
The Intricacies of Renal Phosphate Reabsorption—An Overview


Abstract
1. Introduction
2. The Proximal Renal Tubule
3. Executors of Phosphate Reabsorption: Renal Phosphate Transporters and NHERF1 (Na+/H+ Exchange Regulatory Cofactor 1)
3.1. The Renal Phosphate Transporters
3.1.1. NaPi-2a
NaPi-2a Partitioning into Lipid Rafts
3.1.2. NaPi-2c
3.1.3. PiT-2
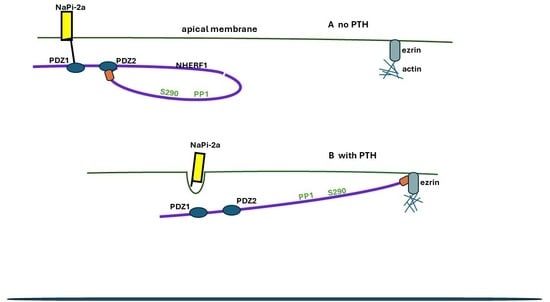
3.2. NHERF1—The Systems Manager
NHERF1
Structure
3.3. Hormonal Regulation of Phosphate Transport by NaPi-2a
4. Regulation of NaPi-2a by Parathyroid Hormone (PTH) and Parathyroid-Hormone-Related Protein PTHrP
4.1. PTH
4.2. PTHrP
4.3. The PTH/PTHrP Receptor (PTH1R)
4.3.1. Structure
4.3.2. PTH/PTHrP Binding
4.3.3. Signalling Pathways and Signalling Bias
4.3.4. Termination of Signalling
RGS14 (Regulator of G Protein Signalling 14)
Receptor-Activity-Modifying Proteins (RAMPs)
4.3.5. Endosomal Signalling
4.3.6. Inherited Defects of PTH/PTHrP Receptor Signalling
4.4. Epac1 (Exchange Protein Directly Activated by cAMP 1)
4.4.1. Structure
4.4.2. Actions of Epac1 in the Proximal Tubules
5. Dopamine
5.1. Sources and Degradation
5.2. Dopamine Receptors and Signalling
6. FGF23 and FGFR1C/KLOTHO Receptor
6.1. Physiological Factors Increasing/Decreasing FGF23 Production
6.2. Structure
6.3. Renal Receptors and α-Klotho
α-Klotho
6.4. Signal Transduction and Signalling
7. Mechanisms of Hormonal Regulation of Phosphate Transport by NaPt-2a
8. Regulator of G Protein Signalling 14 (RGS14)
8.1. Structure
8.2. RGS14 Inhibition of PTH Regulation of NaPi-2a
8.3. RGS14 Polymorphisms in GWAS
9. Growth Hormone and Insulin-like Growth Factor-1 (IGF-1)
10. Dietary Phosphate
10.1. Acute Changes in Dietary Phosphate
10.1.1. Low Phosphate Intake
10.1.2. High Phosphate Intake
10.2. Chronic Changes in Dietary Phosphate
10.3. High Dietary Phosphate and Renal Dopamine Excretion
11. Tumour-Induced Osteomalacia (TIO)
11.1. MEPE
Mechanism Causing Hyper-Phosphaturia
11.2. Secreted Frizzled-Related Protein 4 (sFRP4)
11.3. Fibroblast Growth Factor 7 (FGF7)
12. Clinical Applications of the Expanding Knowledge of Phosphate Transport
13. Summary and Conclusions
- (1)
- Whether interaction of full-length PTH, the PTH1R receptor, and G-signalling are the same as for the PTH1–34 fragment.
- (2)
- Clearer definition of the signalling pathway via cAMP and PKA to NHERF1. It appears speculative at present.
- (3)
- Clearer definition of the signalling pathway of IGFR which increases renal phosphate absorption.
- (4)
- The form of PTH which normally activates proximal tubule apical PTH1R. Activation by filtered intact PTH or N-terminal PTH fragments seems an imprecise regulatory mechanism for such a finely controlled reabsorption system. How far is locally produced PTHrP involved?
- (5)
- The function of PTHrP in the proximal tubules postnatally.
- (6)
- How PTH signalling at the apical and basolateral membranes in the proximal tubule are co-ordinated.
- (7)
- The location of NHERF1 at the BBM. Logically, it should be in the (long) cilia close to apically sited NaPi-2a, but findings are conflicting.
- (8)
- The roles of RAMPS in PTH/PTH1R signalling.
- (9)
- The function of the internal PDZ-binding motif of NaPi-2a.
- (10)
- Whether RGS14 has a role in regulating phosphate transport through inactivation of PTH/PTH1R signalling and/or in humans through blocking NaPi-2a inactivation by PTH.
- (11)
- Whether Epac is stimulated in parallel with PKA by PTH/PTH1R. If it is, do PKA and Epac operate an activation/inhibitory partnership to regulate PTH activity?
- (12)
- The dysfunctional protein activity which is being highlighted in GWASs of calcium stone formers.
- (13)
- Whether MEPE has a physiological role in the kidneys.
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BBM | brush border membrane |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CRF | chronic renal failure |
| D1R | Dopamine 1 receptor |
| Epac | exchange protein directly activated by c-AMP |
| FAM20 | extracellular kinase family member 20C |
| FEPO4 | Fractional excretion of phosphate |
| FGF7 | growth factor 7 |
| FGF23 | fibroblast growth factor 23 |
| FGFR1c | Fibroblast growth factor receptor 1c |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FRS2 | Fibroblast growth factor (FGF) receptor substrate 2 |
| GALNT3 | N-acetylgalactosaminyl transferase 3 |
| GPCRs | G protein-coupled receptors |
| GRK2 | G protein-coupled receptor kinase 2 |
| GRK6A | G protein-coupled receptor kinase |
| HEK | 293 cells human embryonic kidney cells |
| IGF1 | insulin-like growth factor 1 |
| l-DOPA | l-dihydroxyphenylalanine |
| MAPK | mitogen activated protein kinase |
| MEPE | matrix extracellular phosphoglycoprotein |
| NaPi-2a | sodium-dependent phosphate transporter-2a SLC34A1 |
| NaPi-2c | sodium-dependent phosphate transporter-2c SLC34A3 |
| NFAT | nuclear factor of activated T cells |
| NHE3 | sodium/hydrogen exchange factor3 |
| NHERF1 | Na+/H+ exchange regulatory cofactor-1 |
| NHERF3 | (Na+/H+ exchange regulatory cofactor-3, alias PDZK1 |
| OK | cells opossum kidney cells |
| PDZ | PSD-95/Discs-large/ZO1 domain |
| PiT-2 | sodium-dependent phosphate transporter SLC20A2 |
| PKA | protein kinase A |
| PLC | phospholipase C |
| PTH | parathyroid hormone |
| PTH1–34 | Truncated PTH N-terminal |
| PTH1R | PTH/PTHrP 1 receptor |
| PTHrP | PTH-related protein |
| PTHrP1–36 | Truncated PTHrP N-terminal |
| RAMPS | Receptor-activity-modifying proteins |
| RGS14 | Regulator of G protein signalling 14 |
| sFRP4 | secreted frizzled-related protein 4 |
| SGK1 | serum/glucocorticoid-regulated kinase 1 |
| TC/LEF | T-cell factor/lymphoid enhancer factor |
| TIO | Tumour-induced osteomalacia |
References
- Knochel, J.P. Hypophosphatemia and phosphorus deficiency. In The Kidney, 4th ed.; Brenner, B.M., Rector, F.C., Jr., Eds.; W.B. Saunders Company, Harcourt Brace Jovanovich, Inc.: West Philadelphia, PA, USA, 1991; Volume 1, pp. 888–915, Chapter 21. [Google Scholar]
- Fukumoto, S. Phosphate metabolism and vitamin D. Bonekey Rep. 2014, 3, 497. [Google Scholar] [CrossRef] [PubMed]
- Antoniucci, D.M.; Yamashita, T.; Portale, A.A. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J. Clin. Endocrinol. Metab. 2006, 91, 3144–3149. [Google Scholar] [CrossRef] [PubMed]
- Berndt, T.; Kumar, R. Phosphatonins and the regulation of phosphate homeostasis. Annu. Rev. Physiol. 2007, 69, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Gaasbeek, A.; Meinders, A.E. Hypophosphatemia: An update on its etiology and treatment. Am. J. Med. 2005, 118, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.; Debnam, E.S.; Unwin, R.J. Phosphate homeostasis and the renal-gastrointestinal axis. Am. J. Physiol. Renal Physiol. 2010, 299, F285–F296. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Siegel, M.; He, Y.; Nie, B.; Wang, J.; Koo-McCoy, S.; Minassian, N.A.; Jafri, Q.; Pan, D.; Kohler, J.; et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci. Transl. Med. 2018, 10, eaam6474. [Google Scholar] [CrossRef] [PubMed]
- Walker, V. Phosphaturia in kidney stone formers: Still an enigma. Adv. Clin. Chem. 2019, 90, 133–196. [Google Scholar] [CrossRef] [PubMed]
- Bringhurst, F.R.; Demay, M.B.; Kronenberg, H.M. Hormones and Disorders of Mineral Metabolism. In Williams Textbook of Endocrinology, 14th ed.; Melmed, S., Koenig, R., Rosen, C.J., Auchus, R.J., Goldfine, A.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 29; pp. 1196–1255. [Google Scholar]
- Curthoys, N.P.; Moe, O.W. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, K.; Paredes, J.M.; Mamonova, T.; Sneddon, W.B.; Liu, H.; Wang, D.; Li, S.; McGarvey, J.C.; Uehling, D.; et al. Parathyroid hormone initiates dynamic NHERF1 phosphorylation cycling and conformational changes that regulate NPT2A-dependent phosphate transport. J. Biol. Chem. 2019, 294, 4546–4571. [Google Scholar] [CrossRef]
- Levi, M.; Gratton, E.; Forster, I.C.; Hernando, N.; Wagner, C.A.; Biber, J.; Sorribas, V.; Murer, H. Mechanisms of phosphate transport. Nat. Rev. Nephrol. 2019, 15, 482–500. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A. The basics of phosphate metabolism. Nephrol. Dial. Transplant. 2023, 39, gfad188. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.; Turner, R.J. The Renal Proximal Tubule. In Biomembranes; Springer: Boston, MA, USA, 1979; Volume 10. [Google Scholar] [CrossRef]
- Wagner, C.A.; Rubio-Aliaga, I.; Biber, J.; Hernando, N. Genetic diseases of renal phosphate handling. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), iv45–iv54. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A.; Rubio-Aliaga, I.; Hernando, N. Renal phosphate handling and inherited disorders of phosphate reabsorption: An update. Pediatr. Nephrol. 2019, 34, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh Naderi, A.S.; Reilly, R.F. Hereditary disorders of renal phosphate wasting. Nat. Rev. Nephrol. 2010, 6, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Prié, D.; Friedlander, G. Genetic disorders of renal phosphate transport. N. Engl. J. Med. 2010, 362, 2399–2409. [Google Scholar] [CrossRef]
- Gohil, A.; Imel, E.A. FGF23 and Associated Disorders of Phosphate Wasting. Pediatr. Endocrinol. Rev. 2019, 17, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A. Progress in Understanding the Genetics of Calcium-Containing Nephrolithiasis. J. Am. Soc. Nephrol. 2017, 28, 748–759. [Google Scholar] [CrossRef]
- Walker, V.; Stansbridge, E.M.; Griffin, D.G. Demography and biochemistry of 2800 patients from a renal stones clinic. Ann. Clin. Biochem. 2013, 50 Pt 2, 127–139. [Google Scholar] [CrossRef]
- Prié, D.; Ravery, V.; Boccon-Gibod, L.; Friedlander, G. Frequency of renal phosphate leak among patients with calcium nephrolithiasis. Kidney Int. 2001, 60, 272–276. [Google Scholar] [CrossRef]
- Vilardaga, J.P.; Clark, L.J.; White, A.D.; Sutkeviciute, I.; Lee, J.Y.; Bahar, I. Molecular Mechanisms of PTH/PTHrP Class B GPCR Signaling and Pharmacological Implications. Endocr. Rev. 2023, 44, 474–491. [Google Scholar] [CrossRef]
- Murer, H.; Biber, J.; Forster, I.C.; Werner, A. Phosphate transport: From microperfusion to molecular cloning. Pflügers Arch. 2019, 471, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A. Coming out of the PiTs-novel strategies for controlling intestinal phosphate absorption in patients with CKD. Kidney Int. 2020, 98, 273–275. [Google Scholar] [CrossRef]
- Wagner, C.A. Pharmacology of mammalian Na+-dependent transporters of inorganic phosphate. In Anion Channels and Transporters: Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Montanari, A.; Pirini, M.G.; Lotrecchiano, L.; Di Prinzio, L.; Zavatta, G. Phosphaturic mesenchymal tumors with or without Phosphate Metabolism Derangements. Curr. Oncol. 2023, 30, 7478–7488. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S.; McCarthy, E.M.; Yu, A.L.; Stubbs, J.R. Correction of vascular calcification and hyperphosphatemia in CKD Rats treated with ASARM peptide. Kidney360 2022, 3, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Doshi, S.M.; Wish, J.B. Past, present, and future of phosphate management. Kidney Int. Rep. 2022, 7, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Verbueken, D.; Moe, O.W. Strategies to lower fibroblast growth factor 23 bioactivity. Nephrol. Dial. Transplant. 2022, 37, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.V.O.; Moreira, C.A.; Borba, V.Z.C. New treatments for rare bone diseases: Hypophosphatemic rickets/osteomalacia. Arch. Endocrinol. Metab. 2022, 66, 658–665. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Ranch, D.; Pereira, R.C.; Armbrecht, H.J.; Portale, A.A.; Perwad, F. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology 2012, 153, 1806–1816. [Google Scholar] [CrossRef][Green Version]
- Noda, H.; Okazaki, M.; Joyashiki, E.; Tamura, T.; Kawabe, Y.; Khatri, A.; Jueppner, H.; Potts, J.T., Jr.; Gardella, T.J.; Shimizu, M. Optimization of PTH/PTHrP hybrid peptides to derive a long-acting PTH analog (LA-PTH). J. Bone Miner. Res. Plus 2020, 4, e10367. [Google Scholar] [CrossRef]
- Tisher, C.C.; Madsen, K.M. Anatomy of the kidney. In The Kidney, 4th ed.; Brenner, B.M., Rector, F.C., Jr., Eds.; W.B. Saunders Company, Harcourt Brace Jovanovich, Inc.: West Philadelphia, PA, USA, 1991; Volume 1, pp. 3–75, Chapter 1 (proximal tubule pp. 22–35). [Google Scholar]
- McDonough, A.A. Motoring down the microvilli: Focus on “PTH-induced internalization of apical membrane NaPi2a: Role of actin and myosin VI”. Am. J. Physiol. Cell Physiol. 2009, 297, C1331–C1332. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J.; Okamura, K.; Giral, H.; Breusegem, S.; Caldas, Y.; Millard, A.; Barry, N.; Levi, M. PTH-induced internalization of apical membrane NaPi2a: Role of actin and myosin VI. Am. J. Physiol. Cell Physiol. 2009, 297, C1339–C1346. [Google Scholar] [CrossRef] [PubMed]
- Schuh, C.D.; Polesel, M.; Platonova, E.; Haenni, D.; Gassama, A.; Tokonami, N.; Ghazi, S.; Bugarski, M.; Devuyst, O.; Ziegler, U.; et al. Combined Structural and Functional Imaging of the Kidney Reveals Major Axial Differences in Proximal Tubule Endocytosis. J. Am. Soc. Nephrol. 2018, 29, 2696–2712. [Google Scholar] [CrossRef] [PubMed]
- Maddox, D.A.; Gennari, F.J. The early proximal tubule: A high-capacity delivery-responsive reabsorptive site. Am. J. Physiol. 1987, 252 Pt 2, F573–F584. [Google Scholar] [CrossRef] [PubMed]
- DuBose, T.D., Jr.; Pucacco, L.R.; Lucci, M.S.; Carter, N.W. Micropuncture determination of pH, PCO2, and total CO2 concentration in accessible structures of the rat renal cortex. J. Clin. Investig. 1979, 64, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chou, C.L.; Knepper, M.A. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol. 2015, 26, 2669–2677. [Google Scholar] [CrossRef]
- Hato, T.; Winfree, S.; Day, R.; Sandoval, R.M.; Molitoris, B.A.; Yoder, M.C.; Wiggins, R.C.; Zheng, Y.; Dunn, K.W.; Dagher, P.C. Two-Photon Intravital Fluorescence Lifetime Imaging of the Kidney Reveals Cell-Type Specific Metabolic Signatures. J. Am. Soc. Nephrol. 2017, 28, 2420–2430. [Google Scholar] [CrossRef]
- Custer, M.; Lötscher, M.; Biber, J.; Murer, H.; Kaissling, B. Expression of Na-P(i) cotransport in rat kidney: Localization by RT-PCR and immunohistochemistry. Am. J. Physiol. 1994, 266 Pt 2, F767–F774. [Google Scholar] [CrossRef] [PubMed]
- Virkki, L.V.; Biber, J.; Murer, H.; Forster, I.C. Phosphate transporters: A tale of two solute carrier families. Am. J. Physiol. Renal Physiol. 2007, 293, F643–F654. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr. Top. Membr. 2012, 70, 313–356. [Google Scholar] [CrossRef]
- Biber, J.; Hernando, N.; Forster, I. Phosphate transporters and their function. Annu. Rev. Physiol. 2013, 75, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Lederer, E. Renal phosphate transporters. Curr. Opin. Nephrol. Hypertens. 2014, 23, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O. Primary Disorders of Phosphate Metabolism. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279172/ (accessed on 18 April 2024).
- Moser, S.O.; Haykir, B.; Küng, C.J.; Bettoni, C.; Hernando, N.; Wagner, C.A. Expression of phosphate and calcium transporters and their regulators in parotid glands of mice. Pflügers Arch. 2023, 475, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Ravera, S.; Sorribas, V.; Stange, G.; Levi, M.; Murer, H.; Biber, J.; Forster, I.C. The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am. J. Physiol. Renal Physiol. 2009, 296, F691–F699. [Google Scholar] [CrossRef] [PubMed]
- Magagnin, S.; Werner, A.; Markovich, D.; Sorribas, V.; Stange, G.; Biber, J.; Murer, H. Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc. Natl. Acad. Sci. USA 1993, 90, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Forster, I.C. The molecular mechanism of SLC34 proteins: Insights from two decades of transport assays and structure-function studies. Pflügers Arch. 2019, 471, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Fenollar-Ferrer, C.; Patti, M.; Knöpfel, T.; Werner, A.; Forster, I.C.; Forrest, L.R. Structural fold and binding sites of the human Na⁺- phosphate cotransporter NaPi-II. Biophys. J. 2014, 106, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- de La Horra, C.; Hernando, N.; Forster, I.; Biber, J.; Murer, H. Amino acids involved in sodium interaction of murine type II Na+-Pi cotransporters expressed in Xenopus oocytes. J. Physiol. 2001, 531 Pt 2, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Murer, H. Functional domains in the renal type IIa Na/Pi-cotransporter. Kidney Int. 2002, 62, 375–382. [Google Scholar] [CrossRef]
- Shenolikar, S.; Voltz, J.W.; Cunningham, R.; Weinman, E.J. Regulation of ion transport by the NHERF family of PDZ proteins. Physiology 2004, 19, 362–369. [Google Scholar] [CrossRef][Green Version]
- Forster, I.; Hernando, N.; Biber, J.; Murer, H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2). J. Gen. Physiol. 1998, 112, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Waldegger, S.; Herzer, T.; Biber, J.; Markovich, D.; Hayes, G.; Murer, H.; Lang, F. Electrophysiological analysis of Na+/Pi cotransport mediated by a transporter cloned from rat kidney and expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 8205–8208. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Patti, M.; Hany SZinad, H.S.; Fearn, A.; Laude, A.; Forster, I. Molecular determinants of transport function in zebrafish Slc34a Na-phosphate transporters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1213–R1222. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.; Fenollar-Ferrer, C.; Werner, A.; Forrest, L.R.; Forster, I.C. Cation interactions and membrane potential induce conformational changes in NaPi-IIb. Biophys. J. 2016, 111, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Digman, M.A.; Cheng, M.; Breusegem, S.Y.; Halaihel, N.; Sorribas, V.; Mantulin, W.W.; Gratton, E.; Barry, N.P.; Levi, M. Partitioning of NaPi cotransporter in cholesterol-, sphingomyelin-, and glycosphingolipid-enriched membrane domains modulates NaPi protein diffusion, clustering, and activity. J. Biol. Chem. 2004, 279, 49160–49171. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Baird, B.M.; Wilson, P.V. Cholesterol modulates rat renal brush border membrane phosphate transport. J. Clin. Investig. 1990, 85, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, A.I.; Sarasa, M.; Raldúa, D.; Aramayona, J.; Morales, R.; Biber, J.; Murer, H.; Levi, M.; Sorribas, V. Role of thyroid hormone in regulation of renal phosphate transport in young and aged rats. Endocrinology 1999, 140, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Sorribas, V.; Lötscher, M.; Loffing, J.; Biber, J.; Kaissling, B.; Murer, H.; Levi, M. Cellular mechanisms of the age-related decrease in renal phosphate reabsorption. Kidney Int. 1996, 50, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Breusegem, S.Y.; Takahashi, H.; Giral-Arnal, H.; Wang, X.; Jiang, T.; Verlander, J.W.; Wilson, P.; Miyazaki-Anzai, S.; Sutherland, E.; Caldas, Y.; et al. Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am. J. Physiol. Renal Physiol. 2009, 297, F350–F361. [Google Scholar] [CrossRef]
- Zajicek, H.K.; Wang, H.; Puttaparthi, K.; Halaihel, N.; Markovich, D.; Shayman, J.; Béliveau, R.; Wilson, P.; Rogers, T.; Levi, M. Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int. 2001, 60, 694–704. [Google Scholar] [CrossRef]
- Weinman, E.J.; Steplock, D.; Shenolikar, S.; Blanpied, T.A. Dynamics of PTH-induced disassembly of Npt2a/NHERF-1 complexes in living OK cells. Am. J. Physiol. Renal Physiol. 2011, 300, F231–F235. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Segawa, H.; Kaneko, I.; Takahashi, A.; Kuwahata, M.; Ito, M.; Ohkido, I.; Tatsumi, S.; Miyamoto, K. Growth-related renal type II Na/Pi cotransporter. J. Biol. Chem. 2002, 277, 19665–19672. [Google Scholar] [CrossRef]
- Ohkido, I.; Segawa, H.; Yanagida, R.; Nakamura, M.; Miyamoto, K. Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflügers Arch. Eur. J. Physiol. 2003, 446, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.; Karaplis, A.C.; Amizuka, N.; Hewson, A.S.; Ozawa, H.; Tenenhouse, H.S. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 5372–5377. [Google Scholar] [CrossRef] [PubMed]
- Segawa, H.; Onitsuka, A.; Furutani, J.; Kaneko, I.; Aranami, F.; Matsumoto, N.; Tomoe, Y.; Kuwahata, M.; Ito, M.; Matsumoto, M.; et al. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am. J. Physiol. Renal Physiol. 2009, 297, F671–F678. [Google Scholar] [CrossRef] [PubMed]
- Jaureguiberry, G.; Carpenter, T.O.; Forman, S.; Jüppner, H.; Bergwitz, C. A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am. J. Physiol. Renal. Physiol. 2008, 295, F371–F379. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Li, D.; Doyle, D.; Zaritsky, J.; Levine, M.A. Digenic heterozygous mutations in SLC34A3 and SLC34A1 cause dominant hypophosphatemic rickets with hypercalciuria. J. Clin. Endocrinol. Metab. 2020, 105, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Nowik, M.; Picard, N.; Stange, G.; Capuano, P.; Tenenhouse, H.S.; Biber, J.; Murer, H.; Wagner, C.A. Renal phosphaturia during metabolic acidosis revisited: Molecular mechanisms for decreased renal phosphate reabsorption. Pflügers Arch. 2008, 457, 539–549. [Google Scholar] [CrossRef]
- Giral, H.; Lanzano, L.; Caldas, Y.; Blaine, J.; Verlander, J.W.; Lei, T.; Gratton, E.; Levi, M. Role of PDZ domain containing 1 (PDZK1) in apical membrane expression of renal Na-coupled phosphate (Na/Pi) transporters. J. Biol. Chem. 2011, 286, 15032–15042. [Google Scholar] [CrossRef]
- Segawa, H.; Kaneko, I.; Shiozaki, Y.; Ito, M.; Tatsumi, S.; Miyamoto, K.-i. Molecular control of growth-related sodium-phosphate co- transporter (SLC34A3). Curr. Mol. Biol. Rep. 2019, 5, 26–33. [Google Scholar] [CrossRef]
- Segawa, H.; Yamanaka, S.; Ito, M.; Kuwahata, M.; Shono, M.; Yamamoto, T.; Miyamoto, K. Internalization of renal type IIc Na-Pi cotransporter in response to a high-phosphate diet. Am. J. Physiol. Renal Physiol. 2005, 288, F587–F596. [Google Scholar] [CrossRef][Green Version]
- Hori, M.; Shimizu, Y.; Fukumoto, S. Minireview: Fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. Endocrinology 2011, 152, 4–10. [Google Scholar] [CrossRef]
- Tomoe, Y.; Segawa, H.; Shiozawa, K.; Kaneko, I.; Tominaga, R.; Hanabusa, E.; Aranami, F.; Furutani, J.; Kuwahara, S.; Tatsumi, S.; et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am. J. Physiol. Renal Physiol. 2010, 298, F1341–F1350. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Shiozaki, Y.; Segawa, H.; Nishiguchi, S.; Hanazaki, A.; Noguchi, M.; Kirino, R.; Sasaki, S.; Tanifuji, K.; Koike, M.; et al. Analysis of opossum kidney NaPi-IIc sodium-dependent phosphate transporter to understand Pi handling in human kidney. Clin. Exp. Nephrol. 2019, 23, 313–324. [Google Scholar] [CrossRef]
- Bergwitz, C.; Miyamoto, K.I. Hereditary hypophosphatemic rickets with hypercalciuria: Pathophysiology, clinical presentation, diagnosis and therapy. Pflügers Arch. 2019, 471, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Capuano, P.; Stange, G.; Mihailova, M.; Kaissling, B.; Murer, H.; Biber, J.; Wagner, C.A. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflügers Arch. 2010, 460, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Weinman, E.J.; Steplock, D.; Shenolikar, S. CAMP-mediated inhibition of the renal brush border membrane Na+-H+ exchanger requires a dissociable phosphoprotein cofactor. J. Clin. Investig. 1993, 92, 1781–1786. [Google Scholar] [CrossRef]
- Weinman, E.J.; Shenolikar, S. The Na-H exchanger regulatory factor. Exp. Nephrol. 1997, 5, 449–452. [Google Scholar] [PubMed]
- Reczek, D.; Berryman, M.; Bretscher, A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J. Cell Biol. 1997, 139, 169–179. [Google Scholar] [CrossRef]
- Ardura, J.A.; Friedman, P.A. Regulation of G protein-coupled receptor function by Na+/H+ exchange regulatory factors. Pharmacol. Rev. 2011, 63, 882–900. [Google Scholar] [CrossRef] [PubMed]
- Hernando, N.; Gisler, S.M.; Pribanic, S.; Déliot, N.; Capuano, P.; Wagner, C.A.; Moe, O.W.; Biber, J.; Murer, H. NaPi-IIa and interacting partners. J. Physiol. 2005, 567, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Stanley, C.B.; Heller, W.T.; Friedman, P.A.; Bu, Z. Dynamic structure of the full-length scaffolding protein NHERF1 influences signaling complex assembly. J. Biol. Chem. 2019, 294, 11297–11310. [Google Scholar] [CrossRef] [PubMed]
- Hernando, N.; Wagner, C.A.; Gisler, S.M.; Biber, J.; Murer, H.; Hernando, N.; Wagner, C.A.; Gisler, S.M.; Biber, J.; Murer, H. PDZ proteins and proximal ion transport. Curr. Opin. Nephrol. Hypertens. 2004, 13, 569–574. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Bellini, M.; Inuzuka, H.; Xu, J.; Xiong, Y.; Yang, X.; Castleberry, A.M.; Hall, R.A. Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. J. Biol. Chem. 2006, 281, 2820–2827. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A.; Premont, R.T.; Chow, C.W.; Blitzer, J.T.; Pitcher, J.A.; Claing, A.; Stoffel, R.H.; Barak, L.S.; Shenolikar, S.; Weinman, E.J.; et al. The beta2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature 1998, 392, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Mamonova, T.; Friedman, P.A. Noncanonical Sequences Involving NHERF1 Interaction with NPT2A Govern Hormone-Regulated Phosphate Transport: Binding Outside the Box. Int. J. Mol. Sci. 2021, 22, 1087. [Google Scholar] [CrossRef] [PubMed]
- Weinman, E.J.; Lederer, E.D. NHERF-1 and the regulation of renal phosphate reabsoption: A tale of three hormones. Am. J. Physiol. Renal Physiol. 2012, 303, F321–F327. [Google Scholar] [CrossRef] [PubMed]
- Vistrup-Parry, M.; Sneddon, W.B.; Bach, S.; Strømgaard, K.; Friedman, P.A.; Mamonova, T. Multisite NHERF1 phosphorylation controls GRK6A regulation of hormone-sensitive phosphate transport. J. Biol. Chem. 2021, 296, 100473. [Google Scholar] [CrossRef]
- Karim, Z.; Gérard, B.; Bakouh, N.; Alili, R.; Leroy, C.; Beck, L.; Silve, C.; Planelles, G.; Urena-Torres, P.; Grandchamp, B.; et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N. Engl. J. Med. 2008, 359, 1128–1135. [Google Scholar] [CrossRef]
- Li, J.; Callaway, D.J.; Bu, Z. Ezrin induces long-range interdomain allostery in the scaffolding protein NHERF1. J. Mol. Biol. 2009, 392, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Farago, B.; Li, J.; Cornilescu, G.; Callaway, D.J.; Bu, Z. Activation of nanoscale allosteric protein domain motion revealed by neutron spin echo spectroscopy. Biophys. J. 2010, 99, 3473–3482. [Google Scholar] [CrossRef] [PubMed]
- Weinman, E.J.; Steplock, D.; Zhang, Y.; Biswas, R.; Bloch, R.J.; Shenolikar, S. Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J. Biol. Chem. 2010, 285, 25134–25138. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Thompson, J.R. The regulation of parathyroid hormone secretion and synthesis. J. Am. Soc. Nephrol. 2011, 22, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Naveh-Many, T.; ASela-Brown, J. Silver, Protein-RNA interactions in the regulation of PTH gene expression by calcium and phosphate. Nephrol. Dial. Transplant. 1999, 14, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Almaden, Y.; Canalejo, A.; Hernandez, A.; Ballesteros, E.; Garcia-Navarro, S.; Torres, A.; Rodriguez, M. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J. Bone Miner. Res. 1996, 11, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Taketani, Y.; Segawa, H.; Chikamori, M.; Morita, K.; Tanaka, K.; Kido, S.; Yamamoto, H.; Iemori, Y.; Tatsumi, S.; Tsugawa, N.; et al. Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3. Identification of a vitamin D-responsive element in the human NAPi-3 gene. J. Biol. Chem. 1998, 273, 14575–14581. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Naveh-Many, T. FGF23 and the parathyroid. Adv. Exp. Med. Biol. 2012, 728, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Naveh-Many, T.; Silver, J. The Pas de Trois of Vitamin D, FGF23, and PTH. J. Am. Soc. Nephrol. 2017, 28, 393–395. [Google Scholar] [CrossRef]
- Chanakul, A.; Zhang, M.Y.; Louw, A.; Armbrecht, H.J.; Miller, W.L.; Portale, A.A.; Perwad, F. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PLoS ONE 2013, 8, e72816. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sutliff, R.L.; Qian, J.; Lorenz, J.N.; Wang, J.; Tang, H.; Nakayama, T.; Weber, C.; Witte, D.; Strauch, A.R.; et al. Targeted overexpression of parathyroid hormone-related protein (PTHrP) to vascular smooth muscle in transgenic mice lowers blood pressure and alters vascular contractility. Endocrinology 1999, 140, 1815–1825. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Strewler, G.J. The physiology of parathyroid hormone-related protein. N. Engl. J. Med. 2000, 342, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Pioszak, A.A.; Parker, N.R.; Gardella, T.J.; Xu, H.E. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J. Biol. Chem. 2009, 284, 28382–28391. [Google Scholar] [CrossRef]
- Ehrenmann, J.; Schöppe, J.; Klenk, C.; Plückthun, A. New views into class B GPCRs from the crystal structure of PTH1R. FEBS J. 2019, 286, 4852–4860. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Mao, C.; Shen, Q.; Zang, S.; Shen, D.D.; Zhang, H.; Chen, Z.; Wang, G.; Zhang, C.; Zhang, Y.; et al. Molecular insights into the distinct signaling duration for the peptide-induced PTH1R activation. Nat. Commun. 2022, 13, 6276. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.T.; Dimke, H. Effects of parathyroid hormone on renal tubular calcium and phosphate handling. Acta Physiol. 2023, 238, e13959. [Google Scholar] [CrossRef]
- Traebert, M.; Völkl, H.; Biber, J.; Murer, H.; Kaissling, B. Luminal and contraluminal action of 1-34 and 3-34 PTH peptides on renal type IIa Na-Pi cotransporter. Am. J. Physiol. Renal. Physiol. 2000, 278, F792–F798. [Google Scholar] [CrossRef]
- Weinman, E.J.; Lederer, E.D. PTH-mediated inhibition of the renal transport of phosphate. Exp. Cell Res. 2012, 318, 1027–1032. [Google Scholar] [CrossRef]
- Klenk, C.; Hommers, L.; Lohse, M.J. Proteolytic cleavage of the extracellular domain affects signaling of parathyroid hormone 1 receptor. Front. Endocrinol. 2022, 13, 839351. [Google Scholar] [CrossRef]
- Cary, B.P.; Zhang, X.; Cao, J.; Johnson, R.M.; Piper, S.J.; Gerrard, E.J.; Wootten, D.; Sexton, P.M. New Insights into the Structure and Function of Class B1 GPCRs. Endocr. Rev. 2023, 44, 492–517. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Jeong, Y.; Simms, J.; Warner, M.L.; Poyner, D.R.; Chung, K.Y.; Pioszak, A.A. Calcitonin Receptor N-Glycosylation Enhances Peptide Hormone Affinity by Controlling Receptor Dynamics. J. Mol. Biol. 2020, 432, 1996–2014. [Google Scholar] [CrossRef] [PubMed]
- Bisello, A.; Greenberg, Z.; Behar, V.; Rosenblatt, M.; Suva, L.J.; Chorev, M. Role of glycosylation in expression and function of the human parathyroid hormone/parathyroid hormone-related protein receptor. Biochemistry 1996, 35, 15890–15895. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J. Family-B G-protein-coupled receptors. Genome Biol. 2001, 2, reviews3013.1. [Google Scholar] [CrossRef] [PubMed]
- Gardella, T.J.; Luck, M.D.; Fan, M.H.; Lee, C. Transmembrane residues of the parathyroid hormone (PTH)/PTH-related peptide receptor that specifically affect binding and signaling by agonist ligands. J. Biol. Chem. 1996, 271, 12820–12825. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.P.; Vilardarga, J.P.; Baranski, T.J.; Lichtarge, O.; Iiri, T.; Meng, E.C.; Nissenson, R.A.; Bourne, H.R. Similar structures and shared switch mechanisms of the beta2-adrenoceptor and the parathyroid hormone receptor. Zn(II) bridges between helices III and VI block activation. J. Biol. Chem. 1999, 274, 17033–17041. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Leiro, R.; Scheres, S.H. Unravelling biological macromolecules with cryo-electron microscopy. Nature 2016, 537, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Nemec, K.; Schihada, H.; Kleinau, G.; Zabel, U.; Grushevskyi, E.O.; Scheerer, P.; Lohse, M.J.; Maiellaro, I. Functional modulation of PTH1R activation and signaling by RAMP2. Proc. Natl. Acad. Sci. USA 2022, 119, e2122037119. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Bünemann, M.; Krasel, C.; Castro, M.; Lohse, M.J. Measurement of the millisecond activation switch of G protein- coupled receptors in living cells. Nat. Biotechnol. 2003, 21, 807–812. [Google Scholar] [CrossRef]
- Castro, M.; Nikolaev, V.O.; Palm, D.; Lohse, M.J.; Vilardaga, J.P. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 16084–16089. [Google Scholar] [CrossRef]
- Syme, C.A.; Friedman, P.A.; Bisello, A. Parathyroid hormone receptor trafficking contributes to the activation of extracellular signal-regulated kinases but is not required for regulation of cAMP signaling. J. Biol. Chem. 2005, 280, 11281–11288. [Google Scholar] [CrossRef] [PubMed]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M.; et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Romero, G.; Feinstein, T.N.; Wehbi, V.L. Kinetics and dynamics in the G protein-coupled receptor signaling cascade. Methods Enzymol. 2013, 522, 337–363. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Miller, L.J.; Koole, C.; Christopoulos, A.; Sexton, P.M. Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem. Rev. 2017, 117, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Barbash, S.; Kongsamut, S.; Eishingdrelo, A.; Sakmar, T.P.; Eishingdrelo, H. 14-3-3 signal adaptor and scaffold proteins mediate GPCR trafficking. Sci. Rep. 2019, 9, 11156. [Google Scholar] [CrossRef] [PubMed]
- Dicker, F.; Quitterer, U.; Winstel, R.; Honold, K.; Lohse, M.J. Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc. Natl. Acad. Sci. USA 1999, 96, 5476–5481. [Google Scholar] [CrossRef] [PubMed]
- Ferrandon, S.; Feinstein, T.N.; Castro, M.; Wang, B.; Bouley, R.; Potts, J.T.; Gardella, T.J.; Vilardaga, J.P. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 2009, 5, 734–742. [Google Scholar] [CrossRef]
- Maeda, A.; Okazaki, M.; Baron, D.M.; Dean, T.; Khatri, A.; Mahon, M.; Segawa, H.; Abou-Samra, A.B.; Jüppner, H.; Bloch, K.D.; et al. Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH. Proc. Natl. Acad. Sci. USA 2013, 110, 5864–5869. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.P.; Gardella, T.J. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar] [CrossRef]
- Swinney, D.C. Biochemical mechanisms of drug action: What does it take for success? Nat. Rev. Drug Discov. 2004, 3, 801–808. [Google Scholar] [CrossRef]
- Lee, M.; Partridge, N.C. Parathyroid hormone signaling in bone and kidney. Curr. Opin. Nephrol. Hypertens. 2009, 18, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Tawfeek, H.A.; Abou-Samra, A.B. Negative regulation of parathyroid hormone (PTH)-activated phospholipase C by PTH/PTH- related peptide receptor phosphorylation and protein kinase A. Endocrinology 2008, 149, 4016–4023. [Google Scholar] [CrossRef][Green Version]
- Wehbi, V.L.; Stevenson, H.P.; Feinstein, T.N.; Calero, G.; Romero, G.; Vilardaga, J.P. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc. Natl. Acad. Sci. USA 2013, 110, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, T.N.; Wehbi, V.L.; Ardura, J.A.; Wheeler, D.S.; Ferrandon, S.; Gardella, T.J.; Vilardaga, J.P. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat. Chem. Biol. 2011, 7, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Jean-Alphonse, F.G.; Wehbi, V.L.; Chen, J.; Noda, M.; Taboas, J.M.; Xiao, K.; Vilardaga, J.P. β2-adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat. Chem. Biol. 2017, 13, 259–261. [Google Scholar] [CrossRef] [PubMed]
- White, A.D.; Jean-Alphonse, F.G.; Fang, F.; Peña, K.A.; Liu, S.; König, G.M.; Inoue, A.; Aslanoglou, D.; Gellman, S.H.; Kostenis, E.; et al. Gq/11-dependent regulation of endosomal cAMP generation by parathyroid hormone class B GPCR. Proc. Natl. Acad. Sci. USA 2020, 117, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Gidon, A.; Al-Bataineh, M.M.; Jean-Alphonse, F.G.; Stevenson, H.P.; Watanabe, T.; Louet, C.; Khatri, A.; Calero, G.; Pastor-Soler, N.M.; Gardella, T.J.; et al. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat. Chem. Biol. 2014, 10, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Bastepe, M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm. Res. Paediatr. 2013, 80, 229–241. [Google Scholar] [CrossRef]
- Lee, J.H.; Davaatseren, M.; Lee, S. Rare PTH Gene Mutations Causing Parathyroid Disorders: A Review. Endocrinol. Metab. 2020, 35, 64–70. [Google Scholar] [CrossRef]
- Lemos, M.C.; Thakker, R.V. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders. Hum. Mutat. 2015, 36, 11–19. [Google Scholar] [CrossRef]
- Yu, S.; Yu, D.; Lee, E.; Eckhaus, M.; Lee, R.; Corria, Z.; Accili, D.; Westphal, H.; Weinstein, L.S. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein α-subunit (Gsα) knockout mice is due to tissue-specific imprinting of the Gsα gene. Proc. Natl. Acad. Sci. USA 1998, 95, 8715–8720. [Google Scholar] [CrossRef] [PubMed]
- Bastepe, M.; Raas-Rothschild, A.; Silver, J.; Weissman, I.; Wientroub, S.; Jüppner, H.; Gillis, D. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J. Clin. Endocrinol. Metab. 2004, 89, 3595–3600. [Google Scholar] [CrossRef][Green Version]
- Schipani, E.; Kruse, K.; Jüppner, H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science 1995, 268, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Savoldi, G.; Izzi, C.; Signorelli, M.; Bondioni, M.P.; Romani, C.; Lanzi, G.; Moratto, D.; Verdoni, L.; Pinotti, M.; Prefumo, F.; et al. Prenatal presentation and postnatal evolution of a patient with Jansen metaphyseal dysplasia with a novel missense mutation in PTH1R. Am. J. Med. Genet. A 2013, 161A, 2614–2619. [Google Scholar] [CrossRef]
- Parkinson, D.B.; Thakker, R.V. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet. 1992, 1, 149–152. [Google Scholar] [CrossRef]
- Chase, L.R.; Melson, G.L.; Aurbach, G.D. Pseudohypoparathyroidism: Defective excretion of 3′,5′-AMP in response to parathyroid hormone. J. Clin. Investig. 1969, 48, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Burnett, C.H.; Smith, P.H.; Parson, W. Pseudohypoparathyroidism—An example of ‘Seabright-Bantam syndrome’. Endocrinology 1942, 30, 922–932. [Google Scholar]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef]
- Bouvet, M.; Blondeau, J.-P.; Lezoualc’h, F. The Epac1 protein: Pharmacological modulators, cardiac signalosome and pathophysiology. Cells 2019, 8, 1543. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Tomilin, V.N.; Pochynyuk, O. A peek into Epac physiology in the kidney. Am. J. Physiol. Renal. Physiol. 2019, 327, F1094–F1097. [Google Scholar] [CrossRef] [PubMed]
- Honegger, K.J.; Capuano, P.; Winter, C.; Bacic, D.; Stange, G.; Wagner, C.A.; Biber, J.; Murer, H.; Hernando, N. Regulation of sodium- proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC). Proc. Natl. Acad. Sci. USA 2006, 103, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Konings, I.B.; Zhao, J.; Price, L.S.; de Heer, E.; Deen, P.M. Renal expression of exchange protein directly activated by cAMP (Epac) 1 and 2. Am. J. Physiol. Renal. Physiol. 2008, 295, F525–F533. [Google Scholar] [CrossRef] [PubMed]
- Lee, K. Epac: New emerging cAMP-binding protein. BMB Rep. 2021, 54, 149–156. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide- exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Rehmann, H.; van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of regulation of the Epac family of cAMP- dependent RapGEFs. J. Biol. Chem. 2000, 275, 20829–20836. [Google Scholar] [CrossRef] [PubMed]
- Frische, E.W.; Zwartkruis, F.J. Rap1, a mercenary among the Ras-like GTPases. Dev. Biol. 2010, 340, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cherezova, A.; Tomilin, V.; Buncha, V.; Zaika, O.; Ortiz, P.A.; Mei, F.; Cheng, X.; Mamenko, M.; Pochynyuk, O. Urinary concentrating defect in mice lacking Epac1 or Epac2. FASEB J. 2019, 33, 2156–2170. [Google Scholar] [CrossRef]
- Friedman, P.A.; Sneddon, W.B.; Mamonova, T.; Montanez-Miranda, C.; Ramineni, S.; Harbin, N.H.; Squires, K.E.; Gefter, J.V.; Magyar, C.E.; Emlet, D.R.; et al. RGS14 regulates PTH- and FGF23-sensitive NPT2A-mediated renal phosphate uptake via binding to the NHERF1 scaffolding protein. J. Biol. Chem. 2022, 298, 101836. [Google Scholar] [CrossRef]
- Armando, I.; Van Anthony, M.V.; Jose, P.A. Dopamine and Renal Function and Blood Pressure Regulation. Comp. Physiol. 2011, 1, 1075–1117. [Google Scholar] [CrossRef] [PubMed]
- Jose, P.A.; Soares-da-Silva, P.; Eisner, G.M.; Felder, R.A. Dopamine and G protein-coupled receptor kinase 4 in the kidney: Role in blood pressure regulation. Biochim. Biophys. Acta 2010, 1802, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Zhang, M.Z. Dopamine, the kidney, and hypertension. Curr. Hypertens. Rep. 2012, 14, 138–143. [Google Scholar] [CrossRef]
- Adam, W.R.; Adams, B.A. Production and excretion of dopamine by the isolated perfused rat kidney. Kidney Blood Press. Res. 1985, 8, 150–158. [Google Scholar] [CrossRef]
- Baines, A.D. Effects of salt intake and renal denervation on catecholamine catabolism and excretion. Kidney Int. 1982, 21, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Berndt, T.J.; Khraibi, A.A.; Thothathri, V.; Dousa, T.P.; Tyce, G.M.; Knox, F.G. Effect of increased dietary phosphate intake on dopamine excretion in the presence and absence of the renal nerves. Min. Electrolyte Metab. 1994, 20, 158–162. [Google Scholar]
- Stephenson, R.K.; Sole, M.J.; Baines, A.D. Neural and extraneural catecholamine production by rat kidneys. Am. J. Physiol. 1982, 242, F261–F266. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Siragy, H.M.; Felder, R.A.; Carey, R.M. Intrarenal dopamine production and distribution in the rat: Physiological control of sodium excretion. Hypertension 1997, 29 Pt 2, 228–234. [Google Scholar] [CrossRef]
- Suzuki, H.; Nakane, H.; Kawamura, M.; Yoshizawa, M.; Takeshita, E.; Saruta, T. Excretion and metabolism of dopa and dopamine by isolated perfused rat kidney. Am. J. Physiol. 1984, 247 Pt 1, E285–E290. [Google Scholar] [CrossRef]
- Wolfovitz, E.; Grossman, E.; Folio, C.J.; Keiser, H.R.; Kopin, I.J.; Goldstein, D.S. Derivation of urinary dopamine from plasma dihydroxyphenylalanine in humans. Clin. Sci. 1993, 84, 549–557. [Google Scholar] [CrossRef]
- Zimlichman, R.; Levinson, P.D.; Kelly, G.; Stull, R.; Keiser, H.R.; Goldstein, D.S. Derivation of urinary dopamine from plasma dopa. Clin. Sci. 1988, 75, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Eldrup, E.; Hetland, M.L.; Christensen, N.J. Increase in plasma 3,4-dihydroxyphenylalanine (DOPA) appearance rate after inhibition of DOPA decarboxylase in humans. Eur. J. Clin. Investig. 1994, 24, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E.; Hoffman, A.; Armando, I.; Abassi, Z.; Kopin, I.J.; Goldstein, D.S. Sympathoadrenal contribution to plasma dopa (3,4- dihydroxyphenylalanine) in rats. Clin. Sci. 1992, 83, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Soares-Da-Silva, P.; Serrão, M.P.; Vieira-Coelho, M.A. Apical and basolateral uptake and intracellular fate of dopamine precursor L-dopa in LLC-PK1 cells. Am. J. Physiol. 1998, 274, F243–F251. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Soares-da-Silva, P. Na+-independent transporters, LAT-2 and b0,+, exchange L-DOPA with neutral and basic amino acids in two clonal renal cell lines. J. Membr. Biol. 2002, 186, 63–80. [Google Scholar] [CrossRef] [PubMed]
- de Toledo, F.G.; Beers, K.W.; Berndt, T.J.; Thompson, M.A.; Tyce, G.M.; Knox, F.G.; Dousa, T.P. Opposite paracrine effects of 5-HT and dopamine on Na+-Pi cotransport in opossum kidney cells. Kidney Int. 1997, 52, 152–156. [Google Scholar] [CrossRef][Green Version]
- Wassenberg, T.; Monnens, L.A.; Geurtz, B.P.; Wevers, R.A.; Verbeek, M.M.; Willemsen, M.A. The paradox of hyperdopaminuria in aromatic L-amino Acid deficiency explained. JIMD Rep. 2012, 4, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.H.; Pestana, M.; Soares-da-Silva, P. Deamination of newly-formed dopamine in rat renal tissues. Br. J. Pharmacol. 1991, 102, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, J.T.; Soares-da-Silva, P. The activity of MAO A and B in rat renal cells and tubules. Life Sci. 1998, 62, 727–737. [Google Scholar] [CrossRef]
- Xu, J.; Li, G.; Wang, P.; Velazquez, H.; Yao, X.; Li, Y.; Wu, Y.; Peixoto, A.; Crowley, S.; Desir, G.V. Renalase is a novel, soluble monoamine oxidase that regulates cardiac function and blood pressure. J. Clin. Investig. 2005, 115, 1275–1280. [Google Scholar] [CrossRef]
- Isaac, J.; Berndt, T.J.; Chinnow, S.L.; Tyce, G.M.; Dousa, T.P.; Knox, F.G. Dopamine enhances the phosphaturic response to parathyroid hormone in phosphate-deprived rats. J. Am. Soc. Nephrol. 1992, 2, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Berndt, T.J.; MacDonald, A.; Walikonis, R.; Chinnow, S.; Dousa, T.P.; Tyce, G.M.; Knox, F.G. Excretion of catecholamines and metabolites in response to increased dietary phosphate intake. J. Lab. Clin. Med. 1993, 122, 80–84. [Google Scholar]
- Weinman, E.J.; Biswas, R.; Steplock, D.; Wang, P.; Lau, Y.S.; Desir, G.V.; Shenolikar, S. Increased renal dopamine and acute renal adaptation to a high-phosphate diet. Am. J. Physiol. Renal Physiol. 2011, 300, F1123–F1129. [Google Scholar] [CrossRef]
- Sizova, D.; Velazquez, H.; Sampaio-Maia, B.; Quelhas-Santos, J.; Pestana, M.; Desir, G.V. Renalase regulates renal dopamine and phosphate metabolism. Am. J. Physiol. Renal Physiol. 2013, 305, F839–F844. [Google Scholar] [CrossRef]
- Quelhas-Santos, J.; Serrão, M.P.; Soares-Silva, I.; Fernandes-Cerqueira, C.; Simões-Silva, L.; Pinho, M.J.; Remião, F.; Sampaio-Maia, B.; Desir, G.V.; Pestana, M. Renalase regulates peripheral and central dopaminergic activities. Am. J. Physiol. Renal Physiol. 2015, 308, F84–F91. [Google Scholar] [CrossRef]
- O’Connell, D.P.; Botkin, S.J.; Ramos, S.I.; Sibley, D.R.; Ariano, M.A.; Felder, R.A.; Carey, R.M. Localization of dopamine D1A receptor protein in rat kidneys. Am. J. Physiol. 1995, 268 Pt 2, F1185–F1197. [Google Scholar] [CrossRef]
- Felder, C.C.; McKelvey, A.M.; Gitler, M.S.; Eisner, G.M.; Jose, P.A. Dopamine receptor subtypes in renal brush border and basolateral membranes. Kidney Int. 1989, 36, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.; Iwasiow, R.M.; Chaar, Z.Y.; Nantel, M.F.; Tiberi, M. Homologous regulation of the heptahelical D1A receptor responsiveness: Specific cytoplasmic tail regions mediate dopamine-induced phosphorylation, desensitization and endocytosis. J. Neurochem. 2002, 82, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.M.; Sibley, D.R. The role of phosphorylation in D1 dopamine receptor desensitization: Evidence for a novel mechanism of arrestin association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef]
- Tsao, P.; Cao, T.; von Zastrow, M. Role of endocytosis in mediating downregulation of G-protein-coupled receptors. Trends Pharmacol. Sci. 2001, 22, 91–96. [Google Scholar] [CrossRef]
- Weinman, E.J.; Biswas, R.; Steplock, D.; Douglass, T.S.; Cunningham, R.; Shenolikar, S. Sodium-hydrogen exchanger regulatory factor 1 (NHERF-1)transduces signals that mediate dopamine inhibition of sodium-phosphate co-transport in mouse kidney. J. Biol. Chem. 2010, 285, 13454–13460. [Google Scholar] [CrossRef] [PubMed]
- Jose, P.A.; Eisner, G.M.; Felder, R.A. Renal dopamine and sodium homeostasis. Curr. Hypertens. Rep. 2000, 2, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Lachowicz, J.E.; Sibley, D.R. Phenotypic analysis of dopamine receptor knockout mice; recent insights into the functional specificity of dopamine receptor subtypes. Neuropharmacology 2004, 47, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Shultz, P.J.; Sedor, J.R.; Abboud, H.E. Dopaminergic stimulation of cAMP accumulation in cultured rat mesangial cells. Am. J. Physiol. 1987, 253 Pt 2, H358–H364. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; White, B.H.; Sidhu, A. Coupling of human D-1 dopamine receptors to different guanine nucleotide binding proteins. Evidence that D-1 dopamine receptors can couple to both Gs and Go. J. Biol. Chem. 1995, 270, 14672–14678. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Jose, P.A.; Axelrod, J. The dopamine-1 agonist, SKF 82526, stimulates phospholipase-C activity independent of adenylate cyclase. J. Pharmacol. Exp. Ther. 1989, 248, 171–175. [Google Scholar] [PubMed]
- Jin, L.Q.; Wang, H.Y.; Friedman, E. Stimulated D1 dopamine receptors couple to multiple Gα proteins in different brain regions. J. Neurochem. 2001, 78, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.J.; Eichberg, J.; Lokhandwala, M.F. Characterization of receptors involved in dopamine-induced activation of phospholipase-C in rat renal cortex. J. Pharmacol. Exp. Ther. 1992, 260, 134–139. [Google Scholar] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef]
- White, K.E.; Larsson, T.E.; Econs, M.J. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: Frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr. Rev. 2006, 27, 221–241. [Google Scholar] [CrossRef]
- Shimada, T.; Muto, T.; Urakawa, I.; Yoneya, T.; Yamazaki, Y.; Okawa, K.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 2002, 143, 3179–3182. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Maeda, A.; Ohtomo, S.; Hirata, M.; Kusano, K.; Kato, S.; Ogata, E.; Segawa, H.; Miyamoto, K.; Fukushima, N. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus In Vivo. J. Biol. Chem. 2005, 280, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sabbagh, Y.; Davis, S.I.; Demay, M.B.; White, K.E. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone 2005, 36, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Perwad, F.; Azam, N.; Zhang, M.Y.; Yamashita, T.; Tenenhouse, H.S.; Portale, A.A. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 2005, 146, 5358–5364. [Google Scholar] [CrossRef] [PubMed]
- Courbebaisse, M.; Lanske, B. Biology of Fibroblast Growth Factor 23: From Physiology to Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a031260. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Maeda, T.; Kawane, T.; Matsunuma, A.; Horiuchi, N. Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1alpha,25-dihydroxyvitamin D3 synthesis in leptin-deficient mice. J. Bone Miner Res. 2010, 25, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Quarles, L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 2007, 18, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Tenenhouse, H.S.; Gauthier, C.; Chau, H.; St-Arnaud, R. 1alpha-Hydroxylase gene ablation and Pi supplementation inhibit renal calcification in mice homozygous for the disrupted Npt2a gene. Am. J. Physiol. Renal Physiol. 2004, 286, F675–F681. [Google Scholar] [CrossRef]
- Knab, V.M.; Corbin, B.; Andrukhova, O.; Hum, J.M.; Ni, P.; Rabadi, S.; Maeda, A.; White, K.E.; Erben, R.G.; Jüppner, H.; et al. Acute parathyroid hormone injection increases C-terminal but not intact fibroblast growth factor 23 levels. Endocrinology 2017, 158, 1130–1139. [Google Scholar] [CrossRef]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Ni, P.; Noonan, M.L.; White, K.E. Osteocytic FGF23 and Its Kidney Function. Front. Endocrinol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Phan, P.; Saikia, B.B.; Sonnaila, S.; Agrawal, S.; Alraawi, Z.; Kumar, T.K.S.; Iyer, S. The Saga of Endocrine FGFs. Cells 2021, 10, 2418. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [PubMed]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pagès, A.; Lorenz-Depiereux, B.; Bennett, E.P.; Mandel, U.; Strom, T.M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Sitara, D.; Razzaque, M.S.; Hesse, M.; Yoganathan, S.; Taguchi, T.; Erben, R.G.; Jüppner, H.; Lanske, B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004, 23, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, L.; Brady, R. McCune-Albright Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537092/ (accessed on 10 July 2023).
- Leet, A.I.; Collins, M.T. Current approach to fibrous dysplasia of bone and McCune-Albright syndrome. J. Child. Orthop. 2007, 1, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Hori, M.; Taguchi, M.; Fukumoto, S. Functional analysis of mutant FAM20C in Raine syndrome with FGF23-related hypophosphatemia. Bone 2014, 67, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Palma-Lara, I.; Pérez-Ramírez, M.; García Alonso-Themann, P.; Espinosa-García, A.M.; Godinez-Aguilar, R.; Bonilla-Delgado, J.; López-Ornelas, A.; Victoria-Acosta, G.; Olguín-García, M.G.; Moreno, J.; et al. FAM20C Overview: Classic and novel targets, pathogenic variants and Raine Syndrome phenotypes. Int. J. Mol. Sci. 2021, 22, 8039. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef] [PubMed]
- Gattineni, J.; Alphonse, P.; Zhang, Q.; Mathews, N.; Bates, C.M.; Baum, M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am. J. Physiol. Renal Physiol. 2014, 306, F351–F358. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Ni, P.; Agoro, R.; White, K.E.; DiMarchi, R.D. Identification of a second Klotho interaction site in the C terminus of FGF23. Cell Rep. 2021, 34, 108665. [Google Scholar] [CrossRef]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Addo, T.; Cho, H.J.; Barker, S.L.; Ravikumar, P.; Gillings, N.; Bian, A.; Sidhu, S.S.; et al. Renal production, uptake, and handling of circulating αKlotho. J. Am. Soc. Nephrol. 2016, 27, 79–90. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Li, L.; Huang, J.; King, G.; Quarles, L.D. Conditional Deletion of Fgfr1 in the Proximal and distal tubule identifies distinct roles in phosphate and calcium transport. PLoS ONE 2016, 11, e0147845. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Déliot, N.; Hernando, N.; Horst-Liu, Z.; Gisler, S.M.; Capuano, P.; Wagner, C.A.; Bacic, D.; O’Brien, S.; Biber, J.; Murer, H. Parathyroid hormone treatment induces dissociation of type IIa Na+-Pi cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am. J. Physiol. Cell Physiol. 2005, 289, C159–C167. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Ide, N.; Olauson, H.; Sato, T.; Densmore, M.J.; Wang, H.; Hanai, J.I.; Larsson, T.E.; Lanske, B. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int. 2016, 90, 348–362. [Google Scholar] [CrossRef]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef]
- Crabtree, G.R.; Olson, E.N. NFAT signaling: Choreographing the social lives of cells. Cell 2002, 109, S67–S79. [Google Scholar] [CrossRef]
- Farrow, E.G.; Davis, S.I.; Summers, L.J.; White, K.E. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J. Am. Soc. Nephrol. 2009, 20, 955–960. [Google Scholar] [CrossRef]
- Ranch, D.; Zhang, M.Y.; Portale, A.A.; Perwad, F. Fibroblast growth factor 23 regulates renal 1,25-dihydroxyvitamin D and phosphate metabolism via the MAP kinase signaling pathway in Hyp mice. J. Bone Miner Res. 2011, 26, 1883–1890. [Google Scholar] [CrossRef]
- Sneddon, W.B.; Ruiz, G.W.; Gallo, L.I.; Xiao, K.; Zhang, Q.; Rbaibi, Y.; Weisz, O.A.; Apodaca, G.L.; Friedman, P.A. Convergent Signaling Pathways Regulate Parathyroid Hormone and Fibroblast Growth Factor-23 Action on NPT2A-mediated Phosphate Transport. J. Biol. Chem. 2016, 291, 18632–18642. [Google Scholar] [CrossRef] [PubMed]
- Murer, H.; Lötscher, M.; Kaissling, B.; Levi, M.; Kempson, S.A.; Biber, J. Renal brush border membrane Na/Pi-cotransport: Molecular aspects in PTH-dependent and dietary regulation. Kidney Int. 1996, 49, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Murer, H.; Hernando, N.; Forster, I.; Biber, J. Proximal tubular phosphate reabsorption: Molecular mechanisms. Physiol Rev. 2000, 80, 1373–1409. [Google Scholar] [CrossRef] [PubMed]
- Keusch, I.; Traebert, M.; Lötscher, M.; Kaissling, B.; Murer, H.; Biber, J. Parathyroid hormone and dietary phosphate provoke a lysosomal routing of the proximal tubular Na/Pi-cotransporter type II. Kidney Int. 1998, 54, 1224–1232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lötscher, M.; Scarpetta, Y.; Levi, M.; Halaihel, N.; Wang, H.; Zajicek, H.K.; Biber, J.; Murer, H.; Kaissling, B. Rapid downregulation of rat renal Na/Pi cotransporter in response to parathyroid hormone involves microtubule rearrangement. J. Clin. Investig. 1999, 104, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Kendrick-Jones, J.; Buss, F. Myosin VI and its cargo adaptors—Linking endocytosis and autophagy. J. Cell Sci. 2013, 126 Pt 12, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.; E, X.; Steplock, D.; Shenolikar, S.; Weinman, E.J. Defective PTH regulation of sodium-dependent phosphate transport in NHERF-1−/− renal proximal tubule cells and wild-type cells adapted to low-phosphate media. Am. J. Physiol. Renal Physiol. 2005, 289, F933–F938. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.; Biswas, R.; Brazie, M.; Steplock, D.; Shenolikar, S.; Weinman, E.J. Signaling pathways utilized by PTH and dopamine to inhibit phosphate transport in mouse renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 2009, 296, F355–F361. [Google Scholar] [CrossRef]
- Lederer, E.D.; Sohi, S.S.; McLeish, K.R. Dopamine regulates phosphate uptake by opossum kidney cells through multiple counter- regulatory receptors. J. Am. Soc. Nephrol. 1998, 9, 975–985. [Google Scholar] [CrossRef]
- Weinman, E.J.; Steplock, D.; Shenolikar, S.; Biswas, R. Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J. Biol. Chem. 2011, 286, 37216–37221. [Google Scholar] [CrossRef]
- Shenolikar, S.; Voltz, J.W.; Minkoff, C.M.; Wade, J.B.; Weinman, E.J. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc. Natl. Acad. Sci. USA 2002, 99, 11470–11475. [Google Scholar] [CrossRef] [PubMed]
- Voltz, J.W.; Brush, M.; Sikes, S.; Steplock, D.; Weinman, E.J.; Shenolikar, S. Phosphorylation of PDZ1 domain attenuates NHERF-1 binding to cellular targets. J. Biol. Chem. 2007, 282, 33879–33887. [Google Scholar] [CrossRef] [PubMed]
- Mahon, M.J.; Donowitz, M.; Yun, C.C.; Segre, G.V. Na+/H+ exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature 2002, 417, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.; Garrido, J.L.; Bisello, A.; Kim, Y.K.; Friedman, P.A.; Romero, G. Regulation of parathyroid hormone type 1 receptor dynamics, traffic, and signaling by the Na+/H+ exchanger regulatory factor-1 in rat osteosarcoma ROS 17/2.8 cells. Mol. Endocrinol. 2008, 22, 1163–1170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef]
- Dicks, M.; Kock, G.; Kohl, B.; Zhong, X.; Pütz, S.; Heumann, R.; Erdmann, K.S.; Stoll, R. The binding affinity of PTPN13’s tandem PDZ2/3 domain is allosterically modulated. BMC Mol. Cell Biol. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Walker, V.; Vuister, G.W. Biochemistry and pathophysiology of the Transient Potential Receptor Vanilloid 6 (TRPV6) calcium channel. Adv. Clin. Chem. 2023, 113, 43–100. [Google Scholar] [CrossRef] [PubMed]
- Mahon, M.J.; Segre, G.V. Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cβ, and actin increases intracellular calcium in opossum kidney cells. J. Biol. Chem. 2004, 279, 23550–23558. [Google Scholar] [CrossRef] [PubMed]
- Mamonova, T.; Zhang, Q.; Chandra, M.; Collins, B.M.; Sarfo, E.; Bu, Z.; Xiao, K.; Bisello, A.; Friedman, P.A. Origins of PDZ Binding Specificity. A Computational and Experimental Study Using NHERF1 and the Parathyroid Hormone Receptor. Biochemistry 2017, 56, 2584–2593. [Google Scholar] [CrossRef]
- Mamonova, T.; Kurnikova, M.; Friedman, P.A. Structural basis for NHERF1 PDZ domain binding. Biochemistry 2012, 51, 3110–3120. [Google Scholar] [CrossRef]
- Rajagopal, A.; Braslavsky, D.; Lu, J.T.; Kleppe, S.; Clément, F.; Cassinelli, H.; Liu, D.S.; Liern, J.M.; Vallejo, G.; Bergadá, I.; et al. Exome sequencing identifies a novel homozygous mutation in the phosphate transporter SLC34A1 in hypophosphatemia and nephrocalcinosis. J. Clin. Endocrinol. Metab. 2014, 99, E2451–E2456. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Lee, R.; Kim, H.S. Infantile hypercalcemia with novel compound heterozygous mutation in SLC34A1 encoding renal sodium-phosphate cotransporter 2a: A case report. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Oddsson, A.; Sulem, P.; Helgason, H.; Edvardsson, V.O.; Thorleifsson, G.; Sveinbjörnsson, G.; Haraldsdottir, E.; Eyjolfsson, G.I.; Sigurdardottir, O.; Olafsson, I.; et al. Common and rare variants associated with kidney stones and biochemical traits. Nat. Commun. 2015, 6, 7975. [Google Scholar] [CrossRef] [PubMed]
- Howles, S.A.; Wiberg, A.; Goldsworthy, M.; Bayliss, A.L.; Gluck, A.K.; Ng, M.; Grout, E.; Tanikawa, C.; Kamatani, Y.; Terao, C.; et al. Genetic variants of calcium and vitamin D metabolism in kidney stone disease. Nat. Commun. 2019, 10, 5175. [Google Scholar] [CrossRef]
- Urabe, Y.; Tanikawa, C.; Takahashi, A.; Okada, Y.; Morizono, T.; Tsunoda, T.; Kamatani, N.; Kohri, K.; Chayama, K.; Kubo, M.; et al. A genome-wide association study of nephrolithiasis in the Japanese population identifies novel susceptible Loci at 5q35.3, 7p14.3, and 13q14.1. PLoS Genet. 2012, 8, e1002541. [Google Scholar] [CrossRef]
- Chen, W.C.; Chou, W.H.; Chu, H.W.; Huang, C.C.; Liu, X.; Chang, W.P.; Chou, Y.H.; Chang, W.C. The rs1256328 (ALPL) and rs12654812 (RGS14) Polymorphisms are Associated with Susceptibility to Calcium Nephrolithiasis in a Taiwanese population. Sci. Rep. 2019, 9, 17296. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, C.; Kamatani, Y.; Terao, C.; Usami, M.; Takahashi, A.; Momozawa, Y.; Suzuki, K.; Ogishima, S.; Shimizu, A.; Satoh, M.; et al. Novel Risk Loci Identified in a Genome-Wide Association Study of Urolithiasis in a Japanese Population. J. Am. Soc. Nephrol. 2019, 30, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Kestenbaum, B.; Glazer, N.L.; Köttgen, A.; Felix, J.F.; Hwang, S.J.; Liu, Y.; Lohman, K.; Kritchevsky, S.B.; Hausman, D.B.; Petersen, A.K.; et al. Common genetic variants associate with serum phosphorus concentration. J. Am. Soc. Nephrol. 2010, 21, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Laster, M.L.; Rowan, B.; Chen, H.C.; Schwantes-An, T.H.; Sheng, X.; Friedman, P.A.; Ikizler, T.A.; Sinshiemer, J.S.; Ix, J.H.; Susztak, K.; et al. Genetic Variants Associated with Mineral Metabolism Traits in Chronic Kidney Disease. J. Clin. Endocrinol. Metab. 2022, 107, e3866–e3876. [Google Scholar] [CrossRef] [PubMed]
- Alqinyah, M.; Hooks, S.B. Regulating the regulators: Epigenetic, transcriptional, and post-translational regulation of RGS proteins. Cell Signal. 2018, 42, 77–87. [Google Scholar] [CrossRef]
- Willard, F.S.; Willard, M.D.; Kimple, A.J.; Soundararajan, M.; Oestreich, E.A.; Li, X.; Sowa, N.A.; Kimple, R.J.; Doyle, D.A.; Der, C.J.; et al. Regulator of G-protein signaling 14 (RGS14) is a selective H-Ras effector. PLoS ONE 2009, 4, e4884. [Google Scholar] [CrossRef]
- Kimple, R.J.; De Vries, L.; Tronchère, H.; Behe, C.I.; Morris, R.A.; Gist Farquhar, M.; Siderovski, D.P. RGS12 and RGS14 GoLoco motifs are Gαi interaction sites with guanine nucleotide dissociation inhibitor Activity. J. Biol. Chem. 2001, 276, 29275–29281. [Google Scholar] [CrossRef] [PubMed]
- Siderovski, D.P.; Harden, T.K. The RGS Protein Superfamily. In Handbook of Cell Signaling; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press (Elsevier Inc.): Amsterdam, The Netherlands, 2003; Volume 2, pp. 631–638. [Google Scholar] [CrossRef]
- Sjögren, B. The evolution of regulators of G protein signalling proteins as drug targets—20 years in the making: IUPHAR Review 21. Br. J. Pharmacol. 2017, 174, 427–437. [Google Scholar] [CrossRef]
- Brown, N.E.; Goswami, D.; Branch, M.R.; Ramineni, S.; Ortlund, E.A.; Griffin, P.R.; Hepler, J.R. Integration of G protein α (Gα) signaling by the regulator of G protein signaling 14 (RGS14). J. Biol. Chem. 2015, 290, 9037–9049. [Google Scholar] [CrossRef] [PubMed]
- Worcester, E.M.; Coe, F.L. Clinical practice: Calcium kidney stones. N. Engl. J. Med. 2010, 363, 954–963. [Google Scholar] [CrossRef]
- Adeli, K.; Higgins, V.; Nieuwesteeg, M.; Raizman, J.E.; Chen, Y.; Wong, S.L.; Blais, D. Biochemical marker reference values across pediatric, adult, and geriatric ages: Establishment of robust pediatric and adult reference intervals on the basis of the Canadian Health Measures Survey. Clin. Chem. 2015, 61, 1049–1062. [Google Scholar] [CrossRef]
- Saggese, G.; Baroncelli, G.I.; Bertelloni, S.; Cinquanta, L.; Di Nero, G. Effects of long-term treatment with growth hormone on bone and mineral metabolism in children with growth hormone deficiency. J. Pediatr. 1993, 122, 37–45. [Google Scholar] [CrossRef]
- Boot, A.M.; Engels, M.A.; Boerma, G.J.; Krenning, E.P.; De Muinck Keizer-Schrama, S.M. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J. Clin. Endocrinol. Metab. 1997, 82, 2423–2428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haffner, D.; Grund, A.; Leifheit-Nestler, M. Renal effects of growth hormone in health and in kidney disease. Pediatr. Nephrol. 2021, 36, 2511–2530. [Google Scholar] [CrossRef]
- Quigley, R.; Baum, M. Effects of growth hormone and insulin-like growth factor I on rabbit proximal convoluted tubule transport. J. Clin. Investig. 1991, 88, 368–374. [Google Scholar] [CrossRef]
- Hirschberg, R.; Ding, H.; Wanner CHirschberg, R.; Ding, H.; Wanner, C. Effects of insulin-like growth factor I on phosphate transport in cultured proximal tubule cells. J. Lab. Clin. Med. 1995, 126, 428–434. [Google Scholar] [PubMed]
- Jehle, A.W.; Forgo, J.; Biber, J.; Lederer, E.; Krapf, R.; Murer, H. IGF-I and vanadate stimulate Na/Pi-cotransport in OK cells by increasing type II Na/Pi-cotransporter protein stability. Pflügers Arch. 1998, 437, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Bonjour, J.P.; Caverzasio, J. Stimulation of inorganic phosphate transport by insulin-like growth factor I and vanadate in opossum kidney cells is mediated by distinct protein tyrosine phosphorylation processes. Endocrinology 1996, 137, 4699–4705. [Google Scholar] [CrossRef][Green Version]
- Marks, J.; Debnam, E.S.; Unwin, R.J. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Capuano, P.; Bacic, D.; Roos, M.; Gisler, S.M.; Stange, G.; Biber, J.; Kaissling, B.; Weinman, E.J.; Shenolikar, S.; Wagner, C.A.; et al. Defective coupling of apical PTH receptors to phospholipase C prevents internalization of the Na+-phosphate cotransporter NaPi- IIa in Nherf1-deficient mice. Am. J. Physiol. Cell Physiol. 2007, 292, C927–C934. [Google Scholar] [CrossRef]
- Murer, H.; Forster, I.; Biber, J. The sodium phosphate cotransporter family SLC34. Pflügers Arch. 2004, 447, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Lötscher, M.; Sorribas, V.; Custer, M.; Arar, M.; Kaissling, B.; Murer, H.; Biber, J. Cellular mechanisms of acute and chronic adaptation of rat renal Pi transporter to alterations in dietary Pi. Am. J. Physiol. 1994, 267 Pt 2, F900–F908. [Google Scholar] [CrossRef] [PubMed]
- Weinman, E.J.; Boddeti, A.; Cunningham, R.; Akom, M.; Wang, F.; Wang, Y.; Liu, J.; Steplock, D.; Shenolikar, S.; Wade, J.B. NHERF-1 is required for renal adaptation to a low-phosphate diet. Am. J. Physiol. Renal Physiol. 2003, 285, F1225–F1232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cunningham, R.; Steplock, D.; Wang, F.; Huang, H.; E, X.; Shenolikar, S.; Weinman, E.J. Defective parathyroid hormone regulation of NHE3 activity and phosphate adaptation in cultured NHERF-1−/− renal proximal tubule cells. J. Biol. Chem. 2004, 279, 37815–37821. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflügers Arch. 2010, 459, 499–508. [Google Scholar] [CrossRef]
- Capuano, P.; Bacic, D.; Stange, G.; Hernando, N.; Kaissling, B.; Pal, R.; Kocher, O.; Biber, J.; Wagner, C.A.; Murer, H. Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflügers Arch. 2005, 449, 392–402. [Google Scholar] [CrossRef]
- Takahashi, F.; Morita, K.; Katai, K.; Segawa, H.; Fujioka, A.; Kouda, T.; Tatsumi, S.; Nii, T.; Taketani, Y.; Haga, H.; et al. Effects of dietary Pi on the renal Na+-dependent Pi transporter NaPi-2 in thyroparathyroidectomized rats. Biochem J. 1998, 333 Pt 1, 175–181. [Google Scholar] [CrossRef]
- Thomas, L.; Bettoni, C.; Knöpfel, T.; Hernando, N.; Biber, J.; Wagner, C.A. Acute Adaption to Oral or Intravenous Phosphate Requires Parathyroid Hormone. J. Am. Soc. Nephrol. 2017, 28, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Scanni, R.; vonRotz, M.; Jehle, S.; Hulter, H.N.; Krapf, R. The human response to acute enteral and parenteral phosphate loads. J. Am. Soc. Nephrol. 2014, 25, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Kritmetapak, K.; Kumar, R. Phosphate as a Signaling Molecule. Calcif Tissue Int. 2021, 108, 16–31. [Google Scholar] [CrossRef]
- Kido, S.; Miyamoto, K.; Mizobuchi, H.; Taketani, Y.; Ohkido, I.; Ogawa, N.; Kaneko, Y.; Harashima, S.; Takeda, E. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J. Biol. Chem. 1999, 274, 28256–28263. [Google Scholar] [CrossRef]
- Beck, L.; Tenenhouse, H.S.; Meyer, R.A.; Meyer, M.H.; Biber, J.; Murer, H. Renal expression of Na+-phosphate cotransporter mRNA and protein: Effect of the Gy mutation and low phosphate diet. Pflügers Arch. 1996, 431, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Hoag, H.M.; Martel, J.; Gauthier, C.; Tenenhouse, H.S. Effects of Npt2 gene ablation and low-phosphate diet on renal Na+/phosphate cotransport and cotransporter gene expression. J. Clin. Investig. 1999, 104, 679–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kilav, R.; Silver, J.; Biber, J.; Murer, H.; Naveh-Many, T. Coordinate regulation of rat renal parathyroid hormone receptor mRNA and Na-Pi cotransporter mRNA and protein. Am. J. Physiol. 1995, 268 Pt 2, F1017–F1022. [Google Scholar] [CrossRef]
- Conrads, K.A.; Yi, M.; Simpson, K.A.; Lucas, D.A.; Camalier, C.E.; Yu, L.R.; Veenstra, T.D.; Stephens, R.M.; Conrads, T.P.; Beck, G.R., Jr. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol. Cell Proteom. 2005, 4, 1284–1296. [Google Scholar] [CrossRef]
- Julien, M.; Magne, D.; Masson, M.; Rolli-Derkinderen, M.; Chassande, O.; Cario-Toumaniantz, C.; Cherel, Y.; Weiss, P.; Guicheux, J. Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology 2007, 148, 530–537. [Google Scholar] [CrossRef][Green Version]
- Nishino, J.; Yamazaki, M.; Kawai, M.; Tachikawa, K.; Yamamoto, K.; Miyagawa, K.; Kogo, M.; Ozono, K.; Michigami, T. Extracellular Phosphate Induces the Expression of Dentin Matrix Protein 1 Through the FGF Receptor in Osteoblasts. J. Cell. Biochem. 2017, 118, 1151–1163. [Google Scholar] [CrossRef]
- Camalier, C.E.; Yi, M.; Yu, L.R.; Hood, B.L.; Conrads, K.A.; Lee, Y.J.; Lin, Y.; Garneys, L.M.; Bouloux, G.F.; Young, M.R.; et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell. Physiol. 2013, 228, 1536–1550. [Google Scholar] [CrossRef]
- Bansal, N.; Hsu, C.Y.; Whooley, M.; Berg, A.H.; Ix, J.H. Relationship of urine dopamine with phosphorus homeostasis in humans: The heart and soul study. Am. J. Nephrol. 2012, 35, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Boland, J.M.; Tebben, P.J.; Folpe, A.L. Phosphaturic mesenchymal tumors: What an endocrinologist should know. J. Endocrinol. Investig. 2018, 41, 1173–1184. [Google Scholar] [CrossRef]
- Minisola, S.; Fukumoto, S.; Xia, W.; Corsi, A.; Colangelo, L.; Scillitani, A.; Pepe, J.; Cipriani, C.; Thakker, R.V. Tumor-induced Osteomalacia: A Comprehensive Review. Endocr. Rev. 2023, 44, 323–353. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L. Phosphaturic mesenchymal tumors: A review and update. Semin. Diagn. Pathol. 2019, 36, 260–268. [Google Scholar] [CrossRef]
- Rowe, P.S.; de Zoysa, P.A.; Dong, R.; Wang, H.R.; White, K.E.; Econs, M.J.; Oudet, C.L. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 2000, 67, 54–68. [Google Scholar] [CrossRef]
- Imanishi, Y.; Hashimoto, J.; Ando, W.; Kobayashi, K.; Ueda, T.; Nagata, Y.; Miyauchi, A.; Koyano, H.M.; Kaji, H.; Saito, T.; et al. Matrix extracellular phosphoglycoprotein is expressed in causative tumors of oncogenic osteomalacia. J. Bone Miner Metab. 2012, 30, 93–99. [Google Scholar] [CrossRef]
- Imel, E.A.; Peacock, M.; Pitukcheewanont, P.; Heller, H.J.; Ward, L.M.; Shulman, D.; Kassem, M.; Rackoff, P.; Zimering, M.; Dalkin, A.; et al. Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J. Clin. Endocrinol. Metab. 2006, 91, 2055–2061. [Google Scholar] [CrossRef]
- Lee, J.C.; Jeng, Y.M.; Su, S.Y.; Wu, C.T.; Tsai, K.S.; Lee, C.H.; Lin, C.Y.; Carter, J.M.; Huang, J.W.; Chen, S.H.; et al. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J. Pathol. 2015, 235, 539–545. [Google Scholar] [CrossRef]
- Berndt, T.; Craig, T.A.; Bowe, A.E.; Vassiliadis, J.; Reczek, D.; Finnegan, R.; De Beur, S.M.; Schiavi, S.C.; Kumar, R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J. Clin. Investig. 2003, 112, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Su, S.Y.; Changou, C.A.; Yang, R.S.; Tsai, K.S.; Collins, M.T.; Orwoll, E.S.; Lin, C.Y.; Chen, S.H.; Shih, S.R.; et al. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod. Pathol. 2016, 29, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Okuno, Y.; Murakami, N.; Shimoyama, Y.; Imagama, S.; Nishida, Y. Case report: Novel NIPBL-BEND2 fusion gene identified in osteoblastoma-like phosphaturic mesenchymal tumor of the fibula. Front. Oncol. 2023, 12, 956472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rowe, P.S.; Kumagai, Y.; Gutierrez, G.; Garrett, I.R.; Blacher, R.; Rosen, D.; Cundy, J.; Navvab, S.; Chen, D.; Drezner, M.K.; et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004, 34, 303–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schrauwen, I.; Valgaeren, H.; Tomas-Roca, L.; Sommen, M.; Altunoglu, U.; Wesdorp, M.; Beyens, M.; Fransen, E.; Nasir, A.; Vandeweyer, G.; et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet. Med. 2019, 21, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Sprowson, A.P.; McCaskie, A.W.; Birch, M.A. ASARM-truncated MEPE and AC-100 enhance osteogenesis by promoting osteoprogenitor adhesion. J. Orthop. Res. 2008, 26, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S. The wrickkened pathways of FGF23, MEPE and PHEX. Crit. Rev. Oral. Biol. Med. 2004, 15, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86. [Google Scholar] [CrossRef]
- Ogbureke, K.U.; Fisher, L.W. Renal expression of SIBLING proteins and their partner matrix metalloproteinases (MMPs). Kidney Int. 2005, 68, 155–166. [Google Scholar] [CrossRef]
- Ogbureke, K.U.; Koli, K.; Saxena, G. Matrix Metalloproteinase 20 Co-expression With Dentin Sialophosphoprotein in Human and Monkey Kidneys. J. Histochem. Cytochem. 2016, 64, 623–636. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Hedge, A.M.; Rowe, P.S. Matrix extracellular phosphoglycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology 2009, 150, 4012–4023. [Google Scholar] [CrossRef] [PubMed]
- Dobbie, H.; Unwin, R.J.; Faria, N.J.; Shirley, D.G. Matrix extracellular phosphoglycoprotein causes phosphaturia in rats by inhibiting tubular phosphate reabsorption. Nephrol. Dial. Transplant. 2008, 23, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.G.; Faria, N.J.; Unwin, R.J.; Dobbie, H. Direct micropuncture evidence that matrix extracellular phosphoglycoprotein inhibits proximal tubular phosphate reabsorption. Nephrol. Dial. Transplant. 2010, 25, 3191–3195. [Google Scholar] [CrossRef]
- Gowen, L.C.; Petersen, D.N.; Mansolf, A.L.; Qi, H.; Stock, J.L.; Tkalcevic, G.T.; Simmons, H.A.; Crawford, D.T.; Chidsey-Frink, K.L.; Ke, H.Z.; et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J. Biol. Chem. 2003, 278, 1998–2007. [Google Scholar] [CrossRef]
- Zelenchuk, L.V.; Hedge, A.M.; Rowe, P.S. Age dependent regulation of bone-mass and renal function by the MEPE ASARM-motif. Bone 2015, 79, 131–142. [Google Scholar] [CrossRef]
- Marks, J.; Churchill, L.J.; Debnam, E.S.; Unwin, R.J. Matrix extracellular phosphoglycoprotein inhibits phosphate transport. J. Am. Soc. Nephrol. 2008, 19, 2313–2320. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Hedge, A.M.; Drezner, M.K.; Rowe, P.S. ASARM peptides: PHEX-dependent and -independent regulation of serum phosphate. Am. J. Physiol. Renal Physiol. 2011, 300, F783–F791. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef]
- Tenenhouse, H.S.; Lee, J.; Harvey, N.; Potier, M.; Jette, M.; Beliveau, R. Normal molecular size of the Na+-phosphate cotransporter and normal Na+-dependent binding of phosphonoformic acid in renal brush border membranes of X-linked Hyp mice. Biochem Biophys. Res. Commun. 1990, 170, 1288–1293. [Google Scholar] [CrossRef]
- Berndt, T.J.; Bielesz, B.; Craig, T.A.; Tebben, P.J.; Bacic, D.; Wagner, C.A.; O’Brien, S.; Schiavi, S.; Biber, J.; Murer, H.; et al. Secreted frizzled- related protein-4 reduces sodium-phosphate co-transporter abundance and activity in proximal tubule cells. Pflügers Arch. 2006, 451, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Habra, M.A.; Jimenez, C.; Huang, S.C.; Cote, G.J.; Murphy WAJr Gagel, R.F.; Hoff, A.O. Expression analysis of fibroblast growth factor- 23, matrix extracellular phosphoglycoprotein, secreted frizzled-related protein-4, and fibroblast growth factor-7: Identification of fibroblast growth factor-23 and matrix extracellular phosphoglycoprotein as major factors involved in tumor-induced osteomalacia. Endocr. Pract. 2008, 14, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, X.; Wei, L.; Wu, Y.; Xu, L.; Wu, L.; Wei, X.; Zhao, S.; Zhu, X.; Xu, F. A framework for frizzled-G protein coupling and implications to the PCP signaling pathways. Cell Discov. 2024, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling pathways at the plasma membrane and their misregulation in cancer. Front. Cell Dev. Biol. 2021, 9, 631623. [Google Scholar] [CrossRef] [PubMed]
- Pawar, N.M.; Rao, P. Secreted frizzled related protein 4 (sFRP4) update: A brief review. Cell Signal. 2018, 45, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Wawrzak, D.; Métioui, M.; Willems, E.; Hendrickx, M.; de Genst, E.; Leyns, L. Wnt3a binds to several sFRPs in the nanomolar range. Biochem. Biophys. Res. Commun. 2007, 357, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Kiper, P.O.S.; Saito, H.; Gori, F.; Unger, S.; Hesse, E.; Yamana, K.; Kiviranta, R.; Solban, N.; Liu, J.; Brommage, R.; et al. Cortical-Bone Fragility—Insights from sFRP4 Deficiency in Pyle’s Disease. N. Engl. J. Med. 2016, 374, 2553–2562. [Google Scholar] [CrossRef]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116 Pt 13, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Finch, P.W.; He, X.; Kelley, M.J.; Uren, A.; Schaudies, R.P.; Popescu, N.C.; Rudikoff, S.; Aaronson, S.A.; Varmus, H.E.; Rubin, J.S. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc. Natl. Acad. Sci. USA 1997, 94, 6770–6775. [Google Scholar] [CrossRef] [PubMed]
- Rehn, M.; Pihlajaniemi, T.; Hofmann, K.; Bucher, P. The frizzled motif: In how many different protein families does it occur? Trends Biochem Sci. 1998, 23, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Ellis, B.K.; Insogna, K.L.; Philbrick, W.M.; Sterpka, J.; Shimkets, R. Fibroblast growth factor 7: An inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J. Clin. Endocrinol. Metab. 2005, 90, 1012–1020. [Google Scholar] [CrossRef]
- Whyte, M.P.; Zhang, F.; Wenkert, D.; Mumm, S.; Berndt, T.J.; Kumar, R. Hyperphosphatemia with low FGF7 and normal FGF23 and sFRP4 levels in the circulation characterizes pediatric hypophosphatasia. Bone 2020, 134, 115300. [Google Scholar] [CrossRef] [PubMed]
- Kritmetapak, K.; Kumar, R. Phosphatonins: From discovery to therapeutics. Endocr. Pract. 2023, 29, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; Mao, Z.; Shen, X.; Wang, W.; Dai, B.; Tang, B.; Wu, Y.; Cao, Y.; Zhang, S.; Zhao, H.; et al. Role of keratinocyte growth factor in the pathogenesis of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2005, 20, 2368–2375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zinkle, A.; Mohammadi, M. Structural Biology of the FGF7 Subfamily. Front. Genet. 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Moon, C.; Zhang, W.; Yarlagadda, S.; Penmatsa, H.; Ren, A.; Sinha, C.; Naren, A.P. Stabilizing rescued surface-localized ΔF508 CFTR by potentiation of its interaction with Na+/H + exchanger regulatory factor 1. Biochemistry 2014, 53, 4169–4179. [Google Scholar] [CrossRef]
- Loureiro, C.A.; Matos, A.M.; Dias-Alves, Â.; Pereira, J.F.; Uliyakina, I.; Barros, P.; Amaral, M.D.; Matos, P. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci Signal. 2015, 8, ra48. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; Kewalramani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs Cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: A randomized clinical trial. JAMA 2017, 317, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Linstedt, A.D. Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. eLife 2017, 6, e24051. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]












{kind=link}
{kind=link}
{kind=link}
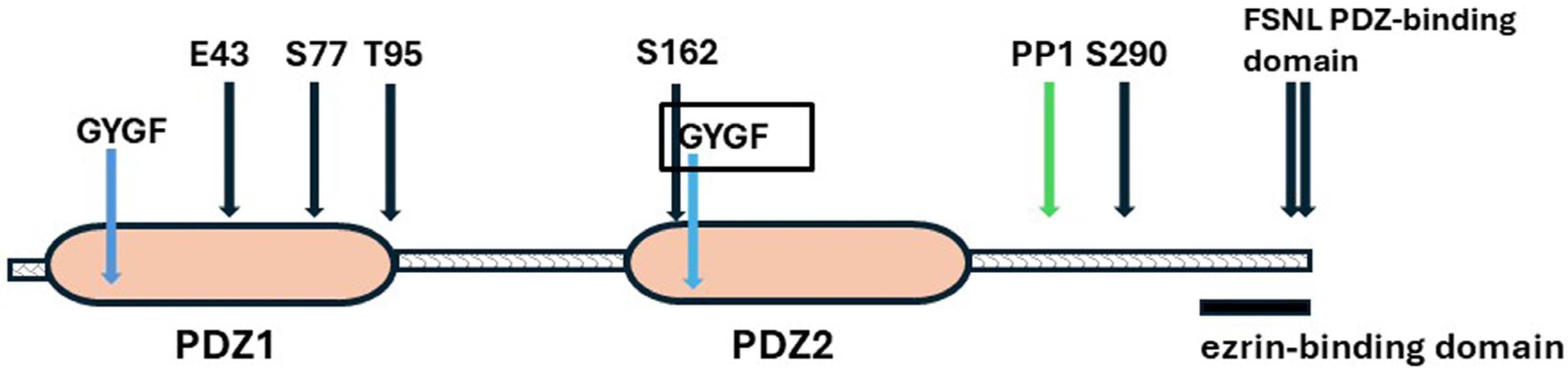{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
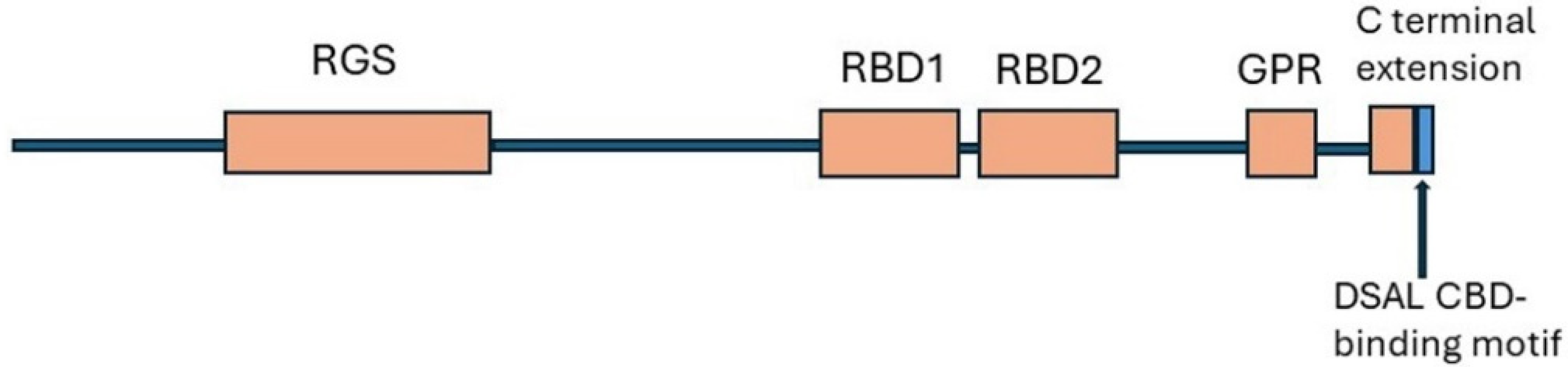{kind=link}
{kind=link}
{kind=link}
| Disorder | Gene | Protein | Plasma: Ca2+ | Pi | PTH | FEPO4 | Urine: cAMP Post PTH | Bone Deformity |
|---|---|---|---|---|---|---|---|---|
| Jansen’s Metaphyseal chondrodysplasia [9,149,150,151] | PTH1R | PTH/PTHrP receptor | ↑ | ↓ | low/ND | ↑ | - | Yes |
| Isolated PTH deficiency [146,152] | PTH | PTH | ↓ | ↑ | low/ND | ↓ | - | No |
| † Pseudohypo-parathyroidism 1A [9,145,147,153,154] | GNAS from mother | Gsα | ↓ | ↑ | ↑ | ↓ | ↓ | Yes |
| † Pseudo-pseudo hypoparathyroidism [9,145,147,153,154] | GNAS from father | Gsα | norm | norm | norm | norm | norm | Yes |
| Disorder | Gene Defect | Mechanism | Clinical Manifestations |
|---|---|---|---|
| Increased intact FGF23 | |||
| Familial X-linked hypophosphatemic rickets (XLH) OMIM 193100 | PHEX deficiency. | Increased FGF23 synthesis. | Hypophosphatemic rickets; dental abnormalities. |
| Autosomal recessive hypophosphatemia OMIM 241520 | DMP1 deficiency. | Increased FGF23 synthesis. | Hypophosphatemic rickets; dental abnormalities. |
| Tumor-induced osteomalacia (TIO) | FN1-FGFR1 fusion gene. | Increased FGF3 synthesis in tumours. | Hypophosphatemia, muscle weakness, osteomalacia, fractures. |
| Linear nevus sebaceous syndrome OMIM 163200 | Sporadic HRAS, KRAS, and NRAS deficiencies. | Increased FGF 23 synthesis possibly by the skin nevi--etc? By the skin nevi-similar to TIO. | Linear sebaceous nevi (face), central nervous system, ocular abnormalities and skeletal defects. |
| McCune-Albright syndrome (fibrous dysplasia) | Somatic GNAS activating mutations of GSα subunit. | Increased FGF23 synthesis in fibrous bone [228,229]. | Bone and endocrine abnormalities, skin pigmentation, 50% have raised FGF23 and hypophosphatemia. |
| Osteoglophonic dysplasia | FGFR1c-activating mutation. | Increased FGF23 synthesis possibly in bone-? in bone [19]. | Craniosynostosis, dwarfism, non-ossifying bone lesions, some have hypophosphatemia. |
| Non-lethal Raine syndrome | FAM20C deficiency—a kinase that normally phosphorylates FGF23 S180 that promotes cleavage. | Decreased FGF23 cleavage [230,231] by furin/subtilisin and increased FGF23 synthesis in bone. | Typically fatal neonatally, bone hyper-density, lung hypoplasia. Survivors have raised FGF23 and hypophosphatemia. |
| Autosomal dominant hypophosphatemic rickets (ADHR) | FGF23 cleavage site mutations. | No cleavage of intact FGF23 by furin/subtilisin pre-secretion. | Presentation in children: hypophosphatemic rickets, dental problems; in adolescents/adults: bone pain, weakness, pseudofractures. |
| Decreased intact FGF23 | |||
| Familial tumoral calcinosis | GALNT3 deficiency. | Loss of glycosyl protection at furin/subtilisin cleavage site, leads to excessive FGF23 degradation. | Low intact FGF23, raised phosphate and 1,25[OH]2D. Periarticular, vascular subcutaneous calcium phosphate deposits. Painful dense bones. |
| Study | Procedure | Relevant Findings |
|---|---|---|
| MEPE infusion | ||
| Rowe et al., 2004 [323] | Wt mice; boluses of human MEPE for 31 h | ↓ serum phosphate, ↓ 1,25[OH]2D ↑ FEPO4 |
| Dobbie et al., 2008 [331] | Wt rats; saline or MEPE infused at three doses for 2 h GFR measured with inulin. | Dose-dependent ↑ FEPO4 |
| Shirley et al., 2010 [332] | Wt rats; 2 h infusion of vehicle or MEPE; fluid from proximal renal tubules | Significant ↑ FEPO4 |
| Animal Models | ||
| Gowen et al., 2002 [333] | Mouse mepe KO at 4 m and 12 m | Homoz−/−, Heteroz−/+: bone mass significant ↑ serum phosphate as wt |
| Zelenchuk et al., 2015 [334] | Group 1: Mouse mepe KO up to 22 m | at 22m ↑ serum phosphate; ↑ FEPO4 Kidney mRNA v. wt: ↑ NaPi-2a; ↑ NaPi-2c ↑ Bone mass |
| David et al., 2009 [330] † | Transgenic mouse with over-expressed mepe up to 19 weeks normal phosphate and vit D intakes | MEPE protein expressed in bone and kidneys ↑ number of renal blood vessels; ↓ diameter ↑ serum phosphate; ↓ FEPO4; ↑ renin/aldosterone Kidney mRNA ↑ NaPi-2a; ↓ NaPi-2c ↑ VEGFR ↑ Bone mass; Bone mineral ↓; osteoid ↑ |
| Renal Cell studies | ||
| Rowe et al., 2004 [323] | 33PO4 uptake: human proximal tubule cells and renal cell line treated with MEPE for 3–4 h | significant ↓ 33PO4 uptake; ↓ Vmax Km no change |
| Marks et al., 2008 [335] | Rats infused with MEPE for 3 h. analyses of BBM vesicles | NaPi-2a: significant dose-dependent ↓ NaPi-2c: trend for an ↑ |
| ASARM infusion | ||
| David et al., 2011 [336] | Wt mice aged 8 to 12 weeks continuous infusion of ASARM or vehicle; normal phosphate diet | Serum: 2-fold ↑ ASARM and 1,25[OH]2D vit D; ↑ FGF23 ↓ phosphate; PTH no change. ↑ FEPO4 |
| Animal Model | ||
| Zelenchuk et al., 2015 [334] †† | Group 2: Transgenic Mouse mepe KO with over-expressed ASARM up to 22 m | ↓ serum phosphate; ↓ 1,25[OH]2D vit D; ↑ PTH; ↑ FGF23; ↑ FEPO4; Kidney mRNA ↑ NaPi-2a; ↑ NaPi-2c; ↓ bone mineral |
| Clinical Aim | Therapeutic Aim | Therapeutic Approach | Therapeutic Agent | Status |
|---|---|---|---|---|
| Reduce cardiovascular risk from high FGF23 in chronic renal failure (CRF) | Decrease excessive bone production of FGF23 in CRF driven by high phosphate. | Reduce intestinal absorption of phosphate | Tenapanor: inhibits NHE3-(Na+/H+ ion exchanger) [7] | Approved for end-stage renal failure |
| Calcimimetics to decrease PTH, bone turnover and FGF23 (secondary unexplained effect) | Cinacalcet Etacalcetide [31,360] | In routine use to decrease PTH in CRF | ||
| Reduce calcium phosphate deposition | Infusion of the MEPE ASARM peptide | ASARM peptide [28] | Experimental. Reduces vascular calcification in Mice with CRF | |
| Correct hypophosphaturia and abnormal bone formation in genetic disorders and TIO with a primary increase in FGF23 | Decrease circulating intact FGF23 | Prevent thr 178 glycosylation of intact FGF23, thereby facilitating proteolytic cleavage of FGF23 by furin | T3 Inh-1 a small molecule inhibitor of GALNT3 [361] | Experimental. Effective in normal mice |
| Decrease the renal response to FGF23 | Block binding of FGF23 to the FGFR1c-αklotho receptor | Burosumab: a recombinant human IgG monoclonal antibody [32] | Approved for X-linked hypophosphatemic rickets and inoperable TIO | |
| Block binding of FGF23 to the FGFR1c-αklotho receptor with C-terminal FGF23 fragments which do not activate signalling | FGF23-C-terminal fragments [29] | Experimental. Effective in cell cultures and Hyp mice | ||
| Inhibit FGFR1c-αklotho /FGF23 signalling | No specific small molecule inhibitors available | Still speculative -clinical safety and merits questionable | ||
| Prolong the action of PTH for treatment of hypoparathyroidism | Prolong cAMP signalling response to PTH | Use a long-acting PTH hybrid to stabilise binding to the PTH1R receptor. in G-protein -independent receptor conformation (R0) | Long-acting PTH ligands [PTH/PTHrP hybrid peptides] [34] | Experimental |
| Reduce effects of increased PTH in primary hyperparathyroidism or PTHrP in hypercalcaemia of malignancy | Reduce activity of the PTH1R receptor | Reduce the PTH1R response by binding with a small molecule modulator | A small modulator, Pitt12, is being optimised [23] | Experimental |
| Improve the efficacy of intermittent treatment with PTHrP to promote bone anabolism in post-menopausal osteoporosis | Prolong the duration of action of PTHrP in bone | Develop analogues to localise in endosomes and generate endosomal cAMP to ensure nuclear delivery | Under investigation [23] | Experimental |
| Increase phosphate reabsorption in phosphate wasting disorders | Increase expression of NaPi-2a in the brush border membrane | Increase the binding affinity between NHERF1 PDZ1 and NaPi-2a and increase its membrane stability | The CFTR corrector VX-809 used to treat cystic fibrosis due to the CFTR F508del increases surface expression and activity of mutant CFTR [358,359] | This is a speculative approach which has not been explored for NaPi-2a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, V. The Intricacies of Renal Phosphate Reabsorption—An Overview. Int. J. Mol. Sci. 2024, 25, 4684. https://doi.org/10.3390/ijms25094684
Walker V. The Intricacies of Renal Phosphate Reabsorption—An Overview. International Journal of Molecular Sciences. 2024; 25(9):4684. https://doi.org/10.3390/ijms25094684
Chicago/Turabian StyleWalker, Valerie. 2024. "The Intricacies of Renal Phosphate Reabsorption—An Overview" International Journal of Molecular Sciences 25, no. 9: 4684. https://doi.org/10.3390/ijms25094684
APA StyleWalker, V. (2024). The Intricacies of Renal Phosphate Reabsorption—An Overview. International Journal of Molecular Sciences, 25(9), 4684. https://doi.org/10.3390/ijms25094684