppression of the Hippo pathway. J. Pathol. 2016, 239, 97–108. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Zeringue, A.; Fu, J.; Rauchman, M.I.; McDonald, J.R.; El-Achkar, T.M.; Balasubramanian, S.; Nurutdinova, D.; Xian, H.; Stroupe, K.; et al. Rate of Kidney Function Decline Associates with Mortality. J. Am. Soc. Nephrol. 2010, 21, 1961–1969. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; Gibney, R.T.N. Conventional markers of kidney function. Crit. Care Med. 2008, 36, S152–S158. [Google Scholar] [CrossRef]
- Ferguson, M.A.; Waikar, S.S. Established and Emerging Markers of Kidney Function. Clin. Chem. 2012, 58, 680–689. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef]
- Naved, B.A.; Bonventre, J.V.; Hubbell, J.A.; Hukriede, N.A.; Humphreys, B.D.; Kesselman, C.; Valerius, M.T.; McMahon, A.P.; Shankland, S.J.; Wertheim, J.A.; et al. Kidney repair and regeneration: Perspectives of the NIDDK (Re)Building a Kidney consortium. Kidney Int. 2022, 101, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Liu, Z.J.; Luo, M.N.; Li, X.Y.; Chen, H.X.; Gong, S.Q.; Zhang, M.J.; Zhang, Y.Z.; Liu, H.F.; Li, X.Y. The pathological role of damaged organelles in renal tubular epithelial cells in the progression of acute kidney injury. Cell Death Discov. 2022, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Schumann-Bischoff, A.; Schmitz, J.; Scheffner, I.; Schmitt, R.; Broecker, V.; Haller, H.; Bräsen, J.H.; Gwinner, W. Distinct morphological features of acute tubular injury in renal allografts correlate with clinical outcome. Am. J. Physiol.-Ren. Physiol. 2018, 315, F701–F710. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Gandolfo, M.T.; Ko, G.J.; Satpute, S.R.; Racusen, L.; Rabb, H. B Cells Limit Repair after Ischemic Acute Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 654–665. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Sutton, T.A. Endothelial injury and dysfunction: Role in the extension phase of acute renal failure. Kidney Int. 2004, 66, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.D.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef]
- Kwon, D.S.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, Y.K. Signal transduction of MEK/ERK and PI3K/Akt activation by hypoxia/reoxygenation in renal epithelial cells. Eur. J. Cell Biol. 2006, 85, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.S.; Ghanim, A.M.H. The Protective Role of Celastrol in Renal Ischemia-Reperfusion Injury by Activating Nrf2/HO-1, PI3K/AKT Signaling Pathways, Modulating NF-kappa b Signaling Pathways, and Inhibiting ERK Phosphorylation. Cell Biochem. Biophys. 2022, 80, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Siroky, M.B.; Yalla, S.V.; Azadzoi, K.M. Mitochondrial stress and activation of PI3K and Akt survival pathway in bladder ischemia. Res. Rep. Urol. 2017, 10, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Du, J. Potential role of Akt signaling in chronic kidney disease. Nephrol. Dial. Transpl. 2015, 30, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Li, L.; Wu, J.C.; Cook, S.A.; Nagoshi, T.; Picard, M.H.; Liao, R.L.; Rosenzweig, A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 2002, 277, 22896–22901. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Kim, H.B.; Kim, M.C.; Lee, J.M.; Lee, J.H.; Kim, J.H.; Kim, J.W.; Park, W.Y.; Kim, S.Y.; Kim, J.B.; et al. Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J. Clin. Investig. 2018, 128, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006, 66, 2576–2583. [Google Scholar] [CrossRef]
- Moon, D.; Padanilam, B.J.; Jang, H.S.; Kim, J. 2-Mercaptoethanol protects against DNA double-strand breaks after kidney ischemia and reperfusion injury through GPX4 upregulation. Pharmacol. Rep. 2022, 74, 1041–1053. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 2015, 87, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Moon, D.; Jung, S.; Lee, J.; Kim, J. Cisplatin nephrotoxicity is induced via poly(ADP-ribose) polymerase activation in adult zebrafish and mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R843–R854. [Google Scholar] [CrossRef] [PubMed]
- Hagdorn, Q.A.J.; Bossers, G.P.L.; Koop, A.M.C.; Piek, A.; Eijgenraam, T.R.; van der Feen, D.E.; Silljé, H.H.W.; de Boer, R.A.; Berger, R.M.F. A novel method optimizing the normalization of cardiac parameters in small animal models: The importance of dimensional indexing. Am. J. Physiol.-Heart C 2019, 316, H1552–H1557. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, D.; Padanilam, B.J.; Park, K.M.; Kim, J. Loss of SAV1 in Kidney Proximal Tubule Induces Maladaptive Repair after Ischemia and Reperfusion Injury. Int. J. Mol. Sci. 2024, 25, 4610. https://doi.org/10.3390/ijms25094610
Moon D, Padanilam BJ, Park KM, Kim J. Loss of SAV1 in Kidney Proximal Tubule Induces Maladaptive Repair after Ischemia and Reperfusion Injury. International Journal of Molecular Sciences. 2024; 25(9):4610. https://doi.org/10.3390/ijms25094610
Chicago/Turabian StyleMoon, Daeun, Babu J. Padanilam, Kwon Moo Park, and Jinu Kim. 2024. "Loss of SAV1 in Kidney Proximal Tubule Induces Maladaptive Repair after Ischemia and Reperfusion Injury" International Journal of Molecular Sciences 25, no. 9: 4610. https://doi.org/10.3390/ijms25094610
APA StyleMoon, D., Padanilam, B. J., Park, K. M., & Kim, J. (2024). Loss of SAV1 in Kidney Proximal Tubule Induces Maladaptive Repair after Ischemia and Reperfusion Injury. International Journal of Molecular Sciences, 25(9), 4610. https://doi.org/10.3390/ijms25094610