
Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides



Abstract
:1. Introduction
Overview of CPPs Classification
2. The CPPs Classification by Their Physicochemical Properties
2.1. Cationic CPPs (cCPPs)
2.1.1. Tat
2.1.2. PTD4
2.1.3. Penetratin
2.1.4. Polyarginines
ATX-101
(R-X-R)4, X = 6-Aminohexanoic Acid
2.1.5. AVB-620
2.1.6. Polylysines
TransMTS®
2.1.7. Z12
2.2. Amphipathic CPPs (aCPPs)
2.2.1. Primary Amphipathic CPPs (paCPPs)
pVEC
bPrPp(1–30)
MPrPp(1–28)
ARF(1–22)
MPG and Pep-1
PEP-010
2.2.2. Secondary Amphipathic CPPs (saCPPs)
MAP
M918
GALA and KALA
p28
2.2.3. Amphipathic -Sheet Peptides
EAK16 and RAD16
MAX1, MAX8 and Q11
E1Y9
2.2.4. Proline-Rich Amphipathic Peptides
SAP
Bac-7
Poly-L-Proline Type II Helix (PPII) Based
2.3. Hydrophobic CPPs
2.3.1. Linear Hydrophobic Peptides Based on Natural Amino Acids
Pentapeptides (CPP5)
Pep-7, SG3 and FGF
2.3.2. Stapled Peptides
SAHBs
NYAD-1
ALRN-6924
2.3.3. Prenylated Peptides
2.3.4. Pepducins
P1pal-7
2.4. Cyclic CPPs (cyCPPs)
2.4.1. BT1718
2.4.2. 177Lu-DOTA0-Tyr3-Octreotate
3. The CPPs Classification by Their Origin
3.1. Protein-Derived CPPs
3.1.1. Homeoprotein-Derived Cell-Penetrating Peptides
3.1.2. Heparin Binding Proteins
3.1.3. Viral Proteins
3.2. CPPs Derived from Animal Venoms and Toxins
3.2.1. Maurocalcine (MCa)
3.2.2. Imperatoxin A
3.2.3. Melittin
3.2.4. Anoplin
3.2.5. Mastoparan
3.2.6. Lycosin-I and Lycosin-II
3.2.7. Pardaxins
4. Types of CPP Attachment to Cargo: Covalent and Non-Covalent Strategies with Recent Advances
4.1. Covalent Conjugation: Stability and Targeted Release
4.1.1. Non-Cleavable Conjugation
4.1.2. Cleavable Conjugation
4.2. Non-Covalent Complexation: Flexibility and Adaptability
4.2.1. Electrostatic Interactions
4.2.2. Hydrophobic Interactions and Adaptor Complexes
4.3. Innovations in CPP–Drug Conjugation Techniques
5. Cell Translocation Mechanisms of CPPs
5.1. Direct Translocation
5.1.1. Inverted Micelle Formation
5.1.2. Direct Translocation via Pore Formation
5.1.3. Carpet-like Model
5.1.4. Direct Translocation Mechanisms Used by Arginine-Rich Peptides
5.2. Endocytosis
5.2.1. Macropinocytosis
5.2.2. Clathrin-Mediated Endocytosis (CME)
5.2.3. Caveolae-Mediated Endocytosis (CvME)
5.2.4. Clathrin- and Caveolae-Independent Endocytosis
6. Computationally-Aided Design and Prediction of New CPPs
7. Discussion
Challenges and Limitations in CPP-Based Therapeutics
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3D | Three-Dimensional |
| aCPP | Amphipathic Cell-Penetrating Peptide |
| ADME | Absorption, Distribution, Metabolism and Excretion |
| AML | Acute Myeloid Leukemia |
| AMP | Antimicrobial Peptide |
| ANN | Artificial Neural Network |
| Bac | Bactenecin |
| BCT | Bicycle Toxin Conjugate |
| BFDV | Beak and Feather Disease Virus |
| BH3 | Bcl-2 Homology 3 |
| BIM | Bcl-2-Interacting Mediator |
| BIP | Bax-Inhibiting Peptide |
| BMP | Bis(Monooleoylglycero) Phophate |
| BPrPp | Bovine Prion Protein |
| cCPP | Cationic Cell-Penetrating Peptide |
| cyCPP | Cyclic Cell-Penetrating Peptide |
| Cav | Caveolin |
| CAV | Chicken Anemia Virus |
| CAPH | Cationic Amphiphilic Polyproline Helix |
| CCV | Clathrin-Coated Vesicle |
| CD | Circular Dichroism |
| CME | Clathrin-Mediated Endocytosis |
| CPP | Cell-Penetrating Peptide |
| CPP5 | Cell-Penetrating Pentapeptide |
| CSFV | Classical Swine Fever Virus |
| CTGF | Connective Tissue Grouwth Factor |
| CTL | Cytotoxic T Lymphocyte |
| CvME | Caveolae-Mediated Endocytosis |
| DENV | Dengue Virus |
| DMBA | 7,12-dimethylbenz[a]anthracene |
| DPVs | Diatos Peptide Vectors |
| ELP | Elastin-Like Polypeptide |
| EN1 | Neural-specific transcription factor Engrailed 1 |
| ERT | Extremely Randomized Tree |
| FGF | Fibroblast Growth Factor |
| FHV | Flock House Virus |
| GAG | Glycosaminoglycan |
| GBDT | Gradient Boost Decision Tree |
| GEP | Gastroenteropancreatic |
| GFP | Green Fluorescent Protein |
| GPCR | G Protein Coupled Receptor |
| hCPP | Hydrophobic Cell-Penetrating Peptide |
| HSPG | Heparan Sulfate Proteoglycan |
| HS | Heparan Sulfate |
| HTX | Hepatocyte transplantation |
| iPep | Interfering Peptide |
| IL-2R | Interleukin-2 Receptor |
| IpTxA | Imperatoxin |
| k-NN | k-Nearest Neighbor |
| LGB | Light Gradient Boosting |
| LightGBM | Light Gradient Boosting Machine |
| MAP | Model Amphipathic Peptide |
| MCa | Maurocalcine |
| MD | Minimal Domain |
| MDR | Multidrug Resistance |
| MIC | Minimum Inhibitory Concentration |
| MPG | N-methyl DNA Glycosylase |
| MPrPp | Mouse Prion Protein |
| MT1-MMP | Membrane Type 1 Matrix Metalloproteinase |
| MTS | Membrane Translocation Sequence |
| NET | Neuroendocrine Tumor |
| NLS | Nuclear Localization Sequence |
| ODN | Oligodeoxynucleotide |
| OSCC | Oral Squamous Cell Carcinoma |
| paCPP | Primary Amphipathic Cell-Penetrating Peptide |
| pHACS | Hemagglutinin Cleavage Site Peptides |
| PAR | Protease Activated Receptor |
| PCNA | Proliferating Cell Nuclear Antigen |
| PCV2 | Porcine Circovirus 2 |
| PDX | Patient-Derived Xenograft |
| PI | Phophatidylinositol |
| PL | Polylysines |
| PNA | Peptide Nucleic Acid |
| PP2A | Protein Phosphatase 2A |
| PPII | Poly-L-Prolyne Type II Helix |
| PRRT | Peptide Receptor Radionuclide Therapy |
| PTD | Protein Transduction Domain |
| PTRF | Polymerase I and Transcript Release Factor |
| pVEC | VE-cadherin-derived Cell-Penetrating Peptide |
| RCM | Ring-Closing Metathesis |
| RF | Random Forest |
| ROS | Reactive Oxygen Species |
| saCPP | Secondary Amphipatic Cell-Penetrating Peptide |
| SAHB | Stabilized -Helix of BCL-2 domains |
| SAP | Sweet Arrow Peptide |
| SAR | Structure–Activity Relationship |
| SCLC | Small Cell Lung Cancer |
| SMO | Sequential Minimal Optimization |
| SSTR | Somatostatin Receptor |
| SSTR2 | Type 2 Somatostatin Receptor |
| SVM | Support Vector Machine |
| TNBC | Triple-Negative Breast Cancer |
References
- Zorko, M.; Langel, U. Cell Penetrating Peptides, Methods and Protocols. Methods Mol. Biol. 2022, 2383, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous Functional Domains of Chemically Synthesized Human lmmunodeficiency Virus Tat Trans-Activator Protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Mäe, M.; Langel, U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr. Opin. Pharmacol. 2006, 6, 509–514. [Google Scholar] [CrossRef]
- Järver, P.; Mäger, I.; Langel, U. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol. Sci. 2010, 31, 528–535. [Google Scholar] [CrossRef]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic Peptides Obtained From Azurin Preferentially Enter Cancer Cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef]
- Johansson, H.J.; El-Andaloussi, S.; Holm, T.; Mäe, M.; Jänes, J.; Maimets, T.; Langel, U. Characterization of a Novel Cytotoxic Cell-penetrating Peptide Derived from p14ARF Protein. Mol. Ther. 2008, 16, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, U.; Andaloussi, S.E. Distinct Uptake Routes of Cell-Penetrating Peptide Conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef]
- Steinman, R.M.; Mellman, I.S.; Müller, W.A.; Cohn, Z.A. Endocytosis and the Recycling of Plasma Membrane. J. Cell Biol. 1983, 96, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- McMahon, H.T.; Gallop, J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Thorén, P.E.G.; Persson, D.; Esbjörner, E.K.; Goksör, M.; Lincoln, P.; Nordén, B. Membrane Binding and Translocation of Cell-Penetrating Peptides. Biochemistry 2004, 43, 3471–3489. [Google Scholar] [CrossRef]
- Jiao, C.Y.; Delaroche, D.; Burlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and Endocytosis for Cell-penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef]
- Ziegler, A.; Seelig, J. Binding and Clustering of Glycosaminoglycans: A Common Property of Mono- and Multivalent Cell-Penetrating Compounds. Biophys. J. 2008, 94, 2142–2149. [Google Scholar] [CrossRef]
- Verdurmen, W.P.; Thanos, M.; Ruttekolk, I.R.; Gulbins, E.; Brock, R. Cationic cell-penetrating peptides induce ceramide formation via acid sphingomyelinase: Implications for uptake. J. Control. Release 2010, 147, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A New Potent Secondary Amphipathic Cell–penetrating Peptide for siRNA Delivery Into Mammalian Cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.A.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide–PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef]
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.M.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim. Biophys. Acta (BBA)—Biomembr. 2013, 1828, 1365–1373. [Google Scholar] [CrossRef]
- Offerman, S.C.; Kadirvel, M.; Abusara, O.H.; Bryant, J.L.; Telfer, B.A.; Brown, G.; Freeman, S.; White, A.; Williams, K.J.; Aojula, H.S. N-tert -Prenylation of the indole ring improves the cytotoxicity of a short antagonist G analogue against small cell lung cancer. MedChemComm 2017, 8, 551–558. [Google Scholar] [CrossRef]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning Receptors On and Off with Intracellular Pepducins: New Insights into G-protein-coupled Receptor Drug Development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef]
- Gowland, C.; Berry, P.; Errington, J.; Jeffrey, P.; Bennett, G.; Godfrey, L.; Pittman, M.; Niewiarowski, A.; Symeonides, S.N.; Veal, G.J. Development of a LC–MS/MS method for the quantification of toxic payload DM1 cleaved from BT1718 in a Phase I study. Bioanalysis 2021, 13, 101–113. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Maupetit, J.; Derreumaux, P.; Tufféry, P. A fast method for large-scale De Novo peptide and miniprotein structure prediction. J. Comput. Chem. 2010, 31, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Peng, H.; Kang, J.; Sun, D. Cell-penetrating peptides: Possible transduction mechanisms and therapeutic applications. Biomed. Rep. 2016, 4, 528–534. [Google Scholar] [CrossRef]
- Mitchell, D.; Steinman, L.; Kim, D.; Fathman, C.; Rothbard, J. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S. Oligoarginine vectors for intracellular delivery: Design and cellular-uptake mechanisms. Pept. Sci. 2006, 84, 241–249. [Google Scholar] [CrossRef]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- Law, M.; Jafari, M.; Chen, P. Physicochemical characterization of siRNA-peptide complexes. Biotechnol. Prog. 2008, 24, 957–963. [Google Scholar] [CrossRef]
- Orzáez, M.; Mondragón, L.; Marzo, I.; Sanclimens, G.; Messeguer, A.; Pérez-Payá, E.; Vicent, M.J. Conjugation of a novel Apaf-1 inhibitor to peptide-based cell-membrane transporters: Effective methods to improve inhibition of mitochondria-mediated apoptosis. Peptides 2007, 28, 958–968. [Google Scholar] [CrossRef]
- Eiríksdóttir, E.; Konate, K.; Langel, U.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta (BBA)—Biomembr. 2010, 1798, 1119–1128. [Google Scholar] [CrossRef]
- Lam, S.L.; Hsu, V.L. NMR identification of left-handed polyproline type II helices. Biopolymers 2003, 69, 270–281. [Google Scholar] [CrossRef]
- Ruzza, P.; Calderan, A.; Guiotto, A.; Osler, A.; Borin, G. Tat cell-penetrating peptide has the characteristics of a poly(proline) II helix in aqueous solution and in SDS micelles. J. Pept. Sci. 2004, 10, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Ruzza, P.; Biondi, B.; Marchiani, A.; Antolini, N.; Calderan, A. Cell-Penetrating Peptides: A Comparative Study on Lipid Affinity and Cargo Delivery Properties. Pharmaceuticals 2010, 3, 1045–1062. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef]
- Chiu, L.S.; Anderton, R.S.; Knuckey, N.W.; Meloni, B.P. The neuroprotective potential of arginine-rich peptides for the acute treatment of traumatic brain injury. Expert Rev. Neurother. 2016, 16, 361–363. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Lamazière, A.; Burlina, F.; Wolf, C.; Chassaing, G.; Trugnan, G.; Ayala-Sanmartin, J. Non-Metabolic Membrane Tubulation and Permeability Induced by Bioactive Peptides. PLoS ONE 2007, 2, e201. [Google Scholar] [CrossRef]
- Yang, S.T.; Zaitseva, E.; Chernomordik, L.V.; Melikov, K. Cell-Penetrating Peptide Induces Leaky Fusion of Liposomes Containing Late Endosome-Specific Anionic Lipid. Biophys. J. 2010, 99, 2525–2533. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Moon, C.; Balthasar, J.P.; Yang, V.C. Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. J. Control. Release 2014, 194, 197–210. [Google Scholar] [CrossRef]
- Lev, N.; Barhum, Y.; Ben-Zur, T.; Aharony, I.; Trifonov, L.; Regev, N.; Melamed, E.; Gruzman, A.; Offen, D. A DJ-1 Based Peptide Attenuates Dopaminergic Degeneration in Mice Models of Parkinson’s Disease via Enhancing Nrf2. PLoS ONE 2015, 10, e0127549. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Chen, C.; Qin, J.; Liu, Q.; Wang, Q.; Xu, X.; Wang, J. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017, 24, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Staecker, H.; Jokovic, G.; Karpishchenko, S.; Kienle-Gogolok, A.; Krzyzaniak, A.; Lin, C.D.; Navratil, P.; Tzvetkov, V.; Wright, N.; Meyer, T. Efficacy and Safety of AM-111 in the Treatment of Acute Unilateral Sudden Deafness—A Double-blind, Randomized, Placebo-controlled Phase 3 Study. Otol. Neurotol. 2019, 40, 584–594. [Google Scholar] [CrossRef]
- Wu, B.; Li, M.; Li, K.; Hong, W.; Lv, Q.; Li, Y.; Xie, S.; Han, J.; Tian, B. Cell penetrating peptide TAT-functionalized liposomes for efficient ophthalmic delivery of flurbiprofen: Penetration and its underlying mechanism, retention, anti-inflammation and biocompatibility. Int. J. Pharm. 2021, 598, 120405. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Park, Y.; Hahm, K.S.; Lee, D.G. Biological activity of Tat (47–58) peptide on human pathogenic fungi. Biochem. Biophys. Res. Commun. 2006, 345, 222–228. [Google Scholar] [CrossRef]
- Zhu, W.L.; Shin, S.Y. Effects of dimerization of the cell-penetrating peptide Tat analog on antimicrobial activity and mechanism of bactericidal action. J. Pept. Sci. 2009, 15, 345–352. [Google Scholar] [CrossRef]
- Kimura, M.; Kosuge, K.; Ko, Y.; Kurosaki, N.; Tagawa, N.; Kato, I.; Uchida, Y. Potent Antibacterial Activity of Synthetic Peptides Designed from Salusin-β and HIV-1 Tat(49–57). Chem. Pharm. Bull. 2020, 68, 810–813. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Nhàn, N.T.T.; Maidana, D.E.; Yamada, K.H. Ocular Delivery of Therapeutic Agents by Cell-Penetrating Peptides. Cells 2023, 12, 1071. [Google Scholar] [CrossRef]
- Ho, A.; Schwarze, S.R.; Mermelstein, S.J.; Waksman, G.; Dowdy, S.F. Synthetic protein transduction domains: Enhanced transduction potential in vitro and in vivo. Cancer Res. 2001, 61, 474–477. [Google Scholar] [PubMed]
- Mucha, P.; Sikorska, E.; Rekowski, P.; Ruczyński, J. Interaction of Arginine-Rich Cell-Penetrating Peptides with an Artificial Neuronal Membrane. Cells 2022, 11, 1638. [Google Scholar] [CrossRef] [PubMed]
- Mazuryk, J.; Puchalska, I.; Koziński, K.; Ślusarz, M.J.; Ruczyński, J.; Rekowski, P.; Rogujski, P.; Płatek, R.; Wiśniewska, M.B.; Piotrowski, A.; et al. PTD4 Peptide Increases Neural Viability in an In Vitro Model of Acute Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 6086. [Google Scholar] [CrossRef]
- Jin, J.l.; Gong, J.; Yin, T.j.; Lu, Y.j.; Xia, J.j.; Xie, Y.y.; Di, Y.; He, L.; Guo, J.l.; Sun, J.; et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur. J. Pharmacol. 2011, 654, 17–25. [Google Scholar] [CrossRef]
- Liu, X.W.; Yuan, P.; Tian, J.; Li, L.J.; Wang, Y.; Huang, S.C.; Liu, L.; Backendorf, C.; Noteborn, M.H.; Sun, J. PTD4-apoptin induces Bcl-2-insensitive apoptosis in human cervical carcinoma in vitro and in vivo. Anti-Cancer Drugs 2016, 27, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.; Chen, Y.; Sun, L.; Li, G.; Zhai, M.; Zhai, W.; Kang, Q.; Gao, Y.; Qi, Y. Antitumor activity of novel chimeric peptides derived from cyclinD/CDK4 and the protein transduction domain 4. Amino Acids 2013, 44, 499–510. [Google Scholar] [CrossRef]
- Lopes, L.B.; Furnish, E.J.; Komalavilas, P.; Flynn, C.R.; Ashby, P.; Hansen, A.; Ly, D.P.; Yang, G.P.; Longaker, M.T.; Panitch, A.; et al. Cell Permeant Peptide Analogues of the Small Heat Shock Protein, HSP20, Reduce TGF-β1-Induced CTGF Expression in Keloid Fibroblasts. J. Investig. Dermatol. 2009, 129, 590–598. [Google Scholar] [CrossRef]
- Flynn, C.R.; Cheung-flynn, J.; Smoke, C.C.; Lowry, D.; Roberson, R.; Sheller, M.R.; Brophy, C.M. Internalization and Intracellular Trafficking of a PTD-Conjugated Anti-Fibrotic Peptide, AZX100, in Human Dermal Keloid Fibroblasts. J. Pharm. Sci. 2010, 99, 3100–3121. [Google Scholar] [CrossRef]
- Tachikawa, K.; Schröder, O.; Frey, G.; Briggs, S.P.; Sera, T. Regulation of the endogenous VEGF-A gene by exogenous designed regulatory proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 15225–15230. [Google Scholar] [CrossRef]
- Asua, D.; Bougamra, G.; Calleja-Felipe, M.; Morales, M.; Knafo, S. Peptides Acting as Cognitive Enhancers. Neuroscience 2018, 370, 81–87. [Google Scholar] [CrossRef]
- Del’Guidice, T.; Lepetit-Stoffaes, J.P.; Bordeleau, L.J.; Roberge, J.; Théberge, V.; Lauvaux, C.; Barbeau, X.; Trottier, J.; Dave, V.; Roy, D.C.; et al. Membrane permeabilizing amphiphilic peptide delivers recombinant transcription factor and CRISPR-Cas9/Cpf1 ribonucleoproteins in hard-to-modify cells. PLoS ONE 2018, 13, e0195558. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.A.; Thapa, R.; Jeong, S.H.; Bae, H.D.; Maeng, J.; Lee, K.; Park, K. Enhanced intranasal insulin delivery by formulations and tumor protein-derived protein transduction domain as an absorption enhancer. J. Control. Release 2019, 294, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Wohlford-Lenane, C.; Kandimalla, S.; Sartre, G.; Meyerholz, D.K.; Théberge, V.; Hallée, S.; Duperré, A.M.; Del’Guidice, T.; Lepetit-Stoffaes, J.P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906. [Google Scholar] [CrossRef] [PubMed]
- Roux, I.L.; Joliot, A.H.; Bloch-Gallego, E.; Prochiantz, A.; Volovitch, M. Neurotrophic activity of the Antennapedia homeodomain depends on its specific DNA-binding properties. Proc. Natl. Acad. Sci. USA 1993, 90, 9120–9124. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Fischer, P.; Zhelev, N.; Wang, S.; Melville, J.; Fåhraeus, R.; Lane, D. Structure–activity relationship of truncated and substituted analogues of the intracellular delivery vector Penetratin. J. Pept. Res. 2000, 55, 163–172. [Google Scholar] [CrossRef]
- Drin, G.; Déméné, H.; Temsamani, J.; Brasseur, R. Translocation of the pAntp Peptide and Its Amphipathic Analogue AP-2AL. Biochemistry 2001, 40, 1824–1834. [Google Scholar] [CrossRef]
- Magzoub, M.; Kilk, K.; Eriksson, L.E.; Langel, U.; Gräslund, A. Interaction and structure induction of cell-penetrating peptides in the presence of phospholipid vesicles. Biochim. Biophys. Acta 2001, 1512, 77–89. [Google Scholar] [CrossRef]
- Magzoub, M.; Eriksson, L.; Gräslund, A. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: Effects of surface charge and peptide concentration. Biochim. Biophys. Acta (BBA)—Biomembr. 2002, 1563, 53–63. [Google Scholar] [CrossRef]
- Prochiantz, A. Getting hydrophilic compounds into cells: Lessons from homeopeptides. Curr. Opin. Neurobiol. 1996, 6, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Mazel, M.; Clair, P.; Mathieu, D.; Kaczorek, M.; Temsamani, J. Physico-chemical requirements for cellular uptake of pAntp peptide. Eur. J. Biochem. 2001, 268, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Balayssac, S.; Burlina, F.; Convert, O.; Bolbach, G.; Chassaing, G.; Lequin, O. Comparison of Penetratin and Other Homeodomain-Derived Cell-Penetrating Peptides: Interaction in a Membrane-Mimicking Environment and Cellular Uptake Efficiency. Biochemistry 2006, 45, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Imesch, P.; Scheiner, D.; Szabo, E.; Fink, D.; Fedier, A. Conjugates of cytochrome c and antennapedia peptide activate apoptosis and inhibit proliferation of HeLa cancer cells. Exp. Ther. Med. 2013, 6, 786–790. [Google Scholar] [CrossRef]
- Alves, I.D.; Carré, M.; Montero, M.P.; Castano, S.; Lecomte, S.; Marquant, R.; Lecorché, P.; Burlina, F.; Schatz, C.; Sagan, S.; et al. A proapoptotic peptide conjugated to penetratin selectively inhibits tumor cell growth. Biochim. Biophys. Acta (BBA)—Biomembr. 2014, 1838, 2087–2098. [Google Scholar] [CrossRef]
- Szabó, I.; Orbán, E.; Schlosser, G.; Hudecz, F.; Bánóczi, Z. Cell-penetrating conjugates of pentaglutamylated methotrexate as potential anticancer drugs against resistant tumor cells. Eur. J. Med. Chem. 2016, 115, 361–368. [Google Scholar] [CrossRef]
- Yin, T.; Xie, W.; Sun, J.; Yang, L.; Liu, J. Penetratin Peptide-Functionalized Gold Nanostars: Enhanced BBB Permeability and NIR Photothermal Treatment of Alzheimer’s Disease Using Ultralow Irradiance. ACS Appl. Mater. Interfaces 2016, 8, 19291–19302. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Luan, J.; Wei, G.; Zhang, X.; Fan, J.; Zai, W.; Wang, S.; Wang, Y.; Liang, Y.; Nan, Y.; et al. In vivo hepatocellular expression of interleukin-22 using penetratin-based hybrid nanoparticles as potential anti-hepatitis therapeutics. Biomaterials 2018, 187, 66–80. [Google Scholar] [CrossRef]
- Ugarte-Alvarez, O.; Muñoz-López, P.; Moreno-Vargas, L.M.; Prada-Gracia, D.; Mateos-Chávez, A.A.; Becerra-Báez, E.I.; Luria-Pérez, R. Cell-Permeable Bak BH3 Peptide Induces Chemosensitization of Hematologic Malignant Cells. J. Oncol. 2020, 2020, 2679046. [Google Scholar] [CrossRef]
- Letoha, T.; Gaál, S.; Somlai, C.; Czajlik, A.; Perczel, A.; Penke, B. Membrane translocation of penetratin and its derivatives in different cell lines. J. Mol. Recognit. 2003, 16, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Buschle, M.; Schmidt, W.; Zauner, W.; Mechtler, K.; Trska, B.; Kirlappos, H.; Birnstiel, M.L. Transloading of tumor antigen-derived peptides into antigen-presenting cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Fleitmann, J.K.; Lingnau, K.; Schmidt, W.; Egyed, A.; Fritz, J.; Zauner, W.; Wittmann, B.; Gorny, I.; Berger, M.; et al. Vaccination with poly-L-arginine as immunostimulant for peptide vaccines: Induction of potent and long-lasting T-cell responses against cancer antigens. Cancer Res. 2002, 62, 1477–1480. [Google Scholar] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A Comprehensive Model for the Cellular Uptake of Cationic Cell-penetrating Peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Nakase, I.; Konishi, Y.; Ueda, M.; Saji, H.; Futaki, S. Accumulation of arginine-rich cell-penetrating peptides in tumors and the potential for anticancer drug delivery in vivo. J. Control. Release 2012, 159, 181–188. [Google Scholar] [CrossRef]
- Sepahi, M.; Jalal, R.; Mashreghi, M. Antibacterial activity of poly-l-arginine under different conditions. Iran. J. Microbiol. 2017, 9, 103–111. [Google Scholar]
- Ahmed, C.M.; Massengill, M.T.; Brown, E.E.; Ildefonso, C.J.; Johnson, H.M.; Lewin, A.S. A cell penetrating peptide from SOCS-1 prevents ocular damage in experimental autoimmune uveitis. Exp. Eye Res. 2018, 177, 12–22. [Google Scholar] [CrossRef]
- Jung, H.E.; Oh, J.E.; Lee, H.K. Cell-Penetrating Mx1 Enhances Anti-Viral Resistance against Mucosal Influenza Viral Infection. Viruses 2019, 11, 109. [Google Scholar] [CrossRef]
- Tünnemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stöckl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-Rich Peptides an Abundant Source of Membrane-Permeable Peptides Having Potential as Carriers for Intracellular Protein Delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular Uptake of Arginine-Rich Peptides: Roles for Macropinocytosis and Actin Rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Takeuchi, T.; Kuwata, K.; Chiba, J.; Hatanaka, Y.; Nakase, I.; Futaki, S. Syndecan-4 Is a Receptor for Clathrin-Mediated Endocytosis of Arginine-Rich Cell-Penetrating Peptides. Bioconjug. Chem. 2016, 27, 1119–1130. [Google Scholar] [CrossRef]
- Futaki, S.; Nakase, I. Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization. Acc. Chem. Res. 2017, 50, 2449–2456. [Google Scholar] [CrossRef]
- Nakase, I.; Hirose, H.; Tanaka, G.; Tadokoro, A.; Kobayashi, S.; Takeuchi, T.; Futaki, S. Cell-surface Accumulation of Flock House Virus-derived Peptide Leads to Efficient Internalization via Macropinocytosis. Mol. Ther. 2009, 17, 1868–1876. [Google Scholar] [CrossRef]
- Tanaka, G.; Nakase, I.; Fukuda, Y.; Masuda, R.; Oishi, S.; Shimura, K.; Kawaguchi, Y.; Takatani-Nakase, T.; Langel, U.; Gräslund, A.; et al. CXCR4 Stimulates Macropinocytosis: Implications for Cellular Uptake of Arginine-Rich Cell-Penetrating Peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. [Google Scholar] [CrossRef]
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting Proliferating Cell Nuclear Antigen and Its Protein Interactions Induces Apoptosis in Multiple Myeloma Cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Müller, R.; Vågbø, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drabløs, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Colapietro, A.; Mancini, A.; Rossetti, A.; Martellucci, S.; Ventura, L.; Franco, M.D.; Marampon, F.; Mattei, V.; Biordi, L.A.; et al. ATX-101, a Peptide Targeting PCNA, Has Antitumor Efficacy Alone or in Combination with Radiotherapy in Murine Models of Human Glioblastoma. Cancers 2022, 14, 289. [Google Scholar] [CrossRef]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of Cell-Penetrating Peptide-Morpholino Oligomer Conjugates in Human Serum and in Cells. Bioconjug. Chem. 2007, 18, 50–60. [Google Scholar] [CrossRef]
- Saleh, A.F.; Arzumanov, A.; Abes, R.; Owen, D.; Lebleu, B.; Gait, M.J. Synthesis and Splice-Redirecting Activity of Branched, Arginine-Rich Peptide Dendrimer Conjugates of Peptide Nucleic Acid Oligonucleotides. Bioconjug. Chem. 2010, 21, 1902–1911. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.; Turner, J.; Clair, P.; Richard, J.P.; Iversen, P.; Gait, M.J.; Lebleu, B. Peptide-based delivery of nucleic acids: Design, mechanism of uptake and applications to splice-correcting oligonucleotides. Biochem. Soc. Trans. 2007, 35, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Abes, R.; Moulton, H.M.; Clair, P.; Yang, S.T.; Abes, S.; Melikov, K.; Prevot, P.; Youngblood, D.S.; Iversen, P.L.; Chernomordik, L.V.; et al. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: Structure–activity studies. Nucleic Acids Res. 2008, 36, 6343–6354. [Google Scholar] [CrossRef] [PubMed]
- Nejad, A.J.; Shahrokhi, N.; Nielsen, P.E. Targeting of the Essential acpP, ftsZ, and rne Genes in Carbapenem-Resistant Acinetobacter baumannii by Antisense PNA Precision Antibacterials. Biomedicines 2021, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Kamiya, M.; Urano, Y. Molecular probes for fluorescence image-guided cancer surgery. Curr. Opin. Chem. Biol. 2022, 67, 102112. [Google Scholar] [CrossRef]
- Miampamba, M.; Liu, J.; Harootunian, A.; Gale, A.J.; Baird, S.; Chen, S.L.; Nguyen, Q.T.; Tsien, R.Y.; González, J.E. Sensitive in vivo Visualization of Breast Cancer Using Ratiometric Protease-activatable Fluorescent Imaging Agent, AVB-620. Theranostics 2017, 7, 3369–3386. [Google Scholar] [CrossRef]
- Liu, R.; Xu, Y.; Xu, K.; Dai, Z. Current trends and key considerations in the clinical translation of targeted fluorescent probes for intraoperative navigation. Aggregate 2021, 2, e23. [Google Scholar] [CrossRef]
- Woo, Y.; Chaurasiya, S.; O’Leary, M.; Han, E.; Fong, Y. Fluorescent imaging for cancer therapy and cancer gene therapy. Mol. Ther.-Oncolytic 2021, 23, 231–238. [Google Scholar] [CrossRef]
- Unkart, J.T.; Chen, S.L.; Wapnir, I.L.; González, J.E.; Harootunian, A.; Wallace, A.M. Intraoperative Tumor Detection Using a Ratiometric Activatable Fluorescent Peptide: A First-in-Human Phase 1 Study. Ann. Surg. Oncol. 2017, 24, 3167–3173. [Google Scholar] [CrossRef]
- Ryser, H.J.P.; Shen, W.C. Conjugation of methotrexate to poly(L-lysine) increases drug transport and overcomes drug resistance in cultured cells. Proc. Natl. Acad. Sci. USA 1978, 75, 3867–3870. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, J.M.; Galivan, Z.N.J.; Galivan, J. Treatment of Reuber H35 Hepatoma Cells with Carrier-Bound M ethotrexate. Mol. Pharmacol. 1981, 19, 505–508. [Google Scholar] [PubMed]
- Ryser, H.J.; Mandel, R.; Hacobian, A.; Shen, W. Methotrexate-poly(lysine) as a selective agent for mutants of chinese hamster ovary cells defective in endocytosis. J. Cell. Physiol. 1988, 135, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Zhuo, R.X. Synthesis, Hydrolysis, and Antitumor Activity of Conjugates of 5-Fluorouracil with Poly(L-lysine). Polym. J. 1993, 25, 401–405. [Google Scholar] [CrossRef]
- Lemaitre, M.; Bayard, B.; Lebleu, B. Specific antiviral activity of a poly(L-lysine)-conjugated oligodeoxyribonucleotide sequence complementary to vesicular stomatitis virus N protein mRNA initiation site. Proc. Natl. Acad. Sci. USA 1987, 84, 648–652. [Google Scholar] [CrossRef]
- Leonetti, J.P.; Degols, G.; Lebleu, B. Biological activity of oligonucleotide-poly(L-lysine) conjugates: Mechanism of cell uptake. Bioconjug. Chem. 1990, 1, 149–153. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, L.; Yu, F.; Pan, W.; Zhang, J. Delivery of different length poly(l-lysine)-conjugated ODN to HepG2 cells using N-stearyllactobionamide-modified liposomes and their enhanced cellular biological effects. Int. J. Pharm. 2006, 311, 82–88. [Google Scholar] [CrossRef]
- Shen, W.C.; Ryser, H.J. Conjugation of poly-L-lysine to albumin and horseradish peroxidase: A novel method of enhancing the cellular uptake of proteins. Proc. Natl. Acad. Sci. USA 1978, 75, 1872–1876. [Google Scholar] [CrossRef]
- Ryser, H.J.; Drummond, I.; Shen, W. The cellular uptake of horseradish peroxidase and its poly(lysine) conjugate by cultured fibroblasts is qualitatively similar despite a 900-fold difference in rate. J. Cell. Physiol. 1982, 113, 167–178. [Google Scholar] [CrossRef]
- Curiel, D.T.; Agarwal, S.; Wagner, E.; Cotten, M. Adenovirus enhancement of transferrin-polylysine-mediated gene delivery. Proc. Natl. Acad. Sci. USA 1991, 88, 8850–8854. [Google Scholar] [CrossRef]
- Curiel, D.T.; Wagner, E.; Cotten, M.; Birnstiel, M.L.; Agarwal, S.; Li, C.M.; Loechel, S.; Hu, P.C. High-Efficiency Gene Transfer Mediated by Adenovirus Coupled to DNAPolylysine Complexes. Hum. Gene Ther. 1992, 3, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Zatloukal, K.; Cotten, M.; Kirlappos, H.; Mechtler, K.; Curiel, D.T.; Birnstiel, M.L. Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes. Proc. Natl. Acad. Sci. USA 1992, 89, 6099–6103. [Google Scholar] [CrossRef]
- Mulders, P.; Pang, S.; Dannull, J.; Kaboo, R.; Hinkel, A.; Michel, K.; Tso, C.L.; Roth, M.; Belldegrun, A. Highly efficient and consistent gene transfer into dendritic cells utilizing a combination of ultraviolet-irradiated adenovirus and poly(L-lysine) conjugates. Cancer Res. 1998, 58, 956–961. [Google Scholar]
- Takechi, Y.; Tanaka, H.; Kitayama, H.; Yoshii, H.; Tanaka, M.; Saito, H. Comparative study on the interaction of cell-penetrating polycationic polymers with lipid membranes. Chem. Phys. Lipids 2012, 165, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Galliher, W.C.; Goun, E.A.; Jones, L.R.; Pillow, T.H. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Deliv. Rev. 2008, 60, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Nervi, P.; Dürrenberger, M.; Seelig, J. The Cationic Cell-Penetrating Peptide CPPTAT Derived from the HIV-1 Protein TAT Is Rapidly Transported into Living Fibroblasts: Optical, Biophysical, and Metabolic Evidence. Biochemistry 2005, 44, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.Y.; Jow, T.; Hantash, B.M. Bioactive oligopeptides in dermatology: Part II. Exp. Dermatol. 2012, 21, 569–575. [Google Scholar] [CrossRef]
- Jankovic, J.; Truong, D.; Patel, A.T.; Brashear, A.; Evatt, M.; Rubio, R.G.; Oh, C.K.; Snyder, D.; Shears, G.; Comella, C. Injectable DaxibotulinumtoxinA in Cervical Dystonia: A Phase 2 Dose-Escalation Multicenter Study. Mov. Disord. Clin. Pract. 2018, 5, 273–282. [Google Scholar] [CrossRef]
- BRANDT, F.; O’CONNELL, C.; CAZZANIGA, A.; WAUGH, J.M. Efficacy and Safety Evaluation of a Novel Botulinum Toxin Topical Gel for the Treatment of Moderate to Severe Lateral Canthal Lines. Dermatol. Surg. 2010, 36, 2111–2118. [Google Scholar] [CrossRef]
- Glogau, R.; Blitzer, A.; Brandt, F.; Kane, M.; Monheit, G.D.; Waugh, J.M. Results of a randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of a botulinum toxin type A topical gel for the treatment of moderate-to-severe lateral canthal lines. J. Drugs Dermatol. JDD 2012, 11, 38–45. [Google Scholar]
- Gallagher, C.J.; Bowsher, R.R.; Clancy, A.; Dover, J.S.; Humphrey, S.; Liu, Y.; Prawdzik, G. Clinical Immunogenicity of DaxibotulinumtoxinA for Injection in Glabellar Lines: Pooled Data from the SAKURA Phase 3 Trials. Toxins 2023, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Marchione, R.; Daydé, D.; Lenormand, J.; Cornet, M. ZEBRA cell-penetrating peptide as an efficient delivery system in Candida albicans. Biotechnol. J. 2014, 9, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Rothe, R.; Liguori, L.; Villegas-Mendez, A.; Marques, B.; Grunwald, D.; Drouet, E.; Lenormand, J.L. Characterization of the Cell-penetrating Properties of the Epstein-Barr Virus ZEBRA trans-Activator. J. Biol. Chem. 2010, 285, 20224–20233. [Google Scholar] [CrossRef] [PubMed]
- Derouazi, M.; Berardino-Besson, W.D.; Belnoue, E.; Hoepner, S.; Walther, R.; Benkhoucha, M.; Teta, P.; Dufour, Y.; Maroun, C.Y.; Salazar, A.M.; et al. Novel Cell-Penetrating Peptide-Based Vaccine Induces Robust CD4+ and CD8+ T Cell–Mediated Antitumor Immunity. Cancer Res. 2015, 75, 3020–3031. [Google Scholar] [CrossRef]
- Belnoue, E.; Berardino-Besson, W.D.; Gaertner, H.; Carboni, S.; Dunand-Sauthier, I.; Cerini, F.; Suso-Inderberg, E.M.; Wälchli, S.; König, S.; Salazar, A.M.; et al. Enhancing Antitumor Immune Responses by Optimized Combinations of Cell-penetrating Peptide-based Vaccines and Adjuvants. Mol. Ther. 2016, 24, 1675–1685. [Google Scholar] [CrossRef]
- Pascolutti, R.; Yeturu, L.; Philippin, G.; Borges, S.C.; Dejob, M.; Santiago-Raber, M.L.; Derouazi, M. ATP128 Clinical Therapeutic Cancer Vaccine Activates NF-κB and IRF3 Pathways through TLR4 and TLR2 in Human Monocytes and Dendritic Cells. Cancers 2022, 14, 5134. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, T.; Du, H. Prospect of cell penetrating peptides in stem cell tracking. Stem Cell Res. Ther. 2021, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Belnoue, E.; Mayol, J.F.; Carboni, S.; Besson, W.D.B.; Dupuychaffray, E.; Nelde, A.; Stevanovic, S.; Santiago-Raber, M.L.; Walker, P.R.; Derouazi, M. Targeting self and neo-epitopes with a modular self-adjuvanting cancer vaccine. JCI Insight 2019, 4, e127305. [Google Scholar] [CrossRef]
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896. [Google Scholar] [CrossRef]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef]
- Zaro, J.L.; Shen, W.C. Cationic and amphipathic cell-penetrating peptides (CPPs): Their structures and in vivo studies in drug delivery. Front. Chem. Sci. Eng. 2015, 9, 407–427. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, U. VE-Cadherin-Derived Cell-Penetrating Peptide, pVEC, with Carrier Functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef]
- Lundberg, P.; Magzoub, M.; Lindberg, M.; Hällbrink, M.; Jarvet, J.; Eriksson, L.; Langel, U.; Gräslund, A. Cell membrane translocation of the N-terminal (1–28) part of the prion protein. Biochem. Biophys. Res. Commun. 2002, 299, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Sandgren, S.; Lundberg, P.; Oglęcka, K.; Lilja, J.; Wittrup, A.; Eriksson, L.G.; Langel, U.; Belting, M.; Gräslund, A. N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem. Biophys. Res. Commun. 2006, 348, 379–385. [Google Scholar] [CrossRef]
- Elmquist, A.; Hansen, M.; Langel, U. Structure–activity relationship study of the cell-penetrating peptide pVEC. Biochim. Biophys. Acta (BBA)—Biomembr. 2006, 1758, 721–729. [Google Scholar] [CrossRef]
- Myrberg, H.; Zhang, L.; Mäe, M.; Langel, U. Design of a Tumor-Homing Cell-Penetrating Peptide. Bioconjug. Chem. 2008, 19, 70–75. [Google Scholar] [CrossRef]
- Kersemans, V.; Cornelissen, B. Targeting the Tumour: Cell Penetrating Peptides for Molecular Imaging and Radiotherapy. Pharmaceuticals 2010, 3, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Regberg, J.; Srimanee, A.; Langel, U. Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies. Pharmaceuticals 2012, 5, 991–1007. [Google Scholar] [CrossRef]
- Biverståhl, H.; Andersson, A.; Gräslund, A.; Mäler, L. NMR Solution Structure and Membrane Interaction of the N-Terminal Sequence (1–30) of the Bovine Prion Protein. Biochemistry 2004, 43, 14940–14947. [Google Scholar] [CrossRef]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003, 31, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Heitz, A.; Morris, M.C.; Charnet, P.; Divita, G.; Heitz, F. Insight into the Mechanism of Internalization of the Cell-Penetrating Carrier Peptide Pep-1 through Conformational Analysis. Biochemistry 2004, 43, 1449–1457. [Google Scholar] [CrossRef]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef]
- Henriques, S.T.; Castanho, M.A.R.B. Translocation or membrane disintegration? Implication of peptide–membrane interactions in pep-1 activity. J. Pept. Sci. 2008, 14, 482–487. [Google Scholar] [CrossRef]
- Veldhoen, S.; Laufer, S.D.; Trampe, A.; Restle, T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: Quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006, 34, 6561–6573. [Google Scholar] [CrossRef]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S.; et al. Oct-3/4 Dose Dependently Regulates Specification of Embryonic Stem Cells toward a Cardiac Lineage and Early Heart Development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Master, G.M.; Coll, J.L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sütlü, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Peptide-Based Strategy for siRNA Delivery into Mammalian Cells. In RNA Silencing: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2005; pp. 251–260. [Google Scholar] [CrossRef]
- Deshayes, S.; Gerbal-Chaloin, S.; Morris, M.C.; Aldrian-Herrada, G.; Charnet, P.; Divita, G.; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta (BBA)—Biomembr. 2004, 1667, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gerbal-Chaloin, S.; Gondeau, C.; Aldrian-Herrada, G.; Heitz, F.; Gauthier-Rouvière, C.; Divita, G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol. Cell 2007, 99, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, J.; Liu, D.; Han, Y.; Xu, H.; Liu, L.; Leng, X.; Kong, D. A cell-penetrating peptide-assisted nanovaccine promotes antigen cross-presentation and anti-tumor immune response. Biomater. Sci. 2019, 7, 5516–5527. [Google Scholar] [CrossRef]
- Deshayes, S.; Plénat, T.; Charnet, P.; Divita, G.; Molle, G.; Heitz, F. Formation of transmembrane ionic channels of primary amphipathic cell-penetrating peptides. Consequences on the mechanism of cell penetration. Biochim. Biophys. Acta (BBA)—Biomembr. 2006, 1758, 1846–1851. [Google Scholar] [CrossRef]
- Kurzawa, L.; Pellerano, M.; Morris, M.C. PEP and CADY-mediated delivery of fluorescent peptides and proteins into living cells. Biochim. Biophys. Acta (BBA)—Biomembr. 2010, 1798, 2274–2285. [Google Scholar] [CrossRef]
- Henriques, S.T.; Costa, J.; Castanho, M.A.R.B. Translocation of β-Galactosidase Mediated by the Cell-Penetrating Peptide Pep-1 into Lipid Vesicles and Human HeLa Cells Is Driven by Membrane Electrostatic Potential. Biochemistry 2005, 44, 10189–10198. [Google Scholar] [CrossRef]
- Henriques, S.T.; Castanho, M.A. Environmental factors that enhance the action of the cell penetrating peptide pep-1 A spectroscopic study using lipidic vesicles. Biochim. Biophys. Acta (BBA)—Biomembr. 2005, 1669, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, A.; Hochstrasser, R.M. Single-Molecule Imaging of the Association of the Cell-Penetrating Peptide Pep-1 to Model Membranes. Biochemistry 2007, 46, 7963–7972. [Google Scholar] [CrossRef] [PubMed]
- Almarwani, B.; Phambu, E.N.; Alexander, C.; Nguyen, H.A.T.; Phambu, N.; Sunda-Meya, A. Vesicles mimicking normal and cancer cell membranes exhibit differential responses to the cell-penetrating peptide Pep-1. Biochim. Biophys. Acta (BBA)—Biomembr. 2018, 1860, 1394–1402. [Google Scholar] [CrossRef]
- Li, G.; Huang, Y.; Feng, Q.; Chen, Y. Tryptophan as a Probe to Study the Anticancer Mechanism of Action and Specificity of α-Helical Anticancer Peptides. Molecules 2014, 19, 12224–12241. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. New Lytic Peptides Based on the d,l-Amphipathic Helix Motif Preferentially Kill Tumor Cells Compared to Normal Cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Chen, Z. Molecular Interactions between Cell Penetrating Peptide Pep-1 and Model Cell Membranes. J. Phys. Chem. B 2012, 116, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, C.; Zheng, S.; Qu, G.; Feng, Z.; Shang, J.; Cheng, Y.; He, N. Insight into the Mechanism of Internalization of the Cell-Penetrating Carrier Peptide Pep-1 by Conformational Analysis. J. Biomed. Nanotechnol. 2020, 16, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G. Making Proteins into Drugs: Assisted Delivery of Proteins and Peptides into Living Neurons. Methods Cell Biol. 2003, 71, 325–338. [Google Scholar] [CrossRef]
- Pandey, A.V.; Mellon, S.H.; Miller, W.L. Protein Phosphatase 2A and Phosphoprotein SET Regulate Androgen Production by P450c17. J. Biol. Chem. 2003, 278, 2837–2844. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, H.K.; Choi, Y.J.; Eum, W.S.; Park, J.; Choi, S.Y.; Kwon, H.Y. PEP-1-paraoxonase 1 fusion protein prevents cytokine-induced cell destruction and impaired insulin secretion in rat insulinoma cells. BMB Rep. 2018, 51, 538–543. [Google Scholar] [CrossRef]
- Gehler, S.; Shaw, A.E.; Sarmiere, P.D.; Bamburg, J.R.; Letourneau, P.C. Brain-Derived Neurotrophic Factor Regulation of Retinal Growth Cone Filopodial Dynamics Is Mediated through Actin Depolymerizing Factor/Cofilin. J. Neurosci. 2004, 24, 10741–10749. [Google Scholar] [CrossRef]
- Garnon, J.; Lachance, C.; Marco, S.D.; Hel, Z.; Marion, D.; Ruiz, M.C.; Newkirk, M.M.; Khandjian, E.W.; Radzioch, D. Fragile X-related Protein FXR1P Regulates Proinflammatory Cytokine Tumor Necrosis Factor Expression at the Post-transcriptional Level. J. Biol. Chem. 2005, 280, 5750–5763. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Riley, N.; Nguyen, V.; Montgomery, R.; French, B.; Li, J.; van Leeuwen, F.; Lungo, W.; McPhaul, L.; French, S. The mechanism of cytokeratin aggresome formation: The role of mutant ubiquitin (UBB+1). Exp. Mol. Pathol. 2003, 74, 160–167. [Google Scholar] [CrossRef]
- Gros, E.; Deshayes, S.; Morris, M.C.; Aldrian-Herrada, G.; Depollier, J.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta (BBA)—Biomembr. 2006, 1758, 384–393. [Google Scholar] [CrossRef]
- Arrouss, I.; Decaudin, D.; Choquet, S.; Azar, N.; Parizot, C.; Zini, J.; Nemati, F.; Rebollo, A. Cell Penetrating Peptides as a Therapeutic Strategy in Chronic Lymphocytic Leukemia. Protein Pept. Lett. 2015, 22, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Arrouss, I.; Nemati, F.; Roncal, F.; Wislez, M.; Dorgham, K.; Vallerand, D.; Rabbe, N.; Karboul, N.; Carlotti, F.; Bravo, J.; et al. Specific Targeting of Caspase-9/PP2A Interaction as Potential New Anti-Cancer Therapy. PLoS ONE 2013, 8, e60816. [Google Scholar] [CrossRef] [PubMed]
- Dorgham, K.; Murail, S.; Tuffery, P.; Savier, E.; Bravo, J.; Rebollo, A. Binding and Kinetic Analysis of Human Protein Phosphatase PP2A Interactions with Caspase 9 Protein and the Interfering Peptide C9h. Pharmaceutics 2022, 14, 2055. [Google Scholar] [CrossRef] [PubMed]
- Lebel-Binay, S.; Nemati, F.; Dominguez-Berrocal, L.; Fleury, J.; Naguez, A.; Decaudin, D.; Rebollo, A. Abstract 3904: PEP-010, a cell penetrating & interfering peptide as a new therapeutic approach in breast cancer. Cancer Res. 2018, 78, 3904. [Google Scholar] [CrossRef]
- Hällbrink, M.; Florén, A.; Elmquist, A.; Pooga, M.; Bartfai, T.; Langel, U. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta (BBA)—Biomembr. 2001, 1515, 101–109. [Google Scholar] [CrossRef]
- Palm-Apergi, C.; Lorents, A.; Padari, K.; Pooga, M.; Hällbrink, M. The membrane repair response masks membrane disturbances caused by cell-penetrating peptide uptake. FASEB J. 2009, 23, 214–223. [Google Scholar] [CrossRef]
- Silva, S.; Alves, C.; Duarte, D.; Costa, A.; Sarmento, B.; Almeida, A.J.; Gomes, P.; Vale, N. Model Amphipathic Peptide Coupled with Tacrine to Improve Its Antiproliferative Activity. Int. J. Mol. Sci. 2021, 22, 242. [Google Scholar] [CrossRef]
- Scheller, A.; Oehlke, J.; Wiesner, B.; Dathe, M.; Krause, E.; Beyermann, M.; Melzig, M.; Bienert, M. Structural requirements for cellular uptake of α-helical amphipathic peptides. J. Pept. Sci. 1999, 5, 185–194. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Johansson, H.J.; Holm, T.; Langel, U. A Novel Cell-penetrating Peptide, M918, for Efficient Delivery of Proteins and Peptide Nucleic Acids. Mol. Ther. 2007, 15, 1820–1826. [Google Scholar] [CrossRef]
- Rostami, B.; Irani, S.; Bolhassani, A.; Cohan, R.A. M918: A Novel Cell Penetrating Peptide for Effective Delivery of HIV-1 Nef and Hsp20-Nef Proteins into Eukaryotic Cell Lines. Curr. HIV Res. 2018, 16, 280–287. [Google Scholar] [CrossRef]
- Li, W.; Nicol, F.; Szoka, F.C. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef]
- Fattal, E.; Nir, S.; Parente, R.A.; Szoka, F.C. Pore-Forming Peptides Induce Rapid Phospholipid Flip-Flop in Membranes. Biochemistry 1994, 33, 6721–6731. [Google Scholar] [CrossRef]
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar] [CrossRef]
- Chen, S.; Zhuo, R.; Cheng, S. Enhanced gene transfection with addition of a cell-penetrating peptide in substrate-mediated gene delivery. J. Gene Med. 2010, 12, 705–713. [Google Scholar] [CrossRef]
- Fominaya, J.; Gasset, M.; García, R.; Roncal, F.; Albar, J.P.; Bernad, A. An optimized amphiphilic cationic peptide as an efficient non-viral gene delivery vector. J. Gene Med. 2000, 2, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Fialho, A.M.; Bernardes, N.; Chakrabarty, A.M. Exploring the anticancer potential of the bacterial protein azurin. AIMS Microbiol. 2016, 2, 292–303. [Google Scholar] [CrossRef]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L.; et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef]
- Yaghoubi, A.; Khazaei, M.; Avan, A.; Hasanian, S.M.; Cho, W.C.; Soleimanpour, S. p28 Bacterial Peptide, as an Anticancer Agent. Front. Oncol. 2020, 10, 1303. [Google Scholar] [CrossRef] [PubMed]
- Garizo, A.R.; Castro, F.; Martins, C.; Almeida, A.; Dias, T.P.; Fernardes, F.; Barrias, C.C.; Bernardes, N.; Fialho, A.M.; Sarmento, B. p28-functionalized PLGA nanoparticles loaded with gefitinib reduce tumor burden and metastases formation on lung cancer. J. Control. Release 2021, 337, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Mander, S.; Gorman, G.S.; Coward, L.U.; Christov, K.; Green, A.; Gupta, T.K.D.; Yamada, T. The brain-penetrant cell-cycle inhibitor p28 sensitizes brain metastases to DNA-damaging agents. Neuro-Oncol. Adv. 2023, 5, vdad042. [Google Scholar] [CrossRef]
- Yaghoubi, A.; Movaqar, A.; Asgharzadeh, F.; Derakhshan, M.; Ghazvini, K.; Hasanian, S.M.; Avan, A.; Mostafapour, A.; Khazaei, M.; Soleimanpour, S. Anticancer activity of Pseudomonas aeruginosa derived peptide with iRGD in colon cancer therapy. Iran. J. Basic Med. Sci. 2023, 26, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, C.J.; Nilsson, B.L. Review self-assembly of amphipathic β-sheet peptides: Insights and applications. Pept. Sci. 2012, 98, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Pujals, S.; Giralt, E. Amphipathic peptides and drug delivery. Pept. Sci. 2004, 76, 196–203. [Google Scholar] [CrossRef]
- Jung, J.P.; Nagaraj, A.K.; Fox, E.K.; Rudra, J.S.; Devgun, J.M.; Collier, J.H. Co-assembling peptides as defined matrices for endothelial cells. Biomaterials 2009, 30, 2400–2410. [Google Scholar] [CrossRef]
- Gray, V.P.; Amelung, C.D.; Duti, I.J.; Laudermilch, E.G.; Letteri, R.A.; Lampe, K.J. Biomaterials via peptide assembly: Design, characterization, and application in tissue engineering. Acta Biomater. 2022, 140, 43–75. [Google Scholar] [CrossRef]
- Zhang, S.; Holmes, T.C.; DiPersio, C.; Hynes, R.O.; Su, X.; Rich, A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 1995, 16, 1385–1393. [Google Scholar] [CrossRef]
- Sankar, S.; O’Neill, K.; D’Arc, M.B.; Rebeca, F.; Buffier, M.; Aleksi, E.; Fan, M.; Matsuda, N.; Gil, E.S.; Spirio, L. Clinical Use of the Self-Assembling Peptide RADA16: A Review of Current and Future Trends in Biomedicine. Front. Bioeng. Biotechnol. 2021, 9, 679525. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.S.; Gilbert, K.P. Synthetic Peptide Hydrogel Formulations for Use as Extracellular Matrix. U.S. Patent US20180023049A1, 25 January 2018. [Google Scholar]
- Ozbas, B.; Kretsinger, J.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Salt-Triggered Peptide Folding and Consequent Self-Assembly into Hydrogels with Tunable Modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar] [CrossRef]
- Kretsinger, J.K.; Haines, L.A.; Ozbas, B.; Pochan, D.J.; Schneider, J.P. Cytocompatibility of self-assembled β-hairpin peptide hydrogel surfaces. Biomaterials 2005, 26, 5177–5186. [Google Scholar] [CrossRef]
- Haines-Butterick, L.; Rajagopal, K.; Branco, M.; Salick, D.; Rughani, R.; Pilarz, M.; Lamm, M.S.; Pochan, D.J.; Schneider, J.P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. USA 2007, 104, 7791–7796. [Google Scholar] [CrossRef]
- Rudra, J.S.; Sun, T.; Bird, K.C.; Daniels, M.D.; Gasiorowski, J.Z.; Chong, A.S.; Collier, J.H. Modulating Adaptive Immune Responses to Peptide Self-Assemblies. ACS Nano 2012, 6, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Rudra, J.S.; Mishra, S.; Chong, A.S.; Mitchell, R.A.; Nardin, E.H.; Nussenzweig, V.; Collier, J.H. Self-assembled peptide nanofibers raising durable antibody responses against a malaria epitope. Biomaterials 2012, 33, 6476–6484. [Google Scholar] [CrossRef] [PubMed]
- Hudalla, G.A.; Modica, J.A.; Tian, Y.F.; Rudra, J.S.; Chong, A.S.; Sun, T.; Mrksich, M.; Collier, J.H. A Self-Adjuvanting Supramolecular Vaccine Carrying a Folded Protein Antigen. Adv. Healthc. Mater. 2014, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Tsutsumi, H.; Mihara, H. Self-Assembling Peptides as Building Blocks of Functional Materials for Biomedical Applications. Bull. Chem. Soc. Jpn. 2018, 92, 391–399. [Google Scholar] [CrossRef]
- Sawada, T.; Tsuchiya, M.; Takahashi, T.; Tsutsumi, H.; Mihara, H. Cell-adhesive hydrogels composed of peptide nanofibers responsive to biological ions. Polym. J. 2012, 44, 651–657. [Google Scholar] [CrossRef]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef]
- Fernández-Carneado, J.; Kogan, M.J.; Castel, S.; Giralt, E. Potential Peptide Carriers: Amphipathic Proline-Rich Peptides Derived from the N-Terminal Domain of γ-Zein. Angew. Chem. Int. Ed. 2004, 43, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-Carneado, J.; Ludevid, M.D.; Giralt, E. D-SAP: A New, Noncytotoxic, and Fully Protease Resistant Cell-Penetrating Peptide. ChemMedChem 2008, 3, 296–301. [Google Scholar] [CrossRef]
- Martín, I.; Teixidó, M.; Giralt, E. Design, Synthesis and Characterization of a New Anionic Cell-Penetrating Peptide: SAP(E). ChemBioChem 2011, 12, 896–903. [Google Scholar] [CrossRef]
- Sadler, K.; Eom, K.D.; Yang, J.L.; Dimitrova, Y.; Tam, J.P. Translocating Proline-Rich Peptides from the Antimicrobial Peptide Bactenecin 7. Biochemistry 2002, 41, 14150–14157. [Google Scholar] [CrossRef]
- Crespo, L.; Sanclimens, G.; Montaner, B.; Pérez-Tomás, R.; Royo, M.; Pons, M.; Albericio, F.; Giralt, E. Peptide Dendrimers Based on Polyproline Helices. J. Am. Chem. Soc. 2002, 124, 8876–8883. [Google Scholar] [CrossRef] [PubMed]
- Geli, M.I.; Torrent, M.; Ludevid, D. Two Structural Domains Mediate Two Sequential Events in [gamma]-Zein Targeting: Protein Endoplasmic Reticulum Retention and Protein Body Formation. Plant Cell 1994, 6, 1911–1922. [Google Scholar] [CrossRef]
- Franz, J.; Lelle, M.; Peneva, K.; Bonn, M.; Weidner, T. SAP(E) – A cell-penetrating polyproline helix at lipid interfaces. Biochim. Biophys. Acta (BBA)—Biomembr. 2016, 1858, 2028–2034. [Google Scholar] [CrossRef] [PubMed]
- Lelle, M.; Frick, S.U.; Steinbrink, K.; Peneva, K. Novel cleavable cell-penetrating peptide–drug conjugates: Synthesis and characterization. J. Pept. Sci. 2014, 20, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Nadas, J.; Sun, D. Anthracyclines as effective anticancer drugs. Expert Opin. Drug Discov. 2006, 1, 549–568. [Google Scholar] [CrossRef]
- Frank, R.; Gennaro, R.; Schneider, K.; Przybylski, M.; Romeo, D. Amino acid sequences of two proline-rich bactenecins. Antimicrobial peptides of bovine neutrophils. J. Biol. Chem. 1990, 265, 18871–18874. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Davis, A.N.; Raucher, D. Targeting a c-Myc inhibitory polypeptide to specific intracellular compartments using cell penetrating peptides. J. Control. Release 2009, 135, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Massodi, I.; Moktan, S.; Rawat, A.; Bidwell, G.L.; Raucher, D. Inhibition of ovarian cancer cell proliferation by a cell cycle inhibitory peptide fused to a thermally responsive polypeptide carrier. Int. J. Cancer 2010, 126, 533–544. [Google Scholar] [CrossRef]
- Cowan, P.M.; McGavin, S. Structure of Poly-L-Proline. Nature 1955, 176, 501–503. [Google Scholar] [CrossRef]
- Garbuio, L.; Lewandowski, B.; Wilhelm, P.; Ziegler, L.; Yulikov, M.; Wennemers, H.; Jeschke, G. Shape Persistence of Polyproline II Helical Oligoprolines. Chem.—Eur. J. 2015, 21, 10747–10753. [Google Scholar] [CrossRef]
- Narwani, T.J.; Santuz, H.; Shinada, N.; Vattekatte, A.M.; Ghouzam, Y.; Srinivasan, N.; Gelly, J.C.; Brevern, A.G.d. Recent advances on polyproline II. Amino Acids 2017, 49, 705–713. [Google Scholar] [CrossRef]
- Farrera-Sinfreu, J.; Zaccaro, L.; Vidal, D.; Salvatella, X.; Giralt, E.; Pons, M.; Albericio, F.; Royo, M. A New Class of Foldamers Based on cis-γ-Amino-l-proline1,2. J. Am. Chem. Soc. 2004, 126, 6048–6057. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sinfreu, J.; Giralt, E.; Royo, M.; Albericio, F. Peptide Characterization and Application Protocols; Humana Press: Totowa, NJ, USA, 2007; pp. 241–267. [Google Scholar] [CrossRef]
- Geisler, I.; Chmielewski, J. Cationic Amphiphilic Polyproline Helices: Side-Chain Variations and Cell-Specific Internalization. Chem. Biol. Drug Des. 2009, 73, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Geisler, I.; Chmielewski, J.; Cheng, J.X. Cationic amphiphilic polyproline helix P11LRR targets intracellular mitochondria. J. Control. Release 2010, 142, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Geisler, I.M.; Chmielewski, J. Dimeric Cationic Amphiphilic Polyproline Helices for Mitochondrial Targeting. Pharm. Res. 2011, 28, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Kalafut, D.; Anderson, T.N.; Chmielewski, J. Mitochondrial targeting of a cationic amphiphilic polyproline helix. Bioorganic Med. Chem. Lett. 2012, 22, 561–563. [Google Scholar] [CrossRef]
- Gomez, J.A.; Gama, V.; Yoshida, T.; Sun, W.; Hayes, P.; Leskov, K.; Boothman, D.; Matsuyama, S. Bax-inhibiting peptides derived from Ku70 and cell-penetrating pentapeptides. Biochem. Soc. Trans. 2007, 35, 797–801. [Google Scholar] [CrossRef]
- Gomez, J.A.; Chen, J.; Ngo, J.; Hajkova, D.; Yeh, I.J.; Gama, V.; Miyagi, M.; Matsuyama, S. Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity. Pharmaceuticals 2010, 3, 3594–3613. [Google Scholar] [CrossRef]
- Tanaka, K.; Kobayashi, N.; Gutierrez, A.S.; Rivas-Carrillo, J.D.; Navarro-Alvarez, N.; Chen, Y.; Narushima, M.; Miki, A.; Okitsu, T.; Noguchi, H.; et al. Prolonged Survival of Mice with Acute Liver Failure with Transplantation of Monkey Hepatocytes Cultured with an Antiapoptotic Pentapeptide V5. Transplantation 2006, 81, 427–437. [Google Scholar] [CrossRef]
- Hui, H.; Dotta, F.; Mario, U.D.; Perfetti, R. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J. Cell. Physiol. 2004, 200, 177–200. [Google Scholar] [CrossRef]
- Rivas-Carrillo, J.D.; Soto-Gutierrez, A.; Navarro-Alvarez, N.; Noguchi, H.; Okitsu, T.; Chen, Y.; Yuasa, T.; Tanaka, K.; Narushima, M.; Miki, A.; et al. Cell-Permeable Pentapeptide V5 Inhibits Apoptosis and Enhances Insulin Secretion, Allowing Experimental Single-Donor Islet Transplantation in Mice. Diabetes 2007, 56, 1259–1267. [Google Scholar] [CrossRef]
- Gao, C.; Mao, S.; Ditzel, H.J.; Farnaes, L.; Wirsching, P.; Lerner, R.A.; Janda, K.D. A cell-penetrating peptide from a novel pVII–pIX phage-displayed random peptide library. Bioorganic Med. Chem. 2002, 10, 4057–4065. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Simon, M.J.; Hue, C.D.; Morrison, B.; Banta, S. An Unusual Cell Penetrating Peptide Identified Using a Plasmid Display-Based Functional Selection Platform. ACS Chem. Biol. 2011, 6, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, F.; Yasuda, T.; Umeda, S.; Asada, M.; Imamura, T.; Meineke, V.; Akashi, M. Fibroblast Growth Factor-12 (FGF12) Translocation into Intestinal Epithelial Cells Is Dependent on a Novel Cell-penetrating Peptide Domain Involvement of Internalization in the In Vivo Role of Exogenous FGF12. J. Biol. Chem. 2011, 286, 25823–25834. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Lau, Y.H.; De Andrade, P.; Wu, Y.; Spring, D.R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2014, 44, 91–102. [Google Scholar] [CrossRef]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol. 2015, 10, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox – Transforming natural peptides into peptide therapeutics. Bioorganic Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Li, X.; Zou, Y.; Hu, H.G. Different stapling-based peptide drug design: Mimicking α-helix as inhibitors of protein–protein interaction. Chin. Chem. Lett. 2018, 29, 1088–1092. [Google Scholar] [CrossRef]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides—A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Zhang, W.D.; Hu, H.G. Stapled Helical Peptides Bearing Different Anchoring Residues. Chem. Rev. 2020, 120, 10079–10144. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, M.; Fu, Y.; Xue, J.; Yuan, F.; Qu, T.; Rissanou, A.N.; Wang, Y.; Li, X.; Hu, H. Therapeutic stapled peptides: Efficacy and molecular targets. Pharmacol. Res. 2024, 203, 107137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Cheng, J.; Zhang, Z.; Kang, F.; Wu, X.; Chu, Q. High-Throughput Screening of Stapled Helical Peptides in Drug Discovery. J. Med. Chem. 2023, 66, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, A.; Josien, H.; Yuen, T.Y.; Duggal, R.; Spiegelberg, D.; Yan, L.; Juang, Y.C.A.; Ge, L.; Aronica, P.G.; Kaan, H.Y.K.; et al. Design-rules for stapled peptides with in vivo activity and their application to Mdm2/X antagonists. Nat. Commun. 2024, 15, 489. [Google Scholar] [CrossRef]
- Ma, B.; Liu, D.; Zheng, M.; Wang, Z.; Zhang, D.; Jian, Y.; Ma, J.; Fan, Y.; Chen, Y.; Gao, Y.; et al. Development of a Double-Stapled Peptide Stabilizing Both α-Helix and β-Sheet Structures for Degrading Transcription Factor AR-V7. JACS Au 2024, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef]
- Bird, G.H.; Bernal, F.; Pitter, K.; Walensky, L.D. Chapter 22 Synthesis and Biophysical Characterization of Stabilized α-Helices of BCL-2 Domains. Methods Enzymol. 2008, 446, 369–386. [Google Scholar] [CrossRef]
- LaBelle, J.L.; Katz, S.G.; Bird, G.H.; Gavathiotis, E.; Stewart, M.L.; Lawrence, C.; Fisher, J.K.; Godes, M.; Pitter, K.; Kung, A.L.; et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Investig. 2012, 122, 2018–2031. [Google Scholar] [CrossRef]
- Stewart, M.L.; Fire, E.; Keating, A.E.; Walensky, L.D. The MCL-1 BH3 Helix is an Exclusive MCL-1 inhibitor and Apoptosis Sensitizer. Nat. Chem. Biol. 2010, 6, 595–601. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, Q.; Bhattacharya, S.; Waheed, A.A.; Tong, X.; Hong, A.; Heck, S.; Curreli, F.; Goger, M.; Cowburn, D.; et al. A Cell-penetrating Helical Peptide as a Potential HIV-1 Inhibitor. J. Mol. Biol. 2008, 378, 565–580. [Google Scholar] [CrossRef]
- Ingelshed, K.; Melssen, M.M.; Kannan, P.; Chandramohan, A.; Partridge, A.W.; Jiang, L.; Wermeling, F.; Lane, D.P.; Nestor, M.; Spiegelberg, D. MDM2/MDMX inhibition by Sulanemadlin synergizes with anti-Programmed Death 1 immunotherapy in wild-type p53 tumors. iScience 2024, 27, 109862. [Google Scholar] [CrossRef] [PubMed]
- Guerlavais, V.; Sawyer, T.K.; Carvajal, L.; Chang, Y.S.; Graves, B.; Ren, J.G.; Sutton, D.; Olson, K.A.; Packman, K.; Darlak, K.; et al. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development. J. Med. Chem. 2023, 66, 9401–9417. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53Phase 1 Trial of ALRN-6924, a Dual MDMX/MDM2 Inhibitor. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Somaiah, N.; DuBois, S.; Dumbrava, E.E.I.; Shapiro, G.; Patel, M.; Goel, S.; Bauer, T.; Pinchasik, D.; Annis, A.; et al. 475P A phase IIa clinical trial combining ALRN-6924 and palbociclib for the treatment of patients with tumours harboring wild-type p53 and MDM2 amplification or MDM2/CDK4 co-amplification. Ann. Oncol. 2019, 30, v179–v180. [Google Scholar] [CrossRef]
- Edwards, A.L.; Wachter, F.; Lammert, M.; Huhn, A.J.; Luccarelli, J.; Bird, G.H.; Walensky, L.D. Cellular Uptake and Ultrastructural Localization Underlie the Pro-apoptotic Activity of a Hydrocarbon-stapled BIM BH3 Peptide. ACS Chem. Biol. 2015, 10, 2149–2157. [Google Scholar] [CrossRef]
- Okamoto, T.; Zobel, K.; Fedorova, A.; Quan, C.; Yang, H.; Fairbrother, W.J.; Huang, D.C.S.; Smith, B.J.; Deshayes, K.; Czabotar, P.E. Stabilizing the Pro-Apoptotic BimBH3 Helix (BimSAHB) Does Not Necessarily Enhance Affinity or Biological Activity. ACS Chem. Biol. 2013, 8, 297–302. [Google Scholar] [CrossRef]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845–852. [Google Scholar] [CrossRef]
- Perry, S.R.; Hill, T.A.; de Araujo, A.D.; Hoang, H.N.; Fairlie, D.P. Contiguous hydrophobic and charged surface patches in short helix-constrained peptides drive cell permeability. Org. Biomol. Chem. 2017, 16, 367–371. [Google Scholar] [CrossRef]
- Walensky, L.D.; Pitter, K.; Morash, J.; Oh, K.J.; Barbuto, S.; Fisher, J.; Smith, E.; Verdine, G.L.; Korsmeyer, S.J. A Stapled BID BH3 Helix Directly Binds and Activates BAX. Mol. Cell 2006, 24, 199–210. [Google Scholar] [CrossRef]
- Leshchiner, E.S.; Braun, C.R.; Bird, G.H.; Walensky, L.D. Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. USA 2013, 110, E986–E995. [Google Scholar] [CrossRef] [PubMed]
- Szlyk, B.; Braun, C.R.; Ljubicic, S.; Patton, E.; Bird, G.H.; Osundiji, M.A.; Matschinsky, F.M.; Walensky, L.D.; Danial, N.N. A phospho-BAD BH3 helix activates glucokinase by a mechanism distinct from that of allosteric activators. Nat. Struct. Mol. Biol. 2014, 21, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Barclay, L.A.; Wales, T.E.; Garner, T.P.; Wachter, F.; Lee, S.; Guerra, R.M.; Stewart, M.L.; Braun, C.R.; Bird, G.H.; Gavathiotis, E.; et al. Inhibition of Pro-Apoptotic BAX by a Noncanonical Interaction Mechanism. Mol. Cell 2015, 57, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted Disruption of the BCL9/β-Catenin Complex Inhibits Oncogenic Wnt Signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; Kerenyi, M.A.; Walensky, L.D.; Orkin, S.H. Targeted Disruption of the EZH2/EED Complex Inhibits EZH2-dependent Cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef]
- Leshchiner, E.S.; Parkhitko, A.; Bird, G.H.; Luccarelli, J.; Bellairs, J.A.; Escudero, S.; Opoku-Nsiah, K.; Godes, M.; Perrimon, N.; Walensky, L.D. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. USA 2015, 112, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, S.; Polak, K.; Fu, A.; Wiwczar, J.; Szlyk, B.; Chang, Y.; Alvarez-Perez, J.; Bird, G.; Walensky, L.; Garcia-Ocaña, A.; et al. Phospho-BAD BH3 Mimicry Protects β Cells and Restores Functional β Cell Mass in Diabetes. Cell Rep. 2015, 10, 497–504. [Google Scholar] [CrossRef]
- Bird, G.H.; Irimia, A.; Ofek, G.; Kwong, P.D.; Wilson, I.A.; Walensky, L.D. Stapled HIV-1 Peptides Recapitulate Antigenic Structures and Engage Broadly Neutralizing Antibodies. Nat. Struct. Mol. Biol. 2014, 21, 1058–1067. [Google Scholar] [CrossRef]
- Bird, G.H.; Boyapalle, S.; Wong, T.; Opoku-Nsiah, K.; Bedi, R.; Crannell, W.C.; Perry, A.F.; Nguyen, H.; Sampayo, V.; Devareddy, A.; et al. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J. Clin. Investig. 2014, 124, 2113–2124. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.T.K.; Hillyer, C.D.; Debnath, A.K. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection In Vitro. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Maas, M.N.; Hintzen, J.C.J.; Löffler, P.M.G.; Mecinović, J. Targeting SARS-CoV-2 spike protein by stapled hACE2 peptides. Chem. Commun. 2021, 57, 3283–3286. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.C.; Morris, C.; Mahindra, A.; Blair, C.M.; Tejeda, G.; Herbert, I.; Turnbull, M.L.; Lieber, G.; Willett, B.J.; Logan, N.; et al. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Pept. Sci. 2021, 113, e24217. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Human Coronaviruses: Counteracting the Damage by Storm. Viruses 2021, 13, 1457. [Google Scholar] [CrossRef] [PubMed]
- Wollack, J.W.; Zeliadt, N.A.; Mullen, D.G.; Amundson, G.; Geier, S.; Falkum, S.; Wattenberg, E.V.; Barany, G.; Distefano, M.D. Multifunctional Prenylated Peptides for Live Cell Analysis. J. Am. Chem. Soc. 2009, 131, 7293–7303. [Google Scholar] [CrossRef] [PubMed]
- Wollack, J.W.; Zeliadt, N.A.; Ochocki, J.D.; Mullen, D.G.; Barany, G.; Wattenberg, E.V.; Distefano, M.D. Investigation of the sequence and length dependence for cell-penetrating prenylated peptides. Bioorganic Med. Chem. Lett. 2010, 20, 161–163. [Google Scholar] [CrossRef]
- Ochocki, J.D.; Mullen, D.G.; Wattenberg, E.V.; Distefano, M.D. Evaluation of a cell penetrating prenylated peptide lacking an intrinsic fluorophore via in situ click reaction. Bioorganic Med. Chem. Lett. 2011, 21, 4998–5001. [Google Scholar] [CrossRef]
- Ochocki, J.D.; Igbavboa, U.; Wood, W.G.; Arriaga, E.A.; Wattenberg, E.V.; Distefano, M.D. Therapeutic Peptides, Methods and Protocols. Methods Mol. Biol. 2013, 1088, 213–223. [Google Scholar] [CrossRef]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 2002, 8, 1161–1165. [Google Scholar] [CrossRef]
- Carr, R.; Du, Y.; Quoyer, J.; Panettieri, R.A.; Janz, J.M.; Bouvier, M.; Kobilka, B.K.; Benovic, J.L. Development and Characterization of Pepducins as Gs-biased Allosteric Agonists. J. Biol. Chem. 2014, 289, 35668–35684. [Google Scholar] [CrossRef]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauß, N.; Choe, H.W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; DeVree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef]
- Dimond, P.; Carlson, K.; Bouvier, M.; Gerard, C.; Xu, L.; Covic, L.; Agarwal, A.; Ernst, O.P.; Janz, J.M.; Schwartz, T.W.; et al. G protein–coupled receptor modulation with pepducins: Moving closer to the clinic. Ann. N. Y. Acad. Sci. 2011, 1226, 34–49. [Google Scholar] [CrossRef]
- Langel, Ü. Cell Penetrating Peptides, Methods and Protocols, 3rd ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2022. [Google Scholar] [CrossRef]
- Janz, J.M.; Ren, Y.; Looby, R.; Kazmi, M.A.; Sachdev, P.; Grunbeck, A.; Haggis, L.; Chinnapen, D.; Lin, A.Y.; Seibert, C.; et al. Direct Interaction between an Allosteric Agonist Pepducin and the Chemokine Receptor CXCR4. J. Am. Chem. Soc. 2011, 133, 15878–15881. [Google Scholar] [CrossRef]
- Quoyer, J.; Janz, J.M.; Luo, J.; Ren, Y.; Armando, S.; Lukashova, V.; Benovic, J.L.; Carlson, K.E.; Hunt, S.W.; Bouvier, M. Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc. Natl. Acad. Sci. USA 2013, 110, E5088–E5097. [Google Scholar] [CrossRef]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 Is a Matrix Metalloprotease-1 Receptor that Promotes Invasion and Tumorigenesis of Breast Cancer Cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the Protease-Activated Receptor 1-4 Heterodimer in Platelet-Mediated Thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef]
- Huang, J.S.; Dong, L.; Kozasa, T.; Breton, G.C.L. Signaling through Gα13 Switch Region I Is Essential for Protease-activated Receptor 1-mediated Human Platelet Shape Change, Aggregation, and Secretion. J. Biol. Chem. 2007, 282, 10210–10222. [Google Scholar] [CrossRef]
- Trivedi, V.; Boire, A.; Tchernychev, B.; Kaneider, N.C.; Leger, A.J.; O’Callaghan, K.; Covic, L.; Kuliopulos, A. Platelet Matrix Metalloprotease-1 Mediates Thrombogenesis by Activating PAR1 at a Cryptic Ligand Site. Cell 2009, 137, 332–343. [Google Scholar] [CrossRef]
- Kimmelstiel, C.; Zhang, P.; Kapur, N.K.; Weintraub, A.; Krishnamurthy, B.; Castaneda, V.; Covic, L.; Kuliopulos, A. Bivalirudin Is a Dual Inhibitor of Thrombin and Collagen-Dependent Platelet Activation in Patients Undergoing Percutaneous Coronary Intervention. Circ. Cardiovasc. Interv. 2011, 4, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gruber, A.; Kasuda, S.; Kimmelstiel, C.; O’Callaghan, K.; Cox, D.H.; Bohm, A.; Baleja, J.D.; Covic, L.; Kuliopulos, A. Suppression of Arterial Thrombosis Without Affecting Hemostatic Parameters with a Cell-Penetrating PAR1 Pepducin. Circulation 2012, 126, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Kamath, L.; Meydani, A.; Foss, F.; Kuliopulos, A. Signaling from Protease-activated Receptor-1 Inhibits Migration and Invasion of Breast Cancer Cells1,2. Cancer Res. 2001, 61, 5933–5940. [Google Scholar] [PubMed]
- Yang, E.; Boire, A.; Agarwal, A.; Nguyen, N.; O’Callaghan, K.; Tu, P.; Kuliopulos, A.; Covic, L. Blockade of PAR1 Signaling with Cell-Penetrating Pepducins Inhibits Akt Survival Pathways in Breast Cancer Cells and Suppresses Tumor Survival and Metastasis. Cancer Res. 2009, 69, 6223–6231. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Tressel, S.L.; Kaimal, R.; Balla, M.; Lam, F.H.; Covic, L.; Kuliopulos, A. Identification of a Metalloprotease-Chemokine Signaling System in the Ovarian Cancer Microenvironment: Implications for Antiangiogenic Therapy. Cancer Res. 2010, 70, 5880–5890. [Google Scholar] [CrossRef]
- Foley, C.J.; Fanjul-Fernández, M.; Bohm, A.; Nguyen, N.; Agarwal, A.; Austin, K.; Koukos, G.; Covic, L.; López-Otín, C.; Kuliopulos, A. Matrix metalloprotease 1a deficiency suppresses tumor growth and angiogenesis. Oncogene 2014, 33, 2264–2272. [Google Scholar] [CrossRef]
- Kaneider, N.C.; Agarwal, A.; Leger, A.J.; Kuliopulos, A. Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat. Med. 2005, 11, 661–665. [Google Scholar] [CrossRef]
- Sevigny, L.M.; Zhang, P.; Bohm, A.; Lazarides, K.; Perides, G.; Covic, L.; Kuliopulos, A. Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc. Natl. Acad. Sci. USA 2011, 108, 8491–8496. [Google Scholar] [CrossRef]
- Tressel, S.L.; Kaneider, N.C.; Kasuda, S.; Foley, C.; Koukos, G.; Austin, K.; Agarwal, A.; Covic, L.; Opal, S.M.; Kuliopulos, A. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol. Med. 2011, 3, 370–384. [Google Scholar] [CrossRef]
- Tchernychev, B.; Ren, Y.; Sachdev, P.; Janz, J.M.; Haggis, L.; O’Shea, A.; McBride, E.; Looby, R.; Deng, Q.; McMurry, T.; et al. Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 22255–22259. [Google Scholar] [CrossRef]
- Kuliopulos, A.; Covic, L. Blocking receptors on the inside: Pepducin-based intervention of PAR signaling and thrombosis. Life Sci. 2003, 74, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Bliden, K.P.; Turner, S.E.; Tantry, U.S.; Gesheff, M.G.; Barr, T.P.; Covic, L.; Kuliopulos, A. Cell-Penetrating Pepducin Therapy Targeting PAR1 in Subjects with Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 36, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Michael, E.; Covic, L.; Kuliopulos, A. Cell Penetrating Peptides, Methods and Protocols. Methods Mol. Biol. 2021, 2383, 307–333. [Google Scholar] [CrossRef]
- Brouillette, R.L.; Besserer-Offroy, E.; Mona, C.E.; Chartier, M.; Lavenus, S.; Sousbie, M.; Belleville, K.; Longpré, J.M.; Marsault, E.; Grandbois, M.; et al. Cell-penetrating pepducins targeting the neurotensin receptor type 1 relieve pain. Pharmacol. Res. 2020, 155, 104750. [Google Scholar] [CrossRef] [PubMed]
- Kuliopulos, A.; Gurbel, P.A.; Rade, J.J.; Kimmelstiel, C.D.; Turner, S.E.; Bliden, K.P.; Fletcher, E.K.; Cox, D.H.; Covic, L.; Investigators, o.b.o.t.T.P. PAR1 (Protease-Activated Receptor 1) Pepducin Therapy Targeting Myocardial Necrosis in Coronary Artery Disease and Acute Coronary Syndrome Patients Undergoing Cardiac Catheterization. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2990–3003. [Google Scholar] [CrossRef]
- Fletcher, E.K.; Ngwenyama, N.; Nguyen, N.; Turner, S.E.; Covic, L.; Alcaide, P.; Kuliopulos, A. Suppression of Heart Failure with PAR1 Pepducin Technology in a Pressure Overload Model in Mice. Circ. Heart Fail. 2023, 16, e010621. [Google Scholar] [CrossRef]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Park, S.E.; Sajid, M.I.; Parang, K.; Tiwari, R.K. Cyclic Cell-Penetrating Peptides as Efficient Intracellular Drug Delivery Tools. Mol. Pharm. 2019, 16, 3727–3743. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Stueber, R.; Park, S.E.; Cho, Y.; Malik, N.u.A.; Tiwari, R.K. Applications of amphipathic and cationic cyclic cell-penetrating peptides: Significant therapeutic delivery tool. Peptides 2021, 141, 170542. [Google Scholar] [CrossRef]
- Oh, D.; Shirazi, A.N.; Northup, K.; Sullivan, B.; Tiwari, R.K.; Bisoffi, M.; Parang, K. Enhanced Cellular Uptake of Short Polyarginine Peptides through Fatty Acylation and Cyclization. Mol. Pharm. 2014, 11, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, E.H.M.; Mandal, D.; Mozaffari, S.; Zahran, M.A.H.; Osman, A.M.; Tiwari, R.K.; Parang, K. Comparative Molecular Transporter Properties of Cyclic Peptides Containing Tryptophan and Arginine Residues Formed through Disulfide Cyclization. Molecules 2020, 25, 2581. [Google Scholar] [CrossRef] [PubMed]
- Tapeinou, A.; Matsoukas, M.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Pept. Sci. 2015, 104, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Reichart, F.; Horn, M.; Neundorf, I. Cyclization of a cell-penetrating peptide via click-chemistry increases proteolytic resistance and improves drug delivery. J. Pept. Sci. 2016, 22, 421–426. [Google Scholar] [CrossRef]
- Tavassoli, A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. [Google Scholar] [CrossRef]
- Grieco, P.; Gitu, P.; Hruby, V. Preparation of ‘side-chain-to-side-chain’ cyclic peptides by Allyl and Alloc strategy: Potential for library synthesis. J. Pept. Res. 2001, 57, 250–256. [Google Scholar] [CrossRef]
- Foster, A.D.; Ingram, J.D.; Leitch, E.K.; Lennard, K.R.; Osher, E.L.; Tavassoli, A. Methods for the Creation of Cyclic Peptide Libraries for Use in Lead Discovery. J. Biomol. Screen. 2014, 20, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. [Google Scholar] [CrossRef]
- Shoushtari, S.K.; Zoghebi, K.; Sajid, M.I.; Tiwari, R.K.; Parang, K. Hybrid Cyclic-Linear Cell-Penetrating Peptides Containing Alternative Positively Charged and Hydrophobic Residues as Molecular Transporters. Mol. Pharm. 2021, 18, 3909–3919. [Google Scholar] [CrossRef]
- Li, X.; Craven, T.W.; Levine, P.M. Cyclic Peptide Screening Methods for Preclinical Drug Discovery. J. Med. Chem. 2022, 65, 11913–11926. [Google Scholar] [CrossRef]
- Madani, F.; Abdo, R.; Lindberg, S.; Hirose, H.; Futaki, S.; Langel, U.; Gräslund, A. Modeling the endosomal escape of cell-penetrating peptides using a transmembrane pH gradient. Biochim. Biophys. Acta (BBA)—Biomembr. 2013, 1828, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Li, X.; Sun, Y.; Cheng, F.; Wang, Q.; Xie, X.; Zhao, W.; Tian, X. Effect of Cationic Cyclopeptides on Transdermal and Transmembrane Delivery of Insulin. Mol. Pharm. 2013, 10, 951–957. [Google Scholar] [CrossRef]
- Shirazi, A.N.; El-Sayed, N.S.; Tiwari, R.K.; Tavakoli, K.; Parang, K. Cyclic Peptide Containing Hydrophobic and Positively Charged Residues as a Drug Delivery System for Curcumin. Curr. Drug Deliv. 2016, 13, 409–417. [Google Scholar] [CrossRef]
- Darwish, S.; Sadeghiani, N.; Fong, S.; Mozaffari, S.; Hamidi, P.; Withana, T.; Yang, S.; Tiwari, R.K.; Parang, K. Synthesis and antiproliferative activities of doxorubicin thiol conjugates and doxorubicin-SS-cyclic peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, S.; Bousoik, E.; Amirrad, F.; Lamboy, R.; Coyle, M.; Hall, R.; Alasmari, A.; Mahdipoor, P.; Parang, K.; Aliabadi, H.M. Amphiphilic Peptides for Efficient siRNA Delivery. Polymers 2019, 11, 703. [Google Scholar] [CrossRef]
- Schneider, A.F.L.; Wallabregue, A.L.D.; Franz, L.; Hackenberger, C.P.R. Targeted Subcellular Protein Delivery Using Cleavable Cyclic Cell-Penetrating Peptides. Bioconjug. Chem. 2019, 30, 400–404. [Google Scholar] [CrossRef]
- Jerath, G.; Goyal, R.; Trivedi, V.; Santhoshkumar, T.R.; Ramakrishnan, V. Conformationally constrained peptides for drug delivery. J. Pept. Sci. 2020, 26, e3244. [Google Scholar] [CrossRef]
- Uhl, P.; Grundmann, C.; Sauter, M.; Storck, P.; Tursch, A.; Özbek, S.; Leotta, K.; Roth, R.; Witzigmann, D.; Kulkarni, J.; et al. Coating of PLA-nanoparticles with cyclic, arginine-rich cell penetrating peptides enables oral delivery of liraglutide. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102132. [Google Scholar] [CrossRef]
- Mozaffari, S.; Salehi, D.; Mahdipoor, P.; Beuttler, R.; Tiwari, R.; Aliabadi, H.M.; Parang, K. Design and application of hybrid cyclic-linear peptide-doxorubicin conjugates as a strategy to overcome doxorubicin resistance and toxicity. Eur. J. Med. Chem. 2021, 226, 113836. [Google Scholar] [CrossRef]
- Park, S.E.; El-Sayed, N.S.; Shamloo, K.; Lohan, S.; Kumar, S.; Sajid, M.I.; Tiwari, R.K. Targeted Delivery of Cabazitaxel Using Cyclic Cell-Penetrating Peptide and Biomarkers of Extracellular Matrix for Prostate and Breast Cancer Therapy. Bioconjug. Chem. 2021, 32, 1898–1914. [Google Scholar] [CrossRef] [PubMed]
- Zoghebi, K.; Aliabadi, H.M.; Tiwari, R.K.; Parang, K. [(WR)8WKβA]-Doxorubicin Conjugate: A Delivery System to Overcome Multi-Drug Resistance against Doxorubicin. Cells 2022, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Melemenidis, S.; Jefferson, A.; Ruparelia, N.; Akhtar, A.M.; Xie, J.; Allen, D.; Hamilton, A.; Larkin, J.R.; Perez-Balderas, F.; Smart, S.C.; et al. Molecular Magnetic Resonance Imaging of Angiogenesis In Vivo using Polyvalent Cyclic RGD-Iron Oxide Microparticle Conjugates. Theranostics 2015, 5, 515–529. [Google Scholar] [CrossRef]
- Yan, B.; Qiu, F.; Ren, L.; Dai, H.; Fang, W.; Zhu, H.; Wang, F. 99mTc-3P-RGD2 molecular imaging targeting integrin αvβ3 in head and neck squamous cancer xenograft. J. Radioanal. Nucl. Chem. 2015, 304, 1171–1177. [Google Scholar] [CrossRef]
- Barth, N.D.; Subiros-Funosas, R.; Mendive-Tapia, L.; Duffin, R.; Shields, M.A.; Cartwright, J.A.; Henriques, S.T.; Sot, J.; Goñi, F.M.; Lavilla, R.; et al. A fluorogenic cyclic peptide for imaging and quantification of drug-induced apoptosis. Nat. Commun. 2020, 11, 4027. [Google Scholar] [CrossRef] [PubMed]
- Mendive-Tapia, L.; Wang, J.; Vendrell, M. Fluorescent cyclic peptides for cell imaging. Pept. Sci. 2021, 113. [Google Scholar] [CrossRef]
- Buck, J.; Perez-Balderas, F.; Zarghami, N.; Johanssen, V.; Khrapitchev, A.A.; Larkin, J.R.; Sibson, N.R. Imaging angiogenesis in an intracerebrally induced model of brain macrometastasis using αvβ3-targeted iron oxide microparticles. NMR Biomed. 2023, 36, e4948. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.; Reichart, F.; Natividad-Tietz, S.; Diaz, D.; Neundorf, I. Tuning the properties of a novel short cell-penetrating peptide by intramolecular cyclization with a triazole bridge. Chem. Commun. 2015, 52, 2261–2264. [Google Scholar] [CrossRef]
- Fajloun, Z.; Kharrat, R.; Chen, L.; Lecomte, C.; Luccio, E.D.; Bichet, D.; Ayeb, M.E.; Rochat, H.; Allen, P.; Pessah, I.; et al. Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca2+ release channel/ryanodine receptors. FEBS Lett. 2000, 469, 179–185. [Google Scholar] [CrossRef]
- Nascimento, F.D.; Hayashi, M.A.; Kerkis, A.; Oliveira, V.; Oliveira, E.B.; Rádis-Baptista, G.; Nader, H.B.; Yamane, T.; Tersariol, I.L.d.S.; Kerkis, I. Crotamine Mediates Gene Delivery into Cells through the Binding to Heparan Sulfate Proteoglycans. J. Biol. Chem. 2007, 282, 21349–21360. [Google Scholar] [CrossRef]
- Lian, W.; Upadhyaya, P.; Rhodes, C.A.; Liu, Y.; Pei, D. Screening Bicyclic Peptide Libraries for Protein–Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-α Antagonist. J. Am. Chem. Soc. 2013, 135, 11990–11995. [Google Scholar] [CrossRef] [PubMed]
- Suojanen, J.; Salo, T.; Koivunen, E.; Sorsa, T.; Pirilä, E. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 2009, 8, 2362–2370. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Sakamoto, T. MT1-MMP as a Key Regulator of Metastasis. Cells 2023, 12, 2187. [Google Scholar] [CrossRef] [PubMed]
- Valkema, R.; Pauwels, M.S.A.; Kvols, M.L.K.; Kwekkeboom, M.D.J.; Jamar, M.F.; de Jong, M.M.; Barone, P.R.; Walrand, M.S.; Kooij, P.P.P.; Bakker, M.W.H.; et al. Long-Term Follow-Up of Renal Function After Peptide Receptor Radiation Therapy with 90Y-DOTA0,Tyr3-Octreotide and 177Lu-DOTA0, Tyr3-Octreotate. J. Nucl. Med. 2005, 46, 83S–91S. [Google Scholar]
- Becx, M.N.; Minczeles, N.S.; Brabander, T.; de Herder, W.W.; Nonnekens, J.; Hofland, J. A Clinical Guide to Peptide Receptor Radionuclide Therapy with 177Lu-DOTATATE in Neuroendocrine Tumor Patients. Cancers 2022, 14, 5792. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Teunissen, J.J.; Bakker, W.H.; Kooij, P.P.; de Herder, W.W.; Feelders, R.A.; van Eijck, C.H.; Esser, J.P.; Kam, B.L.; Krenning, E.P. Radiolabeled Somatostatin Analog [177Lu-DOTA0,Tyr3]Octreotate in Patients with Endocrine Gastroenteropancreatic Tumors. J. Clin. Oncol. 2004, 23, 2754–2762. [Google Scholar] [CrossRef]
- Delker, A.; Ilhan, H.; Zach, C.; Brosch, J.; Gildehaus, F.J.; Lehner, S.; Bartenstein, P.; Böning, G. The Influence of Early Measurements Onto the Estimated Kidney Dose in [177Lu][DOTA0,Tyr3]Octreotate Peptide Receptor Radiotherapy of Neuroendocrine Tumors. Mol. Imaging Biol. 2015, 17, 726–734. [Google Scholar] [CrossRef]
- Brabander, T.; van der Zwan, W.A.; Teunissen, J.J.M.; Kam, B.L.R.; de Herder, W.W.; Feelders, R.A.; Krenning, E.P.; Kwekkeboom, D.J. Pitfalls in the response evaluation after peptide receptor radionuclide therapy with [177Lu-DOTA0,Tyr3]octreotate. Endocr.-Relat. Cancer 2017, 24, 243–251. [Google Scholar] [CrossRef]
- Brabander, T.; van der Zwan, W.A.; Teunissen, J.J.; Kam, B.L.; Feelders, R.A.; de Herder, W.W.; van Eijck, C.H.; Franssen, G.J.; Krenning, E.P.; Kwekkeboom, D.J. Long-Term Efficacy, Survival, and Safety of [177Lu-DOTA0,Tyr3]octreotate in Patients with Gastroenteropancreatic and Bronchial Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Liu, S.V.; Subramaniam, D.S.; Torres, T.; Loda, M.; Esposito, G.; Giaccone, G. Phase I study of the 177Lu-DOTA0,Tyr3-Octreotate (lutathera) in combination with nivolumab in patients with neuroendocrine tumors of the lung. J. ImmunoTherapy Cancer 2020, 8, e000980. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwan, W.A.; Brabander, T.; Kam, B.L.R.; Teunissen, J.J.M.; Feelders, R.A.; Hofland, J.; Krenning, E.P.; De Herder, W.W. Salvage peptide receptor radionuclide therapy with [177Lu-DOTA,Tyr3]octreotate in patients with bronchial and gastroenteropancreatic neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 704–717. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takano, S.; Ito, K.; Sugiura, M.; Ogawa, M.; Takeda, Y.; Okubo, N.; Suzuki, A.; Tokuhisa, M.; Kaneta, T.; et al. Safety and efficacy of peptide receptor radionuclide therapy with 177Lu-DOTA0-Tyr3-octreotate in combination with amino acid solution infusion in Japanese patients with somatostatin receptor-positive, progressive neuroendocrine tumors. Ann. Nucl. Med. 2021, 35, 1332–1341. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Kalantarhormozi, M.; Hassanzadeh, S.; Rekabpour, S.J.; Ravanbod, M.R.; Jafari, E.; Amini, A.; Dadgar, H.; Mahmoudpour, M.; Nabipour, I.; Jokar, N.; et al. Peptide Receptor Radionuclide Therapy Using 177 Lu-DOTATATE in Advanced Neuroendocrine Tumors (NETs) in a Limited-Resource Environment. World J. Nucl. Med. 2022, 21, 215–221. [Google Scholar] [CrossRef]
- Sagan, S.; Burlina, F.; Alves, I.D.; Bechara, C.; Dupont, E.; Joliot, A. Homeoproteins and Homeoprotein-derived Peptides: Going in and Out. Curr. Pharm. Des. 2013, 19, 2851–2862. [Google Scholar] [CrossRef]
- Gehring, W.J.; Qian, Y.Q.; Billeter, M.; Furukubo-Tokunaga, K.; Schier, A.F.; Resendez-Perez, D.; Affolter, M.; Otting, G.; Wüthrich, K. Homeodomain-DNA recognition. Cell 1994, 78, 211–223. [Google Scholar] [CrossRef]
- Furukubo-Tokunaga, K.; Flister, S.; Gehring, W.J. Functional specificity of the Antennapedia homeodomain. Proc. Natl. Acad. Sci. USA 1993, 90, 6360–6364. [Google Scholar] [CrossRef]
- Maniti, O.; Alves, I.; Trugnan, G.; Ayala-Sanmartin, J. Distinct Behaviour of the Homeodomain Derived Cell Penetrating Peptide Penetratin in Interaction with Different Phospholipids. PLoS ONE 2010, 5, e15819. [Google Scholar] [CrossRef]
- Chatelin, L.; Volovitch, M.; Joliot, A.H.; Perez, F.; Prochiantz, A. Transcription factor Hoxa-5 is taken up by cells in culture and conveyed to their nuclei. Mech. Dev. 1996, 55, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Joliot, A.; Maizel, A.; Rosenberg, D.; Trembleau, A.; Dupas, S.; Volovitch, M.; Prochiantz, A. Identification of a signal sequence necessary for the unconventional secretion of Engrailed homeoprotein. Curr. Biol. 1998, 8, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Kilk, K.; Magzoub, M.; Pooga, M.; Eriksson, L.E.G.; Langel, U.; Gräslund, A. Cellular Internalization of a Cargo Complex with a Novel Peptide Derived from the Third Helix of the Islet-1 Homeodomain. Comparison with the Penetratin Peptide. Bioconjug. Chem. 2001, 12, 911–916. [Google Scholar] [CrossRef]
- Noguchi, H.; Kaneto, H.; Weir, G.C.; Bonner-Weir, S. PDX-1 Protein Containing Its Own Antennapedia-Like Protein Transduction Domain Can Transduce Pancreatic Duct and Islet Cells. Diabetes 2003, 52, 1732–1737. [Google Scholar] [CrossRef]
- Beltran, A.S.; Graves, L.M.; Blancafort, P. Novel role of Engrailed 1 as a prosurvival transcription factor in basal-like breast cancer and engineering of interference peptides block its oncogenic function. Oncogene 2014, 33, 4767–4777. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- De Coupade, C.; Fittipaldi, A.; Chagnas, V.; Michel, M.; Carlier, S.; Tasciotti, E.; Darmon, A.; Ravel, D.; Kearsey, J.; Giacca, M.; et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem. J. 2005, 390, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Losic, F.; Quinonero, J.; Dubois, V.; Alluis, B.; Dechambre, M.; Michel, M.; Cailler, F.; Fernandez, A.M.; Trouet, A.; Kearsey, J. Improved Therapeutic Efficacy of Doxorubicin through Conjugation with a Novel Peptide Drug Delivery Technology (Vectocell). J. Med. Chem. 2006, 49, 6908–6916. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arterioscler. Off. J. Am. Heart Assoc. Inc. 2018, 9, 21–32. [Google Scholar] [CrossRef]
- Friedrich, U.; Blom, A.M.; Dahlbäck, B.; Villoutreix, B.O. Structural and Energetic Characteristics of the Heparin-binding Site in Antithrombotic Protein C. J. Biol. Chem. 2001, 276, 24122–24128. [Google Scholar] [CrossRef]
- Chen, C.J.; Tsai, K.C.; Kuo, P.H.; Chang, P.L.; Wang, W.C.; Chuang, Y.J.; Chang, M.D.T. A Heparan Sulfate-Binding Cell Penetrating Peptide for Tumor Targeting and Migration Inhibition. BioMed Res. Int. 2015, 2015, 237969. [Google Scholar] [CrossRef] [PubMed]
- Bahadoran, A.; Ebrahimi, M.; Yeap, S.K.; Safi, N.; Moeini, H.; Hair-Bejo, M.; Hussein, M.Z.; Omar, A.R. Induction of a robust immune response against avian influenza virus following transdermal inoculation with H5-DNA vaccine formulated in modified dendrimer-based delivery system in mouse model. Int. J. Nanomed. 2017, 12, 8573–8585. [Google Scholar] [CrossRef]
- Zou, L.; Peng, Q.; Wang, P.; Zhou, B. Progress in Research and Application of HIV-1 TAT-Derived Cell-Penetrating Peptide. J. Membr. Biol. 2017, 250, 115–122. [Google Scholar] [CrossRef]
- Backendorf, C.; Visser, A.E.; De Boer, A.; Zimmerman, R.; Visser, M.; Voskamp, P.; Zhang, Y.H.; Noteborn, M. Apoptin: Therapeutic Potential of an Early Sensor of Carcinogenic Transformation. Pharmacol. Toxicol. 2008, 48, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Panigrahi, S.; Rashedi, I.; Mandal, S.; Stetefeld, J.; Essmann, F.; Schulze-Osthoff, K. Apoptin, a tumor-selective killer. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zheng, W.; Li, A.; Mu, Y.; Shi, M.; Li, T.; Zou, H.; Shao, H.; Qin, A.; Ye, J. A novel CAV derived cell-penetrating peptide efficiently delivers exogenous molecules through caveolae-mediated endocytosis. Vet. Res. 2018, 49, 16. [Google Scholar] [CrossRef]
- Hu, G.; Miao, Y.; Luo, X.; Chu, W.; Fu, Y. Identification of a novel cell-penetrating peptide derived from the capsid protein of chicken anemia virus and its application in gene delivery. Appl. Microbiol. Biotechnol. 2020, 104, 10503–10513. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef]
- Freire, J.M.; Veiga, A.S.; Rego de Figueiredo, I.; de la Torre, B.G.; Santos, N.C.; Andreu, D.; Da Poian, A.T.; Castanho, M.A. Nucleic acid delivery by cell penetrating peptides derived from dengue virus capsid protein: Design and mechanism of action. FEBS J. 2014, 281, 191–215. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tamiya, S.; Shibuya, M.; Nakase, I.; Yoshioka, Y. Peptides with the multibasic cleavage site of the hemagglutinin from highly pathogenic influenza viruses act as cell-penetrating via binding to heparan sulfate and neuropilins. Biochem. Biophys. Res. Commun. 2019, 512, 453–459. [Google Scholar] [CrossRef]
- Sitinjak, M.C.; Chen, J.K.; Wang, C.Y. Characterization of novel cell-penetrating peptides derived from the capsid protein of beak and feather disease virus. Virus Res. 2023, 330, 199109. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, S.; Behzadipour, Y.; Haddad, M. Decoding the proteome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for cell-penetrating peptides involved in pathogenesis or applicable as drug delivery vectors. Infect. Genet. Evol. 2020, 85, 104474. [Google Scholar] [CrossRef] [PubMed]
- Montrose, K.; Yang, Y.; Krissansen, G.W. X-pep, a novel cell-penetrating peptide motif derived from the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 453, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.M.; Eroglu, E.; Bawage, S.S.; Vig, K.; Miller, M.E.; Pillai, S.; Dennis, V.A.; Singh, S.R. Enhanced intracellular translocation and biodistribution of gold nanoparticles functionalized with a cell-penetrating peptide (VG-21) from vesicular stomatitis virus. Biomaterials 2014, 35, 9484–9494. [Google Scholar] [CrossRef]
- Yu, W.; Zhan, Y.; Xue, B.; Dong, Y.; Wang, Y.; Jiang, P.; Wang, A.; Sun, Y.; Yang, Y. Highly efficient cellular uptake of a cell-penetrating peptide (CPP) derived from the capsid protein of porcine circovirus type 2. J. Biol. Chem. 2018, 293, 15221–15232. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated Arginine-Rich Peptides: A New Class of Transfection Systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Langedijk, J.P. Translocation Activity of C-terminal Domain of Pestivirus Erns and Ribotoxin L3 Loop. J. Biol. Chem. 2002, 277, 5308–5314. [Google Scholar] [CrossRef]
- Rádis-Baptista, G. Cell-Penetrating Peptides Derived from Animal Venoms and Toxins. Toxins 2021, 13, 147. [Google Scholar] [CrossRef]
- Aroui, S.; Ram, N.; Appaix, F.; Ronjat, M.; Kenani, A.; Pirollet, F.; Waard, M.D. Maurocalcine as a Non Toxic Drug Carrier Overcomes Doxorubicin Resistance in the Cancer Cell Line MDA-MB 231. Pharm. Res. 2009, 26, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Aroui, S.; Brahim, S.; Waard, M.D.; Bréard, J.; Kenani, A. Efficient induction of apoptosis by doxorubicin coupled to cell-penetrating peptides compared to unconjugated doxorubicin in the human breast cancer cell line MDA-MB 231. Cancer Lett. 2009, 285, 28–38. [Google Scholar] [CrossRef]
- Ram, N.; Weiss, N.; Texier-Nogues, I.; Aroui, S.; Andreotti, N.; Pirollet, F.; Ronjat, M.; Sabatier, J.M.; Darbon, H.; Jacquemond, V.; et al. Design of a Disulfide-less, Pharmacologically Inert, and Chemically Competent Analog of Maurocalcine for the Efficient Transport of Impermeant Compounds into Cells. J. Biol. Chem. 2008, 283, 27048–27056. [Google Scholar] [CrossRef] [PubMed]
- Tisseyre, C.; Bahembera, E.; Dardevet, L.; Sabatier, J.M.; Ronjat, M.; Waard, M.D. Cell Penetration Properties of a Highly Efficient Mini Maurocalcine Peptide. Pharmaceuticals 2013, 6, 320–339. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, F.Z.; Gurrola, G.B.; Arévalo, C.; Sreekumar, R.; Walker, J.W.; Valdivia, H.H.; Possani, L.D. Primary structure and synthesis of Imperatoxin A (IpTxa), a peptide activator of Ca2+ release channels/ryanodine receptors. FEBS Lett. 1997, 405, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Seo, I.R.; Choi, M.R.; Park, C.S.; Kim, D.H. Effects of Recombinant Imperatoxin A (lpTxa) Mutants on the Rabbit Ryanodine Receptor. Mol. Cells 2006, 22, 328–335. [Google Scholar] [CrossRef]
- Chen, L.; Estève, E.; Sabatier, J.M.; Ronjat, M.; Waard, M.D.; Allen, P.D.; Pessah, I.N. Maurocalcine and Peptide A Stabilize Distinct Subconductance States of Ryanodine Receptor Type 1, Revealing a Proportional Gating Mechanism. J. Biol. Chem. 2003, 278, 16095–16106. [Google Scholar] [CrossRef]
- Green, D.; Pace, S.; Curtis, S.M.; Sakowska, M.; Lamb, G.D.; Dulhunty, A.F.; Casarotto, M.G. The three-dimensional structural surface of two beta-sheet scorpion toxins mimics that of an alpha-helical dihydropyridine receptor segment. Biochem. J. 2003, 370, 517–527. [Google Scholar] [CrossRef]
- El-Hayek, R.; Lokuta, A.J.; Arévalo, C.; Valdivia, H.H. Peptide Probe of Ryanodine Receptor Function Imperatoxin A, A Peptide from the Venom of the Scorpion Pandinus Imperator, Selectively Activates Skeletal-Type Ryanodine Receptor Isoforms. J. Biol. Chem. 1995, 270, 28696–28704. [Google Scholar] [CrossRef]
- Gurrola, G.B.; Capes, E.M.; Zamudio, F.Z.; Possani, L.D.; Valdivia, H.H. Imperatoxin A, a Cell-Penetrating Peptide from Scorpion Venom, as a Probe of Ca2+-Release Channels/Ryanodine Receptors. Pharmaceuticals 2010, 3, 1093–1107. [Google Scholar] [CrossRef]
- Rady, I.; Siddiqui, I.A.; Rady, M.; Mukhtar, H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017, 402, 16–31. [Google Scholar] [CrossRef]
- Rajabnejad, S.H.; Mokhtarzadeh, A.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Razavi, B.M. Targeted delivery of melittin to cancer cells by AS1411 anti-nucleolin aptamer. Drug Dev. Ind. Pharm. 2018, 44, 982–987. [Google Scholar] [CrossRef]
- Ceremuga, M.; Stela, M.; Janik, E.; Gorniak, L.; Synowiec, E.; Sliwinski, T.; Sitarek, P.; Saluk-Bijak, J.; Bijak, M. Melittin—A Natural Peptide from Bee Venom Which Induces Apoptosis in Human Leukaemia Cells. Biomolecules 2020, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.; Sorolla, A.; Wang, E.; Golden, E.; Woodward, E.; Davern, K.; Ho, D.; Johnstone, E.; Pfleger, K.; Redfern, A.; et al. Honeybee venom and melittin suppress growth factor receptor activation in HER2-enriched and triple-negative breast cancer. Npj Precis. Oncol. 2020, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Cardarelli, F.; Luca, M.D.; Boccardi, C.; Nifosì, R.; Bardi, G.; Bari, L.D.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. ImmunoTherapy Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.A.; Quinn, J.M.; Roach, S.T.; Palisoul, M.; Nguyen, M.; Noia, H.; Guo, L.; Fazal, J.; Mutch, D.G.; Wickline, S.A.; et al. p5RHH nanoparticle-mediated delivery of AXL siRNA inhibits metastasis of ovarian and uterine cancer cells in mouse xenografts. Sci. Rep. 2019, 9, 4762. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, R.; Jin, J.M.; Zhang, L.J.; Zhang, H.; Chen, H.Z.; Chen, L.L.; Luan, X. Advances in the Study of Structural Modification and Biological Activities of Anoplin. Front. Chem. 2020, 8, 519. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Macyszyn, J.; Miszkiewicz, J.; Grzela, R.; Trylska, J. Stapled Anoplin as an Antibacterial Agent. Front. Microbiol. 2021, 12, 772038. [Google Scholar] [CrossRef]
- Stergiou, V.; Krikorian, D.; Koukkou, A.; Sakarellos-Daitsiotis, M.; Panou-Pomonis, E. Novel anoplin-based (lipo)-peptide models show potent antimicrobial activity. J. Pept. Sci. 2021, 27, e3303. [Google Scholar] [CrossRef]
- Munk, J.K.; Uggerhøj, L.E.; Poulsen, T.J.; Frimodt-Møller, N.; Wimmer, R.; Nyberg, N.T.; Hansen, P.R. Synthetic analogs of anoplin show improved antimicrobial activities. J. Pept. Sci. 2013, 19, 669–675. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.J.; Liu, Y.Y.; Li, H.; Guo, L.X.; Liu, Z.H.; Shi, X.L.; Hu, M. In Vitro Potential of Lycosin-I as an Alternative Antimicrobial Drug for Treatment of Multidrug-Resistant Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 2014, 58, 6999–7002. [Google Scholar] [CrossRef]
- Chionis, K.; Krikorian, D.; Koukkou, A.; Sakarellos-Daitsiotis, M.; Panou-Pomonis, E. Synthesis and biological activity of lipophilic analogs of the cationic antimicrobial active peptide anoplin. J. Pept. Sci. 2016, 22, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, N.; Zhong, C.; Zhu, Y.; Gou, S.; Chang, L.; Bao, H.; Liu, H.; Zhang, Y.; Ni, J. Effect of N-methylated and fatty acid conjugation on analogs of antimicrobial peptide Anoplin. Eur. J. Pharm. Sci. 2020, 152, 105453. [Google Scholar] [CrossRef] [PubMed]
- Gou, S.; Li, B.; Ouyang, X.; Ba, Z.; Zhong, C.; Zhang, T.; Chang, L.; Zhu, Y.; Zhang, J.; Zhu, N.; et al. Novel Broad-Spectrum Antimicrobial Peptide Derived from Anoplin and Its Activity on Bacterial Pneumonia in Mice. J. Med. Chem. 2021, 64, 11247–11266. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Hällbrink, M.; Zorko, M.; Langel, Ü. From galanin and mastoparan to galparan and transportan. Curr. Top. Pept. Protein Res. 1997, 2, 83–113. [Google Scholar]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hällbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta (BBA)—Biomembr. 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Andaloussi, S.E.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Andaloussi, S.E.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef]
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Vasconcelos, L.D.F.; Veiman, K.L.; Uusna, J.; Margus, H.; Garcia-Sosa, A.T.; Pooga, M.; Langel, U. Optimization of in vivo DNA delivery with NickFect peptide vectors. J. Control. Release 2016, 241, 135–143. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Penetrating Peptides and Transportan. Pharmaceutics 2021, 13, 987. [Google Scholar] [CrossRef]
- Jones, S.; Martel, C.; Belzacq-Casagrande, A.S.; Brenner, C.; Howl, J. Mitoparan and target-selective chimeric analogues: Membrane translocation and intracellular redistribution induces mitochondrial apoptosis. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2008, 1783, 849–863. [Google Scholar] [CrossRef]
- Liu, Z.; Deng, M.; Xiang, J.; Ma, H.; Hu, W.; Zhao, Y.; Li, D.W.C.; Liang, S. A Novel Spider Peptide Toxin Suppresses Tumor Growth Through Dual Signaling Pathways. Curr. Mol. Med. 2012, 12, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Luo, W.; Wei, L.; Chen, B.; Li, W.; Xiao, L.; Manzhos, S.; Liu, Z.; Liang, S. Quantifying the Distribution of the Stoichiometric Composition of Anticancer Peptide Lycosin-I on the Lipid Membrane with Single Molecule Spectroscopy. J. Phys. Chem. B 2016, 120, 3081–3088. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Ding, X.; Meng, S.; Liu, C.; Wang, H.; Xia, L.; Liu, Z.; Liang, S. Antimicrobial Potential of Lycosin-I, a Cationic and Amphiphilic Peptide from the Venom of the Spider Lycosa singorensis. Curr. Mol. Med. 2013, 13, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xie, Y.; Ye, S.; He, K.; Yi, L.; Cui, R. Spider peptide toxin lycosin-I induces apoptosis and inhibits migration of prostate cancer cells. Exp. Biol. Med. 2018, 243, 725–735. [Google Scholar] [CrossRef]
- Zhang, X.; Brossas, J.Y.; Parizot, C.; Zini, J.M.; Rebollo, A. Identification and characterization of novel enhanced cell penetrating peptides for anti-cancer cargo delivery. Oncotarget 2017, 9, 5944–5957. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Yang, H.; Xiao, H.; Farooq, A.; Liu, Z.; Hu, M.; Shi, X. The Spider Venom Peptide Lycosin-II Has Potent Antimicrobial Activity against Clinically Isolated Bacteria. Toxins 2016, 8, 119. [Google Scholar] [CrossRef]
- Lazarovici, P.; Primor, N.; Loew, L.M. Purification and pore-forming activity of two hydrophobic polypeptides from the secretion of the Red Sea Moses sole (Pardachirus marmoratus). J. Biol. Chem. 1986, 261, 16704–16713. [Google Scholar] [CrossRef]
- Thompson, S.A.; Tachibana, K.; Nakanishi, K.; Kubota, I. Melittin-Like Peptides from the Shark-Repelling Defense Secretion of the Sole Pardachirus pavoninus. Science 1986, 233, 341–343. [Google Scholar] [CrossRef]
- Shai, Y.; Fox, J.; Caratsch, C.; Shih, Y.L.; Edwards, C.; Lazarovici, P. Sequencing and synthesis of pardaxin, a polypeptide from the Red Sea Moses sole with ionophore activity. FEBS Lett. 1988, 242, 161–166. [Google Scholar] [CrossRef]
- Shai, Y. Pardaxin: Channel formation by a shark repellant peptide from fish. Toxicology 1994, 87, 109–129. [Google Scholar] [CrossRef]
- Hsu, J.C.; Lin, L.C.; Tzen, J.T.; Chen, J.Y. Pardaxin-induced apoptosis enhances antitumor activity in HeLa cells. Peptides 2011, 32, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Uen, W.C.; Choong, C.Y.; Tai, C.J.; Tai, C.J. Pardaxin Promoted Differentiation and Maturation of Leukemic Cells via Regulating TLR2/MyD88 Signal against Cell Proliferation. Evid.-Based Complement. Altern. Med. 2019, 2019, 7035087. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Shih, P.C.; Feng, C.W.; Wu, C.C.; Tsui, K.H.; Lin, Y.H.; Kuo, H.M.; Wen, Z.H. Pardaxin Activates Excessive Mitophagy and Mitochondria-Mediated Apoptosis in Human Ovarian Cancer by Inducing Reactive Oxygen Species. Antioxidants 2021, 10, 1883. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Cui, Z.; Li, Y.H.; Hsu, W.H.; Lee, B.H. In Vitro and in Vivo Anticancer Activity of Pardaxin against Proliferation and Growth of Oral Squamous Cell Carcinoma. Mar. Drugs 2016, 14, 2. [Google Scholar] [CrossRef]
- Böhmová, E.; Machová, D.; Pechar, M.; Pola, R.; Venclíková, K.; JanouškovÁ, O.; Etrych, T. Cell-Penetrating Peptides: A Useful Tool for the Delivery of Various Cargoes Into Cells. Physiol. Res. 2018, 67, S267–S279. [Google Scholar] [CrossRef]
- Behzadipour, Y.; Hemmati, S. Covalent conjugation and non-covalent complexation strategies for intracellular delivery of proteins using cell-penetrating peptides. Biomed. Pharmacother. 2024, 176, 116910. [Google Scholar] [CrossRef]
- Shoari, A.; Tooyserkani, R.; Tahmasebi, M.; Löwik, D.W.P.M. Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade. Pharmaceutics 2021, 13, 1391. [Google Scholar] [CrossRef]
- Gayraud, F.; Klußmann, M.; Neundorf, I. Recent Advances and Trends in Chemical CPP–Drug Conjugation Techniques. Molecules 2021, 26, 1591. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides A Reevaluation of the Mechanism of Cellular Uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular Uptake, Intracellular Trafficking, and Cytotoxicity of Nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef]
- Duchardt, F.; Ruttekolk, I.R.; Verdurmen, W.P.; Lortat-Jacob, H.; Bürck, J.; Hufnagel, H.; Fischer, R.; Van den Heuvel, M.; Löwik, D.W.; Vuister, G.W.; et al. A Cell-penetrating Peptide Derived from Human Lactoferrin with Conformation-dependent Uptake Efficiency. J. Biol. Chem. 2009, 284, 36099–36108. [Google Scholar] [CrossRef] [PubMed]
- Oehlke, J.; Krause, E.; Wiesner, B.; Beyermann, M.; Bienert, M. Extensive cellular uptake into endothelial cells of an amphipathic β-sheet forming peptide. FEBS Lett. 1997, 415, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ruczynski, J.; Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Mucha, P.; Siedlecka-Kroplewska, K.; Rekowski, P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem. Cytobiol. 2014, 52, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Yoneyama, S.; Murase, O.; Miyajima, K. Transbilayer Transport of Ions and Lipids Coupled with Mastoparan X Translocation. Biochemistry 1996, 35, 8450–8456. [Google Scholar] [CrossRef]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of Antimicrobial Dermaseptin and Its Fluorescently Labeled Analogues with Phospholipid Membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Many-Body Effect of Antimicrobial Peptides: On the Correlation Between Lipid’s Spontaneous Curvature and Pore Formation. Biophys. J. 2005, 89, 4006–4016. [Google Scholar] [CrossRef]
- Wallbrecher, R.; Ackels, T.; Olea, R.A.; Klein, M.J.; Caillon, L.; Schiller, J.; Bovée-Geurts, P.H.; van Kuppevelt, T.H.; Ulrich, A.S.; Spehr, M.; et al. Membrane permeation of arginine-rich cell-penetrating peptides independent of transmembrane potential as a function of lipid composition and membrane fluidity. J. Control. Release 2017, 256, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient Focal Membrane Deformation Induced by Arginine-rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef]
- Jones, A.T. Macropinocytosis: Searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 2007, 11, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Haucke, V.; Kozlov, M.M. Membrane remodeling in clathrin-mediated endocytosis. J. Cell Sci. 2018, 131, jcs216812. [Google Scholar] [CrossRef]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a Family of Scaffolding Proteins for Organizing “Preassembled Signaling Complexes” at the Plasma Membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef]
- Machleidt, T.; Li, W.P.; Liu, P.; Anderson, R.G. Multiple Domains in Caveolin-1 Control Its Intracellular Traffic. J. Cell Biol. 2000, 148, 17–28. [Google Scholar] [CrossRef]
- Kiss, A.L.; Botos, E. Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J. Cell. Mol. Med. 2009, 13, 1228–1237. [Google Scholar] [CrossRef]
- Pelkmans, L.; Püntener, D.; Helenius, A. Local Actin Polymerization and Dynamin Recruitment in SV40-Induced Internalization of Caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Damm, E.M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzchalia, T.; Helenius, A. Clathrin- and caveolin-1–independent endocytosis. J. Cell Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Kobzev, E.; Chan, J.; de Zoysa, G.H.; Sarojini, V.; Piggot, T.J.; Allison, J.R. Molecular Dynamics Simulation of the Interaction of Two Linear Battacin Analogs with Model Gram-Positive and Gram-Negative Bacterial Cell Membranes. ACS Omega 2020, 6, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Choong, F.H.; Yap, B.K. Cell-Penetrating Peptides: Correlation between Peptide-Lipid Interaction and Penetration Efficiency. ChemPhysChem 2021, 22, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Strandberg, E.; van den Berg, J.; Mink, C.; Bürck, J.; Ciriello, R.A.; Ulrich, A.S. Dynamical structure of the short multifunctional peptide BP100 in membranes. Biochim. Biophys. Acta (BBA)—Biomembr. 2014, 1838, 940–949. [Google Scholar] [CrossRef]
- Strandberg, E.; Wadhwani, P.; Bürck, J.; Anders, P.; Mink, C.; van den Berg, J.; Ciriello, R.A.M.; Melo, M.N.; Castanho, M.A.R.B.; Bardaji, E.; et al. Temperature-dependent re-alignment of the short multifunctional peptide BP100 in membranes revealed by solid-state NMR and molecular dynamics simulations. ChemBioChem 2022, 24, e202200602. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Moreno-Vargas, L.M. History and Anatomy of Cell-Penetrating Peptides Predictors. in preparation.
- Hällbrink, M.; Kilk, K.; Elmquist, A.; Lundberg, P.; Lindgren, M.; Jiang, Y.; Pooga, M.; Soomets, U.; Langel, U. Prediction of Cell-Penetrating Peptides. Int. J. Pept. Res. Ther. 2005, 11, 249–259. [Google Scholar] [CrossRef]
- Hansen, M.; Kilk, K.; Langel, U. Predicting cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 572–579. [Google Scholar] [CrossRef]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef]
- Dobchev, D.A.; Mager, I.; Tulp, I.; Karelson, G.; Tamm, T.; Tamm, K.; Janes, J.; Langel, U.; Karelson, M. Prediction of Cell-Penetrating Peptides Using Artificial Neural Networks. Curr. Comput. Aided-Drug Des. 2010, 6, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Sanders, W.S.; Johnston, C.I.; Bridges, S.M.; Burgess, S.C.; Willeford, K.O. Prediction of Cell Penetrating Peptides by Support Vector Machines. PLoS Comput. Biol. 2011, 7, e1002101. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ke, L.; Cai, L.; Chen, X.; Ren, X.; Gao, M. Improved Prediction of Cell-Penetrating Peptides via Effective Orchestrating Amino Acid Composition Feature Representation. IEEE Access 2019, 7, 163547–163555. [Google Scholar] [CrossRef]
- Tang, J.; Ning, J.; Liu, X.; Wu, B.; Hu, R. A Novel Amino Acid Sequence-based Computational Approach to Predicting Cell-penetrating Peptides. Curr. Comput.-Aided Drug Des. 2019, 15, 206–211. [Google Scholar] [CrossRef]
- Tang, H.; Su, Z.D.; Wei, H.H.; Chen, W.; Lin, H. Prediction of cell-penetrating peptides with feature selection techniques. Biochem. Biophys. Res. Commun. 2016, 477, 150–154. [Google Scholar] [CrossRef]
- Holton, T.A.; Pollastri, G.; Shields, D.C.; Mooney, C. CPPpred: Prediction of cell penetrating peptides. Bioinformatics 2013, 29, 3094–3096. [Google Scholar] [CrossRef]
- Chen, L.; Chu, C.; Huang, T.; Kong, X.; Cai, Y.D. Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids 2015, 47, 1485–1493. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. SkipCPP-Pred: An improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genom. 2018, 18, 742. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Zhou, C.; Ye, X.; Du, P.F.; Su, R.; Wei, L. CPPred-FL: A sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief. Bioinform. 2018, 21, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Patel, V.; George, N.V.; Mallajosyula, S.S. KELM-CPPpred: Kernel Extreme Learning Machine Based Prediction Model for Cell-Penetrating Peptides. J. Proteome Res. 2018, 17, 3214–3222. [Google Scholar] [CrossRef]
- Arif, M.; Ahmad, S.; Ali, F.; Fang, G.; Li, M.; Yu, D.J. TargetCPP: Accurate prediction of cell-penetrating peptides from optimized multi-scale features using gradient boost decision tree. J. Comput.-Aided Mol. Des. 2020, 34, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, S.; Zou, Q.; Ding, Y. Prediction of Cell-Penetrating Peptides Using a Novel HSIC-Based Multiview TSK Fuzzy System. Appl. Sci. 2022, 12, 5383. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Garg, A.; Keizer, D.; Pires, D.E.V.; Ascher, D.B. CSM-peptides: A computational approach to rapid identification of therapeutic peptides. Protein Sci. 2022, 31, e4442. [Google Scholar] [CrossRef]
- Maroni, G.; Stojceski, F.; Pallante, L.; Deriu, M.A.; Piga, D.; Grasso, G. LightCPPgen: An Explainable Machine Learning Pipeline for Rational Design of Cell Penetrating Peptides. arXiv 2024, arXiv:2406.01617. [Google Scholar] [CrossRef]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Patra, M.C. MLCPP 2.0: An Updated Cell-penetrating Peptides and Their Uptake Efficiency Predictor. J. Mol. Biol. 2022, 434, 167604. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Agrawal, P.; Kumar, R.; Bhalla, S.; Usmani, S.S.; Varshney, G.C.; Raghava, G.P.S. Prediction of Cell-Penetrating Potential of Modified Peptides Containing Natural and Chemically Modified Residues. Front. Microbiol. 2018, 9, 725. [Google Scholar] [CrossRef]
- Fu, X.; Cai, L.; Zeng, X.; Zou, Q. StackCPPred: A stacking and pairwise energy content-based prediction of cell-penetrating peptides and their uptake efficiency. Bioinformatics 2020, 36, 3028–3034. [Google Scholar] [CrossRef]
- de Oliveira, E.C.L.; Santana, K.; Josino, L.; Lima e Lima, A.H.; de Souza de Sales Júnior, C. Predicting cell-penetrating peptides using machine learning algorithms and navigating in their chemical space. Sci. Rep. 2021, 11, 7628. [Google Scholar] [CrossRef]
- Guo, Y.; Yan, K.; Lv, H.; Liu, B. PreTP-EL: Prediction of therapeutic peptides based on ensemble learning. Brief. Bioinform. 2021, 22, bbab358. [Google Scholar] [CrossRef]
- Yan, K.; Lv, H.; Wen, J.; Guo, Y.; Liu, B. TP-MV: Therapeutic Peptides Prediction by Multi-view Learning. Curr. Bioinform. 2022, 17, 174–183. [Google Scholar] [CrossRef]
- Yan, K.; Lv, H.; Guo, Y.; Chen, Y.; Wu, H.; Liu, B. TPpred-ATMV: Therapeutic peptide prediction by adaptive multi-view tensor learning model. Bioinformatics 2022, 38, 2712–2718. [Google Scholar] [CrossRef]
- Arif, M.; Kabir, M.; Ahmed, S.; Khan, A.; Ge, F.; Khelifi, A.; Yu, D.J.; Kabir, M. DeepCPPred: A Deep Learning Framework for the Discrimination of Cell-Penetrating Peptides and Their Uptake Efficiencies. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Serebrennikova, M.; Grafskaia, E.; Maltsev, D.; Ivanova, K.; Bashkirov, P.; Kornilov, F.; Volynsky, P.; Efremov, R.; Bocharov, E.; Lazarev, V. TriplEP-CPP: Algorithm for Predicting the Properties of Peptide Sequences. Int. J. Mol. Sci. 2024, 25, 6869. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A.; Open Source Drug Discovery Consortium Info@osdd.net; Raghava, G.P.S. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Su, R.; Shi, G.; Ma, Z.S.; Zou, Q. CPPred-RF: A Sequence-based Predictor for Identifying Cell-Penetrating Peptides and Their Uptake Efficiency. J. Proteome Res. 2017, 16, 2044–2053. [Google Scholar] [CrossRef]
- Yan, K.; Lv, H.; Wen, J.; Guo, Y.; Xu, Y.; Liu, B. PreTP-Stack: Prediction of Therapeutic Peptides Based on the Stacked Ensemble Learing. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wang, L.; Fu, X.; Xia, C.; Zeng, X.; Zou, Q. ITP-Pred: An interpretable method for predicting, therapeutic peptides with fused features low-dimension representation. Brief. Bioinform. 2021, 22, bbaa367. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.; Ye, X.; Zhang, K.; Teng, S.; Li, Z.; Jin, J.; Kim, M.J.; Sakurai, T.; Cui, L.; et al. SiameseCPP: A sequence-based Siamese network to predict cell-penetrating peptides by contrastive learning. Brief. Bioinform. 2022, 24, bbac545. [Google Scholar] [CrossRef]
- Yan, W.; Tang, W.; Wang, L.; Bin, Y.; Xia, J. PrMFTP: Multi-functional therapeutic peptides prediction based on multi-head self-attention mechanism and class weight optimization. PLoS Comput. Biol. 2022, 18, e1010511. [Google Scholar] [CrossRef]
- Cui, Z.; Wang, S.G.; He, Y.; Chen, Z.H.; Zhang, Q.H. DeepTPpred: A Deep Learning Approach with Matrix Factorization for Predicting Therapeutic Peptides by Integrating Length Information. IEEE J. Biomed. Health Inform. 2023, 27, 4611–4622. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, J.H.; Kim, M.S.; Cho, K.; Shin, J.M. In Silico Screening and Optimization of Cell-Penetrating Peptides Using Deep Learning Methods. Biomolecules 2023, 13, 522. [Google Scholar] [CrossRef]
- Shi, K.; Xiong, Y.; Wang, Y.; Deng, Y.; Wang, W.; Jing, B.; Gao, X. PractiCPP: A deep learning approach tailored for extremely imbalanced datasets in cell-penetrating peptide prediction. Bioinformatics 2024, 40, btae058. [Google Scholar] [CrossRef]
- Cao, L.; Xu, Z.; Shang, T.; Zhang, C.; Wu, X.; Wu, Y.; Zhai, S.; Zhan, Z.; Duan, H. Multi_CycGT: A Deep Learning-Based Multimodal Model for Predicting the Membrane Permeability of Cyclic Peptides. J. Med. Chem. 2024, 67, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.M.; Fadzen, C.M.; Choo, Z.N.; Holden, R.L.; Yao, M.; Hanson, G.J.; Pentelute, B.L. Machine Learning To Predict Cell-Penetrating Peptides for Antisense Delivery. ACS Cent. Sci. 2018, 4, 512–520. [Google Scholar] [CrossRef]
- Diener, C.; Martínez, G.G.R.; Blas, D.M.; González, D.A.C.; Corzo, G.; Castro-Obregon, S.; Rio, G.D. Effective Design of Multifunctional Peptides by Combining Compatible Functions. PLoS Comput. Biol. 2016, 12, e1004786. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Su, R.; Zou, Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019, 35, 4272–4280. [Google Scholar] [CrossRef] [PubMed]
- Cherene, M.B.; Taveira, G.B.; Almeida-Silva, F.; da Silva, M.S.; Cavaco, M.C.; da Silva-Ferreira, A.T.; Perales, J.E.A.; de Oliveira Carvalho, A.; Venâncio, T.M.; da Motta, O.V.; et al. Structural and Biochemical Characterization of Three Antimicrobial Peptides from Capsicum annuum L. var. annuum Leaves for Anti-Candida Use. Probiotics Antimicrob. Proteins 2024, 16, 1270–1287. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Banerjee, T.; Swamy, K.V.; Ghosh, P.; Deshpande, M. An in silico scientific basis for LL-37 as a therapeutic for Covid-19. Proteins: Struct. Funct. Bioinform. 2022, 90, 1029–1043. [Google Scholar] [CrossRef]
- Tomazou, M.; Oulas, A.; Anagnostopoulos, A.K.; Tsangaris, G.T.; Spyrou, G.M. In Silico Identification of Antimicrobial Peptides in the Proteomes of Goat and Sheep Milk and Feta Cheese. Proteomes 2019, 7, 32. [Google Scholar] [CrossRef]
- Hsueh, H.T.; Chou, R.T.; Rai, U.; Liyanage, W.; Kim, Y.C.; Appell, M.B.; Pejavar, J.; Leo, K.T.; Davison, C.; Kolodziejski, P.; et al. Machine learning-driven multifunctional peptide engineering for sustained ocular drug delivery. Nat. Commun. 2023, 14, 2509. [Google Scholar] [CrossRef] [PubMed]
- Farajnia, S.; Rahbarnia, L.; Khajehnasiri, N.; Zarredar, H. Design of a hybrid peptide derived from Melittin and CXCL14 –C17: A molecular dynamics simulation study. Biologia 2022, 77, 2269–2280. [Google Scholar] [CrossRef]
- Shin, M.K.; Jang, B.Y.; Bu, K.B.; Lee, S.H.; Han, D.H.; Oh, J.W.; Sung, J.S. De Novo Design of AC-P19M, a Novel Anticancer Peptide with Apoptotic Effects on Lung Cancer Cells and Anti-Angiogenic Activity. Int. J. Mol. Sci. 2022, 23, 15594. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Robbins, P.D. Cell-Type Specific Penetrating Peptides: Therapeutic Promises and Challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef]
- Komin, A.; Russell, L.; Hristova, K.; Searson, P. Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: Mechanisms and challenges. Adv. Drug Deliv. Rev. 2017, 110, 52–64. [Google Scholar] [CrossRef]
- PEP-Therapy Press Note. Available online: https://presse.curie.fr/pep-therapy-and-institut-curie-announce-first-patients-dosed-in-phase-i-clinical-trial-of-pep-010-for-the-treatment-of-advanced-solid-tumors/ (accessed on 30 November 2024).

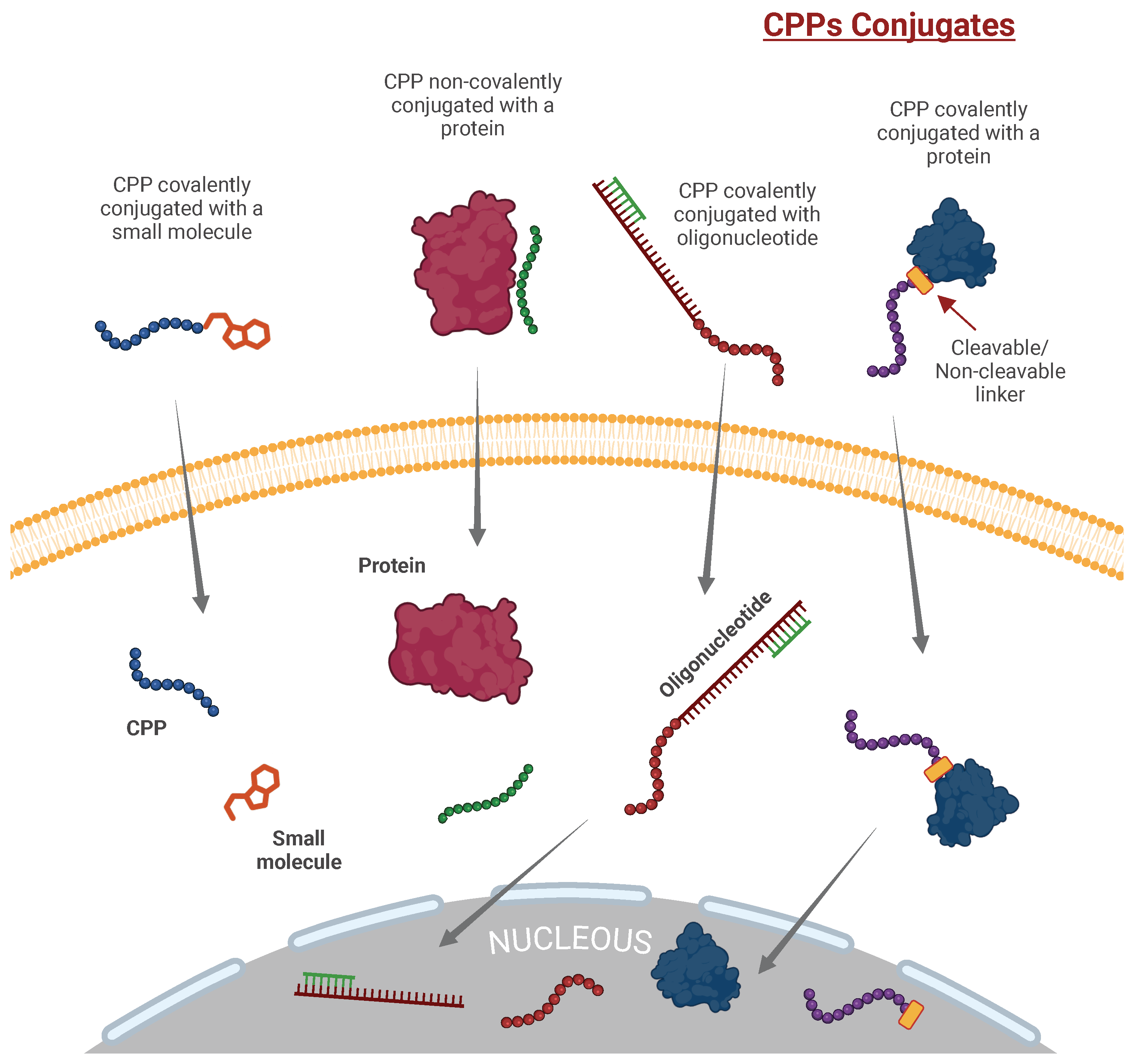
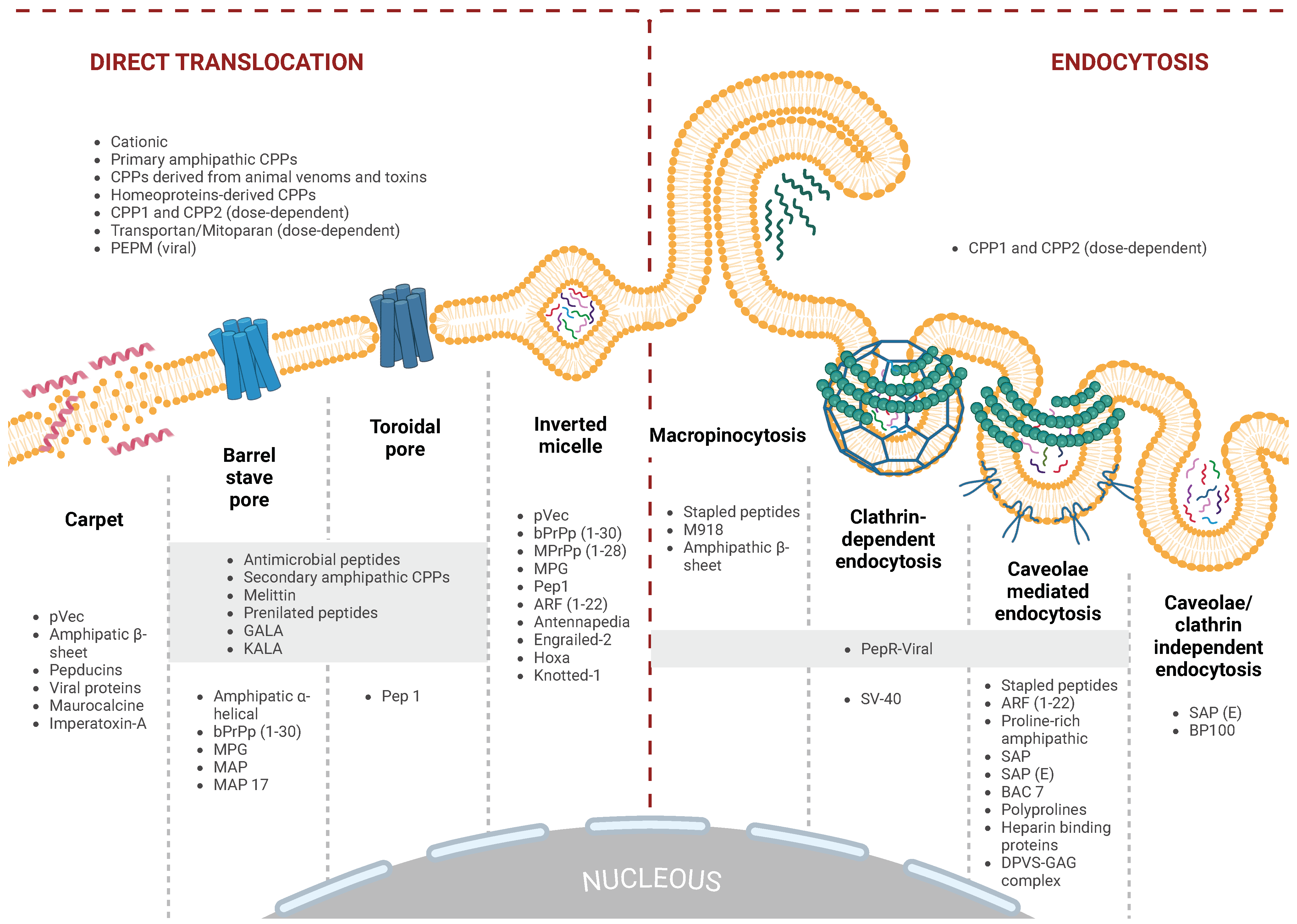

{kind=link}
{kind=link}
{kind=link}
| CPP | Cargo | Compound | Application | Status | ClinicalTrial.gov ID |
|---|---|---|---|---|---|
| Cationic CPPs | |||||
| HIV-1 Tat-protein-derived Tat peptide | PKC inhibitor | KAI-9803 | Acute myocardial infarction | Phase II completed 2004 | NCT00093197 |
| HIV-1 Tat-protein-derived Tat peptide | MAGE-A3/HPV-16 | Head and neck carcinoma | Phase I completed 2005 | NCT00257738 | |
| HIV-1 Tat-protein-derived Tat peptide | -PKC inhibitor | Pain: postherpetic neuralgia, spinal cord injury, postoperative | Phase II completed 2010 | NCT01106716 | |
| HIV-1 Tat-protein-derived Tat peptide | PKC inhibitor | KAI-9803 | Heart attack | Phase II completed 2011 | NCT00785954 |
| HIV-1 Tat-protein-derived Tat peptide | -PKC inhibitor | Pain: postherpetic neuralgia, spinal cord injury, postoperative | Phase II completed 2011 | NCT01135108 | |
| HIV-1 Tat-protein-derived Tat peptide | Dextrogyre peptide | Intraocular inflammation and pain | Phase I completed 2012 | NCT01570205 | |
| HIV-1 Tat-protein-derived Tat peptide | Botulinum toxin A | Cervical dystonia | Phase III completed 2012 | NCT01753310 | |
| HIV-1 Tat-protein-derived Tat peptide | -PKC inhibitor | Pain: postherpetic neuralgia, spinal cord injury, postoperative | Phase II completed 2013 | NCT01015235 | |
| HIV-1 Tat-protein-derived Tat peptide | D-JNKI-1 gel | Hearing loss, idiopathic sudden sensorineural | Phase III completed 2015 | NCT02561091 | |
| HIV-1 Tat-protein-derived Tat peptide | D-JNKI-1 gel | Hearing loss, idiopathic sudden sensorineural | Phase III completed 2016 | NCT02809118 | |
| HIV-1 Tat-protein-derived Tat peptide | JNK-1 | XG-102 | Intraocular inflammation and pain | Phase III completed 2016 | NCT02235272 |
| HIV-1 Tat-protein-derived Tat peptide | PSD-95 protein inhibitor | Ischemic stroke | Phase III completed 2016 | NCT02930018 | |
| HIV-1 Tat-protein-derived Tat peptide | Botulinum toxin A | Cervical dystonia | Phase II completed 2016 | NCT02706795 | |
| HIV-1 Tat-protein-derived Tat peptide | Dextrogyre peptide | XG-102 | Postoperative ocular inflammation | Phase III completed 2017 | NCT02508337 |
| HIV-1 Tat-protein-derived Tat peptide | JNK-1 | AM-111 | Acute inner ear hearing loss | Phase III completed 2017 | NCT02561091 |
| ATX-101 | Advanced dedifferentiated liposarcoma and leiomyosarcoma | Phase II completed 2023 | NCT05116683 | ||
| ATX-101 | Carboplatin | Fallopian Tube and Primary Peritoneal Cancer | Phase I/II terminated 2024 | NCT04814875 | |
| (R-X-R)4, X = 6-Aminohexanoic acid | PMO targeted to human c-Myc | AVI-5126 | Obstruction of vein graft after cardiovascular bypass surgery | Phase II completed 2009 | NCT00451256 |
| TransMTS | Botulinumtoxin A | Cervical dystonia | Phase III completed 2022 | NCT03608397 | |
| MTS | Botulinumtoxin A | Lateral canthal lines | Phase III completed 2016 | NCT02580370 | |
| MTS | Botulinumtoxin A | Primary Axillary Hyperhidrosis | Phase II completed 2016 | NCT02565732 | |
| AVB-620 | Tetramethylindo(di)-carbocyanines (Cy5 and Cy7) | Breast cancer | Phase I completed 2015 | NCT02391194 | |
| AVB-620 | Tetramethylindo(di)-carbocyanines (Cy5 and Cy7) | Breast cancer | Phase II completed 2021 | NCT03113825 | |
| Z12 | BI754091 | BI754091/ATP128/VSV-GP128 | Stage IV Colorectal Cancer | Phase I active, not recruiting, 2024 | NCT04046445 |
| R7 | Cyclosporine A | PsorBan | Psoriasis | Phase II terminated 2003 | Not Applicable |
| PEP-010 | Paclitaxel | Metastatic solid tumor cancer | Phase I recruiting (2024) | NCT04733027 | |
| Amphipatic CPPs | |||||
| p28 | p28 Non-HDM2-mediated peptide inhibitor of p53 | p28 | Solid tumors | Phase I completed 2014 | NCT00914914 |
| p28 | p28 | p28 | Central Nervous System Tumors | Phase I completed 2017 | NCT01975116 |
| p28 | Valganciclovir (VGCV) | RZ-001 | Glioblastoma | Phase I/II recruiting | NCT06102525 |
| p28 | SRF388 | Atezolizumab/Bevazizumab | Phase II active | NCT05359861 | |
| PTD4 | AZX100 (a synthetic 24-amino acid peptide analog of heat shock protein 20 (Hsp20)) | Excision of Keloid Scars | Phase II completed 2012 | NCT00825916 | |
| Hydrophobic CPPs: Stapled Peptides | |||||
| Sulanemadlin (ALRN-6924) | Cytarabine | Acute Myeloid Leukemia or Advanced Myelodysplastic Syndrome | Phase I completed 2019 | NCT02909972 | |
| Sulanemadlin (ALRN-6924) | Palbociclib | Solid Tumor/Lymphoma/Peripheral T-Cell Lymphoma | Phase I/Phase II completed 2020 | NCT02264613 | |
| Sulanemadlin (ALRN-6924) | Topotecan | Small cell lung cancer | Phase I active (Study completion (Actual) 2022) | NCT04022876 | |
| Sulanemadlin (ALRN-6924) | Cytarabine | Solid tumor, brain tumor, leukemia or pediatric lymphoma | Phase I active (Study completion (Actual) 2023) | NCT03654716 | |
| Sulanemadlin (ALRN-6924) | Doxorubicin/Cyclophosphamide/Docetaxel | TP53-Mutant Breast Cancer | Phase I active (Study completion (Actual) 2023) | NCT05622058 | |
| Sulanemadlin (ALRN-6924) | Palbociclib | Advanced, Metastatic, or Unresectable Solid Tumors | Phase I active (Study completion (Estimated) 2025) | NCT02264613 | |
| Pepducins | |||||
| Pepducin PZ-128 (P1pal-7) | Multiple Coronary Artery Disease Risk Factors | Phase I completed 2016 | NCT01806077 | ||
| Pepducin PZ-128 (P1pal-7) | Coronary artery disease | Phase II completed 2021 | NCT02561000 | ||
| Cyclic CPPs | |||||
| 177Lu-DOTA0-Tyr3-octreotate | Lutetium Lu 177 | Neuroendocrine Tumors | Not Applicable completed 2019 | NCT02125474 | |
| 177Lu-DOTA0-Tyr3-octreotate | Lutetium Lu 177 | Neuroendocrine Carcinoma | Phase II active 2023 | NCT02236910 | |
| BT1718 | Solid tumors | Phase I/Phase II completed 2024 | NCT03486730 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Vargas, L.M.; Prada-Gracia, D. Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides. Int. J. Mol. Sci. 2025, 26, 59. https://doi.org/10.3390/ijms26010059
Moreno-Vargas LM, Prada-Gracia D. Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides. International Journal of Molecular Sciences. 2025; 26(1):59. https://doi.org/10.3390/ijms26010059
Chicago/Turabian StyleMoreno-Vargas, Liliana Marisol, and Diego Prada-Gracia. 2025. "Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides" International Journal of Molecular Sciences 26, no. 1: 59. https://doi.org/10.3390/ijms26010059
APA StyleMoreno-Vargas, L. M., & Prada-Gracia, D. (2025). Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides. International Journal of Molecular Sciences, 26(1), 59. https://doi.org/10.3390/ijms26010059