
Single and Combined Impact of Semaglutide, Tirzepatide, and Metformin on β-Cell Maintenance and Function Under High-Glucose–High-Lipid Conditions: A Comparative Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
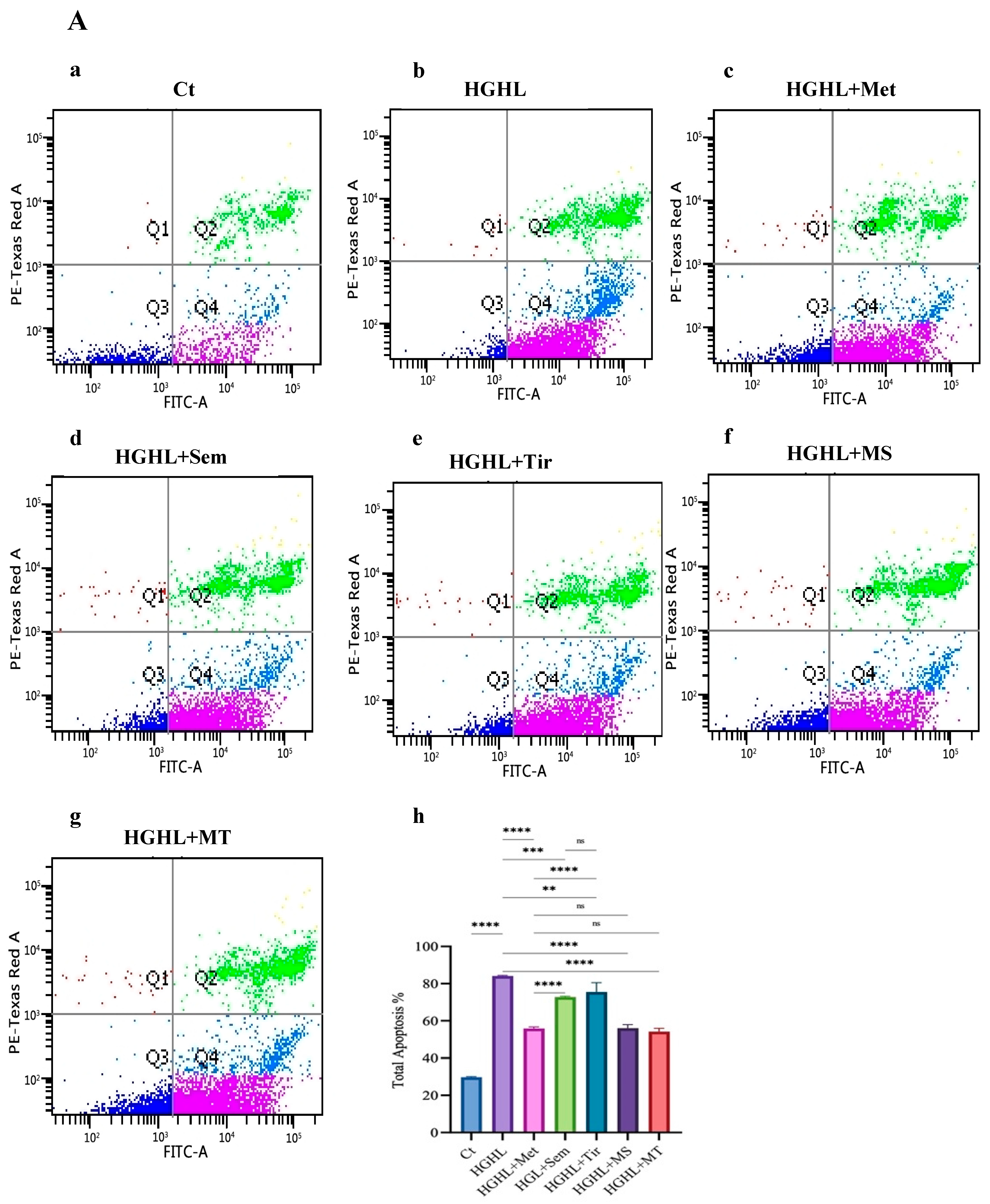
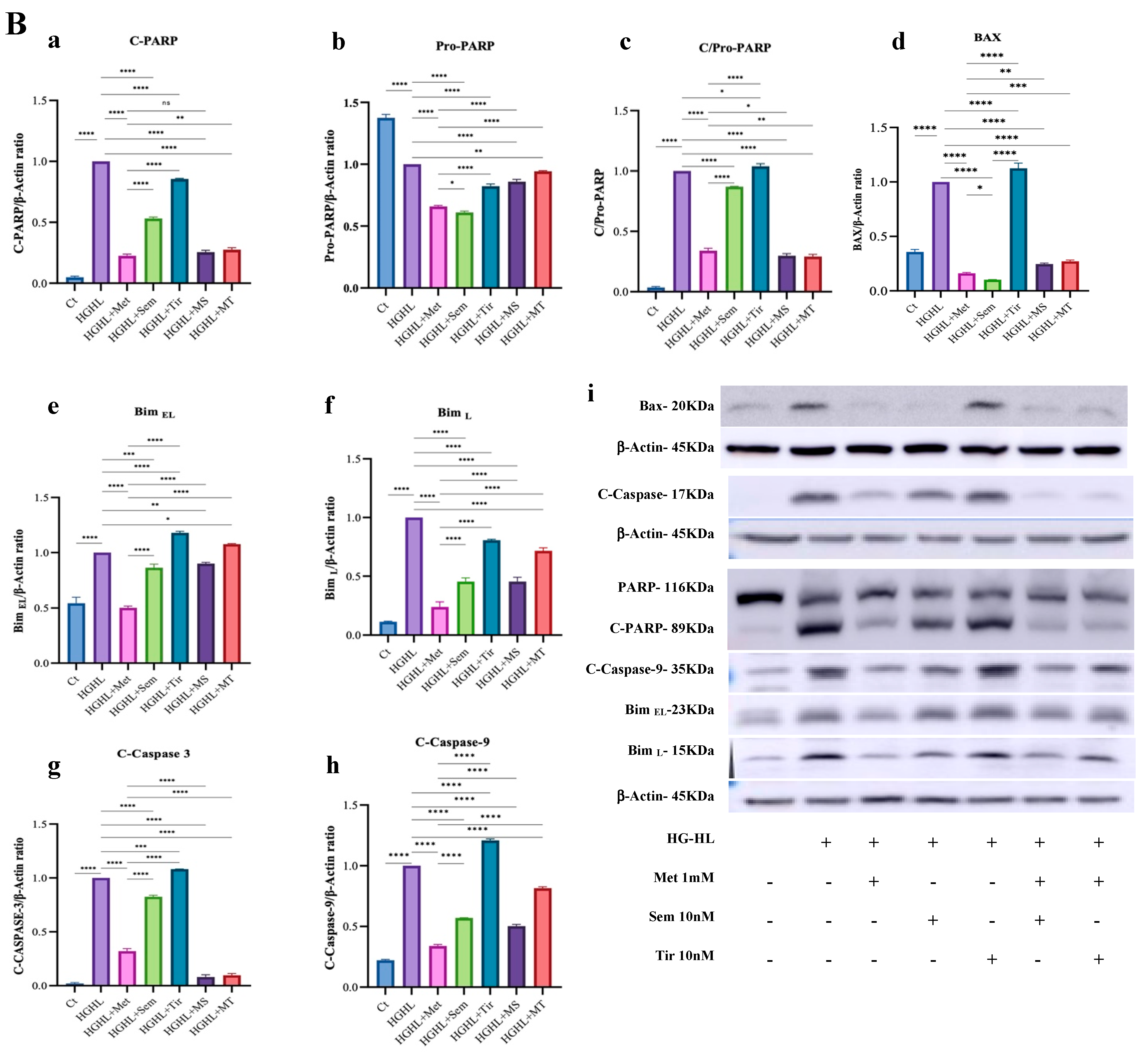
2.1. 1 mM Metformin and Its Combinations with 10 nM Semaglutide and 10 nM Tirzepatide Exhibit Enhanced Modulatory Effects on HG-HL-Induced Apoptosis and Apoptotic Biomarkers
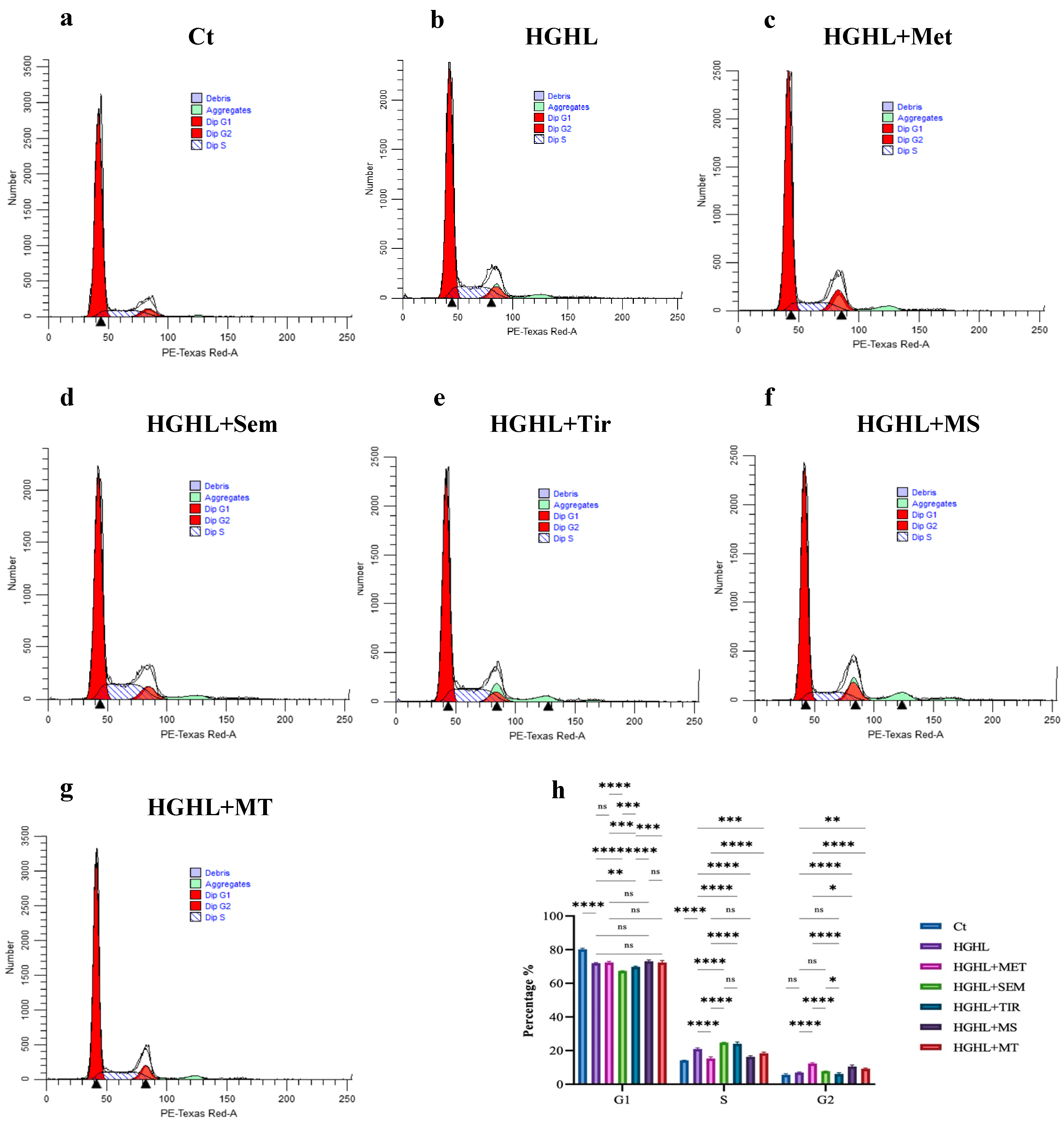
2.2. 1 mM Metformin and Its Combinations with Both 10 nM Semaglutide and 10 nM Tirzepatide Modulate HG-HL-Induced Cell Cycle Dysregulation
2.3. 1 mM Metformin and Its Combinations with Both 10 nM Semaglutide and 10 nM Tirzepatide Improve HG-HL-Reduced GSIS in INS-1 β-Cells
2.4. All Treatments Significantly Reduce TXNIP and P-STAT1 Levels in HG-HL-Induced Cells
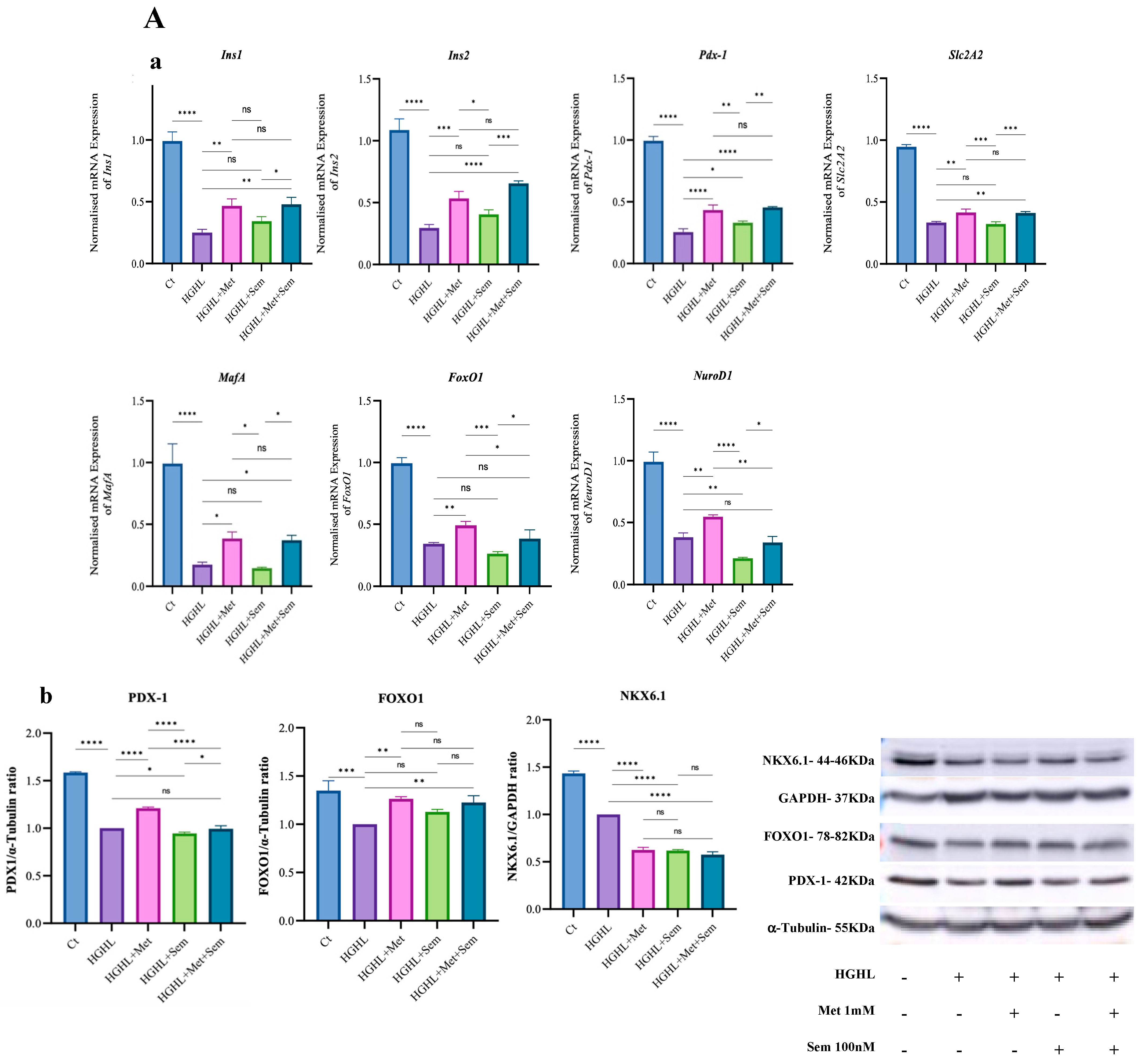
2.5. 1 mM Metformin and MS Restore the Suppressed Expression of Genes Crucial for INS-1 β-Cell Maintenance and Identity Under HG-HL Conditions
3. Discussion
3.1. The Impact of the Treatments on HG-HL-Induced Apoptosis
3.2. The Impact of the Treatments on HG-HL Cell Cycle Dysregulation
3.3. The Impact of the Treatments on HG-HL-Impaired GSIS
3.4. The Impact of the Treatments on the Response of β-enriched Genes and Proteins
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Treatments
4.3. Apoptosis Assay
4.4. Cell Cycle Assay
4.5. Western Blotting
4.6. qRT-PCR
4.7. Glucose-Stimulated Insulin Secretion (GSIS) and Potassium-Stimulated Insulin Secretion (KSIS)
4.8. Statistics
5. Conclusions
6. Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magliano, D.J.; Boyko, E.J. IDF Diabetes Atlas; International Diabetes Federation: Brussel, Belgium, 2022. [Google Scholar]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel Excess and β-Cell Dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflügers Arch.-Eur. J. Physiol. 2010, 460, 70–718. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; E Mabhida, S.; Ziqubu, K.; Nkambule, B.B.; E Mazibuko-Mbeje, S.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic β-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes 2023, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Vela-Guajardo, J.E.; Garza-González, S.; García, N. Glucolipotoxicity-induced oxidative stress is related to mitochondrial dys-function and apoptosis of pancreatic β-cell. Curr. Diabetes Rev. 2021, 17, 46–56. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Butler, A.E. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr. Diabetes Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE−/− mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ische-mia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef]
- E LaMoia, T.; I Shulman, G. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- The RISE Consortium. Impact of insulin and metformin versus metformin alone on β-cell function in youth with impaired glucose tolerance or recently diagnosed type 2 diabetes. Diabetes Care 2018, 41, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, Z.; Zhang, C.; Cai, Z.; Zhang, J. Metformin, beyond an insulin sensitizer, targeting heart and pancreatic β cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1984–1990. [Google Scholar] [CrossRef]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic Med. Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.-K.; Lee, H.W.; Kim, Y.-W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress–induced CD36 expression in pancreatic beta cells. J. Diabetes Its Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef]
- Li, Q.; Jia, S.; Xu, L.; Li, B.; Chen, N. Metformin-induced autophagy and irisin improves INS-1 cell function and survival in high-glucose environment via AMPK/SIRT1/PGC-1α signal pathway. Food Sci. Nutr. 2019, 7, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002. [Google Scholar] [CrossRef]
- Li, J.; Jiang, Q.; Wang, X.; Hou, L.; Wang, L.; Lou, K.; Pang, S. Metformin Preserves Insulin Secretion in Pancreatic β-cells through FGF21/Akt Pathway In vitro and In vivo. Comb. Chem. High Throughput Screen. 2024, 27, 2691–2698. [Google Scholar] [CrossRef]
- Christou, G.A.; Katsiki, N.; Blundell, J.; Fruhbeck, G.; Kiortsis, D.N. Semaglutide as a promising antiobesity drug. Obes. Rev. 2019, 20, 805–815. [Google Scholar] [CrossRef]
- Mather, K.J.; Mari, A.; Heise, T.; DeVries, J.H.; Hua, M.; Urva, S.; Coskun, T.; Haupt, A.; Heine, R.J.; Pratt, E. Effects of Tir-zepatide vs. Semaglutide on β-cell Function, Insulin Sensitivity, and Glucose Control During a Meal Test. J. Clin. Endocrinol. Metab. 2024, 109, dgae319. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.K.; Nikooienejad, A.; Bray, R.; Cui, X.; Wilson, J.; Duffin, K.; Milicevic, Z.; Haupt, A.; A Robins, D. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2020, 106, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Mao, H.; Thieu, V.T.; Landó, L.F.; Thomas, M.K. Tirzepatide as Monotherapy Improved Markers of Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes (SURPASS-1). J. Endocr. Soc. 2023, 7, bvad056. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, Y.; Kimura, T.; Dan, K.; Iwamoto, H.; Sanada, J.; Fushimi, Y.; Katakura, Y.; Shimoda, M.; Yamasaki, Y.; Nogami, Y. Tirzepatide, a dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide 1 receptor agonist, exhibits favourable effects on pancreatic β-cells and hepatic steatosis in obese type 2 diabetic db/db mice. Diabetes Obes. Metab. 2024, 26, 5982–5994. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.; Cummings, M.H.; Ustyugova, A.; Raza, S.I.; de Silva, S.U.; Lamotte, M. Long-Term Cost-Effectiveness Analyses of Empagliflozin Versus Oral Semaglutide, in Addition to Metformin, for the Treatment of Type 2 Diabetes in the UK. Diabetes Ther. 2020, 11, 2041–2055. [Google Scholar] [CrossRef]
- Kellerer, M.; Kaltoft, M.S.; Lawson, J.; Nielsen, L.L.; Strojek, K.; Tabak, Ö.; Jacob, S. Effect of once-weekly semaglutide versus thrice-daily insulin aspart, both as add-on to metformin and optimized insulin glargine treatment in participants with type 2 diabetes (SUSTAIN 11): A randomized, open-label, multinational, phase 3b trial. Diabetes Obes. Metab. 2022, 24, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Dong, X.; Li, Y.; Li, Y.; Lim, S.; Liu, M.; Ning, Z.; Rasmussen, S.; Skjøth, T.V.; Yuan, G. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as add-on to metformin in patients with type 2 diabetes in SUSTAIN China: A 30-week, double-blind, phase 3a, randomized trial. Diabetes Obes. Metab. 2021, 23, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ludvik, B.; Giorgino, F.; Jódar, E.; Frias, J.P.; Landó, L.F.; Brown, K.; Bray, R.; Rodríguez, Á. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): A randomised, open-label, parallel-group, phase 3 trial. The Lancet 2021, 398, 583–598. [Google Scholar] [CrossRef]
- Sassin, A.M.; Sangi-Haghpeykar, H.; Aagaard, K.M.; Detti, L. Effects of metformin alone versus metformin and tirzepatide on weight loss in patients with polycystic ovarian syndrome (PCOS). Fertil. Steril. 2023, 120, e222. [Google Scholar] [CrossRef]
- Gojani, E.G.; Wang, B.; Li, D.-P.; Kovalchuk, O.; Kovalchuk, I. The Impact of Psilocybin on High Glucose/Lipid-Induced Changes in INS-1 Cell Viability and Dedifferentiation. Genes 2024, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty acid-induced lipotoxicity in pancreatic beta-cells during devel-opment of type 2 diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- You, S.; Zheng, J.; Chen, Y.; Huang, H. Research progress on the mechanism of beta-cell apoptosis in type 2 diabetes mellitus. Front. Endocrinol. 2022, 13, 976465. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, Y.; Yang, Q.; Xu, C.; Zheng, Y.; Wang, L.; Wu, J.; Zeng, M.; Luo, M. Metformin prevents methylglyoxal-induced apoptosis by suppressing oxidative stress in vitro and in vivo. Cell Death Dis. 2022, 13, 29. [Google Scholar] [CrossRef]
- Chen, D.; Xia, D.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef]
- Gallardo-Villanueva, P.; Fernández-Marcelo, T.; Villamayor, L.; Valverde, A.M.; Ramos, S.; Fernández-Millán, E.; Martín, M.A. Synergistic Effect of a Flavonoid-Rich Cocoa–Carob Blend and Metformin in Preserving Pancreatic Beta Cells in Zucker Diabetic Fatty Rats. Nutrients 2024, 16, 273. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-I.; Park, M.-J.; Heo, Y.-R.; Park, S.-H. Metformin ameliorates lipotoxicity-induced mesangial cell apoptosis partly via upregulation of glucagon like peptide-1 receptor (GLP-1R). Arch. Biochem. Biophys. 2015, 584, 90–97. [Google Scholar] [CrossRef]
- Dai, Y.-L.; Huang, S.-L.; Leng, Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms. Int. J. Biol. Sci. 2015, 11, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.; Kreutz, R.; Eisenreich, A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. J. Nephrol. 2016, 29, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 2414–2422. [Google Scholar] [CrossRef]
- Shaked, M.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. AMP-Activated Protein Kinase (AMPK) Mediates Nutrient Regulation of Thioredoxin-Interacting Protein (TXNIP) in Pancreatic Beta-Cells. PLoS ONE 2011, 6, e28804. [Google Scholar] [CrossRef]
- Roohi, T.F.; Krishna, K.; Shakeel, F. Synergistic modulation of endoplasmic reticulum stress pathway, oxidative DNA damage and apoptosis by β-amyrin and metformin in mitigating hyperglycemia-induced renal damage using adult zebrafish model. BMC Pharmacol. Toxicol. 2024, 25, 66. [Google Scholar] [CrossRef]
- Geng, Y.; Villanueva, A.H.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165621. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, X.-F.; Gao, L.-L.; Ding, Z.-R.; Huang, X.-P.; Li, Y.-Y.; Xie, D.-Z. Molecular characterization of thioredoxin-interacting protein (TXNIP) from Megalobrama amblycephala and its potential roles in high glucose-induced inflammatory response. Int. J. Biol. Macromol. 2021, 188, 460–472. [Google Scholar] [CrossRef]
- Li, X.; Kover, K.L.; Heruth, D.P.; Watkins, D.J.; Moore, W.V.; Jackson, K.; Zang, M.; Clements, M.A.; Yan, Y. New Insight into Metformin Action: Regulation of ChREBP and FOXO1 Activities in Endothelial Cells. Mol. Endocrinol. 2015, 29, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, C.; Chen, J.; Xu, G.; Jing, G.; Shalev, A. FOXO1 Competes with Carbohydrate Response Element-binding Protein (ChREBP) and Inhibits Thioredoxin-interacting Protein (TXNIP) Transcription in Pancreatic Beta Cells. J. Biol. Chem. 2013, 288, 23194–23202. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.; El-Assaad, W.; Rhodes, C.; Rosenberg, L.; Joly, E.; Prentki, M. Glucagon-like peptide-1 prevents beta cell glucolipo-toxicity. Diabetologia 2004, 47, 806–815. [Google Scholar]
- Farilla, L.; Bulotta, A.; Hirshberg, B.; Calzi, S.L.; Khoury, N.; Noushmehr, H.; Bertolotto, C.; Di Mario, U.; Harlan, D.M.; Perfetti, R. Glucagon-Like Peptide 1 Inhibits Cell Apoptosis and Improves Glucose Responsiveness of Freshly Isolated Human Islets. Endocrinology 2003, 144, 5149–5158. [Google Scholar] [CrossRef]
- Li, Y.; Hansotia, T.; Yusta, B.; Ris, F.; Halban, P.A.; Drucker, D.J. Glucagon-like Peptide-1 Receptor Signaling Modulates β Cell Apoptosis. J. Biol. Chem. 2003, 278, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.E.; Marinho, T.S.; Martins, F.F.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Treatment with semaglutide, a GLP-1 receptor agonist, improves extracellular matrix remodeling in the pancreatic islet of diet-induced obese mice. Life Sci. 2023, 319, 121502. [Google Scholar] [CrossRef] [PubMed]
- Moffett, R.C.; Patterson, S.; Irwin, N.; Flatt, P.R. Positive effects of GLP-1 receptor activation with liraglutide on pancreatic islet morphology and metabolic control in C57BL/KsJ db/db mice with degenerative diabetes. Diabetes/Metabolism Res. Rev. 2014, 31, 248–255. [Google Scholar] [CrossRef]
- Killion, E.A.; Lu, S.-C.; Fort, M.; Yamada, Y.; Véniant, M.M.; Lloyd, D.J. Glucose-dependent insulinotropic polypeptide receptor therapies for the treatment of obesity, do agonists= antagonists? Endocr. Rev. 2020, 41, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Chen, M.; Falsey, J.R.; Sivits, G.; Hager, T.; Atangan, L.; Helmering, J.; Lee, J.; Li, H.; Wu, B. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat. Commun. 2020, 11, 4981. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Wang, J.; Yie, J.; Shi, S.D.-H.; Bates, D.; Min, X.; Komorowski, R.; Hager, T.; Deng, L.; Atangan, L. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 2018, 10, eaat3392. [Google Scholar] [CrossRef]
- Campbell, J.E. Targeting the GIPR for obesity: To agonize or antagonize? Potential mechanisms. Mol. Metab. 2021, 46, 101139. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-C.; Chen, M.; Atangan, L.; Killion, E.A.; Komorowski, R.; Cheng, Y.; Netirojjanakul, C.; Falsey, J.R.; Stolina, M.; Dwyer, D. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep. Med. 2021, 2, 100263. [Google Scholar] [CrossRef]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): Implication of lysosomal proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Mahfouz, R.Z.; Sharma, R.K.; Sarkar, O.; Mangrola, D.; Mathur, P.P. Potential biological role of poly (ADP-ribose) polymerase (PARP) in male gametes. Reprod. Biol. Endocrinol. 2009, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly (ADP-ribose) polymerase (PARP) cleavage in apoptosis: Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.-Y.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P. Gluco-lipotoxicity impairs ceramide flow from the endoplasmic reticulum to the Golgi apparatus in INS-1 β-cells. PLoS ONE 2014, 9, e110875. [Google Scholar] [CrossRef]
- Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 1. Cell Intrinsic Hallmarks. Cancers 2021, 13, 2042. [Google Scholar] [CrossRef]
- Huang, H.; Lorenz, B.R.; Zelmanovitz, P.H.; Chan, C.B. Metformin Preserves β-Cell Compensation in Insulin Secretion and Mass Expansion in Prediabetic Nile Rats. Int. J. Mol. Sci. 2021, 22, 421. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, C.; Dahl, K.; Jacobsen, J.B.; Axelsen, M.B.; Flint, A. Effects of semaglutide on beta cell function and glycaemic control in participants with type 2 diabetes: A randomised, double-blind, placebo-controlled trial. Diabetologia 2017, 60, 1390–1399. [Google Scholar] [CrossRef]
- Hao, Y.; Wei, M.; Zhang, N.; Zhang, X. Novel glucagon-like peptide-1 analogue exhibits potency-driven G-protein biased ag-onism with promising effects on diabetes and diabetic dry eye syndrome. Bioengineered 2022, 13, 5467–5479. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, L.; Li, H. Phytochemicals modulate pancreatic islet β cell function through glucagon-like peptide-1-related mechanisms. Biochem. Pharmacol. 2021, 197, 114817. [Google Scholar] [CrossRef] [PubMed]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. J. Clin. Investig. 2020, 5, e133429. [Google Scholar] [CrossRef] [PubMed]
- Pickford, P.; Lucey, M.; Rujan, R.-M.; McGlone, E.R.; Bitsi, S.; Ashford, F.B.; Corrêa, I.R.; Hodson, D.J.; Tomas, A.; Deganutti, G.; et al. Partial agonism improves the anti-hyperglycaemic efficacy of an oxyntomodulin-derived GLP-1R/GCGR co-agonist. Mol. Metab. 2021, 51, 101242. [Google Scholar] [CrossRef]
- de Mesquita, Y.L.L.; Calvi, I.P.; Marques, I.R.; Cruz, S.A.; Padrao, E.M.H.; Carvalho, P.E.d.P.; da Silva, C.H.A.; Cardoso, R.; Moura, F.A.; Rafalskiy, V.V. Efficacy and safety of the dual GIP and GLP-1 receptor agonist tirzepatide for weight loss: A meta-analysis of randomized controlled trials. Int. J. Obes. 2023, 47, 883–892. [Google Scholar] [CrossRef]
- Tysoe, O. Tirzepatide highly effective for weight loss. Nat. Rev. Endocrinol. 2022, 18, 520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; He, S.-S.; Wan, T.-T.; Li, Y.-B. Liraglutide protects palmitate-induced INS-1 cell injury by enhancing autophagy me-diated via FoxO1. Mol. Med. Rep. 2021, 23, 1. [Google Scholar]
- Hinds, C.E.; Peace, E.; Chen, S.; Davies, I.; El Eid, L.; Tomas, A.; Tan, T.; Minnion, J.; Jones, B.; Bloom, S.R. Abolishing β-arrestin recruitment is necessary for the full metabolic benefits of G protein-biased glucagon-like peptide-1 receptor agonists. Diabetes Obes. Metab. 2024, 26, 65–77. [Google Scholar] [CrossRef]
- Chavda, V.P.; Ajabiya, J.; Teli, D.; Bojarska, J.; Apostolopoulos, V. Tirzepatide, a New Era of Dual-Targeted Treatment for Diabetes and Obesity: A Mini-Review. Molecules 2022, 27, 4315. [Google Scholar] [CrossRef]
- Willard, F.S.; Douros, J.D.; Gabe, M.B.; Showalter, A.D.; Wainscott, D.B.; Suter, T.M.; Capozzi, M.E.; van der Velden, W.J.; Stutsman, C.; Cardona, G.R.; et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. J. Clin. Investig. 2020, 5, e140532. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- MacDonald, P.E.; El-Kholy, W.; Riedel, M.J.; Salapatek, A.M.F.; Light, P.E.; Wheeler, M.B. The Multiple Actions of GLP-1 on the Process of Glucose-Stimulated Insulin Secretion. Diabetes 2002, 51, S434–S442. [Google Scholar] [CrossRef]
- Straub, S.G.; Sharp, G.W.G. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes/Metabolism Res. Rev. 2002, 18, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Bratanova-Tochkova, T.K.; Cheng, H.; Daniel, S.; Gunawardana, S.; Liu, Y.-J.; Mulvaney-Musa, J.; Schermerhorn, T.; Straub, S.G.; Yajima, H.; Sharp, G.W. Triggering and Augmentation Mechanisms, Granule Pools, and Biphasic Insulin Secretion. Diabetes 2002, 51, S83–S90. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kaneto, H. Metformin induces insulin secretion by preserving pancreatic aquaporin 7 expression in type 2 diabetes mellitus. J. Diabetes Investig. 2021, 13, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Del Guerra, S.; Marselli, L.; Lupi, R.; Masini, M.; Pollera, M.; Bugliani, M.; Boggi, U.; Vistoli, F.; Mosca, F.; et al. Pancreatic Islets from Type 2 Diabetic Patients Have Functional Defects and Increased Apoptosis That Are Ameliorated by Metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5535–5541. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Li, Q.; You, W.; Wen, S.; Chen, T.; Su, J.; Zhao, W.; Hu, J. Complement C3 promotes islet β-cell dedifferentiation by activating Wnt/β-catenin pathway. iScience 2024, 27, 111064. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Shirakawa, J.; Okuyama, T.; Kyohara, M.; Yamazaki, S.; Togashi, Y.; Terauchi, Y. Effects of metformin on com-pensatory pancreatic β-cell hyperplasia in mice fed a high-fat diet. Am. J. Physiol.-Endocrinol. Metab. 2017, 313, E367–E380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, H.; Lu, J.; Dong, Y.; Feng, J. Design, synthesis, and biological evaluation of a potential long-acting glucagon-like peptide-1 (GLP-1) analog. Bioorg. Med. Chem. 2023, 85, 117291. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, F.; Wan, D.; Liu, Y.; Yang, L.; Feng, H.; Cui, X.; Gao, X.; Song, H. Expression and Characterization of a Potent Long-Acting GLP-1 Receptor Agonist, GLP-1-IgG2σ-Fc. PLoS ONE 2016, 11, e0156449. [Google Scholar] [CrossRef]
- Lee, S.P.; Qi, J.; Xu, G.; Rankin, M.M.; Littrell, J.; Xu, J.Z.; Bakaj, I.; Pocai, A. GRK Inhibition Potentiates Glucagon-Like Peptide-1 Action. Front. Endocrinol. 2021, 12, 652628. [Google Scholar] [CrossRef]
- Marqués, P.; Kamitz, A.; Bartolomé, A.; Burillo, J.; Martínez, H.; Jiménez, B.; Fernández-Rhodes, M.; Guillén, C.; Benito, M. Essential role of glucokinase in the protection of pancreatic β cells to the glucose energetic status. Cell Death Discov. 2019, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Liu, L.; Huh, E.; Gbahou, F.; Cecon, E.; Oshima, M.; Houzé, L.; Katsonis, P.; Hegron, A.; Fan, Z.; et al. Human GLP1R variants affecting GLP1R cell surface expression are associated with impaired glucose control and increased adiposity. Nat. Metab. 2023, 5, 1673–1684. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gojani, E.G.; Wang, B.; Li, D.; Kovalchuk, O.; Kovalchuk, I. Single and Combined Impact of Semaglutide, Tirzepatide, and Metformin on β-Cell Maintenance and Function Under High-Glucose–High-Lipid Conditions: A Comparative Study. Int. J. Mol. Sci. 2025, 26, 421. https://doi.org/10.3390/ijms26010421
Gojani EG, Wang B, Li D, Kovalchuk O, Kovalchuk I. Single and Combined Impact of Semaglutide, Tirzepatide, and Metformin on β-Cell Maintenance and Function Under High-Glucose–High-Lipid Conditions: A Comparative Study. International Journal of Molecular Sciences. 2025; 26(1):421. https://doi.org/10.3390/ijms26010421
Chicago/Turabian StyleGojani, Esmaeel Ghasemi, Bo Wang, Dongping Li, Olga Kovalchuk, and Igor Kovalchuk. 2025. "Single and Combined Impact of Semaglutide, Tirzepatide, and Metformin on β-Cell Maintenance and Function Under High-Glucose–High-Lipid Conditions: A Comparative Study" International Journal of Molecular Sciences 26, no. 1: 421. https://doi.org/10.3390/ijms26010421
APA StyleGojani, E. G., Wang, B., Li, D., Kovalchuk, O., & Kovalchuk, I. (2025). Single and Combined Impact of Semaglutide, Tirzepatide, and Metformin on β-Cell Maintenance and Function Under High-Glucose–High-Lipid Conditions: A Comparative Study. International Journal of Molecular Sciences, 26(1), 421. https://doi.org/10.3390/ijms26010421