
Oncoviruses in the Oral Cavity: Recent Advances in Understanding Viral Infections and Tumorigenesis

 ,
,  ,
,  ,
,  and
and {kind=link}
Abstract
1. Introduction
2. Epstein–Barr Virus
2.1. How EBV Triggers Tumorigenesis
2.2. EBV and Oral Cavity Cancer
2.3. Recent Advances and Future Perspectives in EBV (Vaccines and/or Treatment)
3. Kaposi-Sarcoma-Associated Herpesvirus (KSHV)
3.1. How KSHV Triggers Tumorigenesis
3.2. KSHV and Oral Cavity Cancer
3.3. Recent Advances and Future Perspectives in KSHV (Vaccines and/or Treatment)
4. Hepatitis B Virus (HBV)
4.1. How HBV Triggers Tumorigenesis
4.2. HBV and Oral Cavity Cancer
4.3. Recent Advances and Future Perspectives in HBV (Vaccines and/or Treatment)
5. Hepatitis C Virus (HCV)
5.1. How HCV Triggers Tumorigenesis
5.2. HCV and Oral Cavity Cancer
5.3. Recent Advances and Future Perspectives in HCV (Vaccines and/or Treatment)
6. Human Papillomaviruses (HPVs)
6.1. How HPV Triggers Tumorigenesis
6.2. HPV and Oral Cavity Cancer
6.3. Recent Advances and Future Perspectives in HPV (Vaccines and/or Treatment)
7. Human Polyomaviruses (HPyVs)
7.1. How HPyVs Triggers Tumorigenesis
7.2. HPyVs and Oral Cavity Cancer
7.3. Recent Advances and Future Perspectives in HPyVs (Vaccines and/or Treatment)
8. Human T-Lymphotropic Virus (HTLV)
8.1. How HTLV Triggers Tumorigenesis
8.2. HTLV and Oral Cavity Cancer
8.3. Recent Advances and Future Perspectives in HTLV (Vaccines and/or Treatment)
9. Herpes Simplex Virus Type 1 (HSV-1)
9.1. How HSV-1 Triggers Tumorigenesis
9.2. HSV-1 and Oral Cavity Cancer
9.3. Recent Advances and Future Perspectives in HSV-1 (Vaccines and/or Treatment)
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| AMP | Doxorubicin, ranimustine, prednisone |
| ATLL | Adult T-cell leukemia-lymphoma |
| BKPyV | Betapolyomavirus secuhominis polyoma virus |
| BL | Burkitt’s lymphoma |
| BLV | Bovine leukemia virus |
| CCC | Covalently closed circular DNA |
| CDKs | Cyclin-dependent kinases |
| CTCL | Cutaneous T-cell lymphoma |
| CTLs | Cytotoxic T lymphocytes |
| DAAs | Direct-acting antivirals |
| DLPCL | Diffuse large B-cell lymphoma |
| DNA | Deoxyribonucleic acid |
| EBNA | EBV nuclear antigen |
| EBV | Epstein–Barr virus |
| GC | Gastric carcinoma |
| GCF | Gingival crevicular fluid |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| HAAP | HTLV-I-associated arthropathy |
| HAM/TSP | Myelopathy/tropical spastic paraparesis |
| HAV | Hepatitis A virus |
| HBV | Hepatitis B virus |
| HBZ | HTLV-1 basic leucine zipper factor |
| HBcAg | Core antigens |
| HBsAg | Surface antigens |
| HBxAg | X proteins |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HHHV-8 | Human rhadinovirus gamma-8 |
| HIV | Human immunodeficiency virus |
| HL | Hodgkin’s lymphoma |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| HPyV | Human polyomaviruses |
| HSV-1 | Herpes simplex virus 1 |
| HTLV | Human T-lymphotropic virus |
| IARC | International Agency for Research on Cancer |
| ICTV | International Committee of Taxonomy of Viruses |
| IFN-a | Interferon-alpha |
| IM | Infectious mononucleosis |
| IR | Insulin resistance |
| IRIS | Immune reconstitution inflammatory syndrome |
| JAK/STAT | Janus tyrosine kinase/signal transducer and transcription activator |
| JCPyV | John Cunningham polyomavirus |
| KICS | Inflammatory cytokine syndrome |
| KS | Kaposi sarcoma |
| KSHV | Kaposi-sarcoma-associated herpesvirus |
| LPS | Lipopolysaccharide |
| LTA | Lipoteichoic acid |
| LTRs | Long terminal repeats |
| MAPKs | Mitogen-activated protein kinases |
| MCC | Merkel cell carcinoma |
| MCD | Multicentric Castleman disease |
| MCPyV | Alphapolyomavirus quintihominis polyomavirus |
| MLL | Mixed-lineage leukemia |
| NF-κB | Nuclear factor-κB |
| NPC | Nasopharyngeal carcinoma |
| OLP | Oral lichen planus |
| ORFs | Open reading frames |
| OSCC | Oral squamous cell carcinomas |
| PEL | Primary effusion lymphoma |
| PML | Progressive multifocal leukoencephalopathy |
| PVL | Proliferative verrucous leukoplakia |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SCT | Stem cell transplants |
| SSRNA | Single-stranded RNA |
| SV40 | Simian vacuolating virus 40 |
| SVR | Sustained virological response |
| T-VEC | Talimogene laherparepvec |
| TLRs | Toll-like receptors |
| VCAP | Vincristine, cyclophosphamide, doxorubicin, and prednisone |
| VCEP | Vindesine, etoposide, carboplatin, and prednisone |
| VLP | Virus-like particle |
| WHO | World Health Organization |
| c-ART | Combined antiretroviral treatment |
| cccDNA | Covalently closed circular DNA |
| pgRNA | Pregenomic RNA |
| sgRNA | Subgenomic RNA |
References
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 283, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Jaffe, E.S.; Dale, J.K.; Pittaluga, S.; Heslop, H.E.; Rooney, C.M.; Gottschalk, S.; Bollard, C.M.; Rao, V.K.; Marques, A.; et al. Characterization and treatment of chronic active Epstein-Barr virus disease: A 28-year experience in the United States. Blood 2011, 117, 5835–5849. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://ictv.global/ (accessed on 11 April 2025).
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [PubMed]
- Kieff, E.D.; Rickinson, A.B. Epstein-Barr Virus and Its Replication. In Fields’ Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 1, pp. 2603–2654. [Google Scholar]
- Naughton, P.; Healy, M.; Enright, F.; Lucey, B. Infectious Mononucleosis: Diagnosis and clinical interpretation. Br. J. Biomed. Sci. 2021, 78, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Bakkalci, D.; Jia, Y.; Winter, J.R.; Lewis, J.E.; Taylor, G.S.; Stagg, H.R. Risk factors for Epstein Barr virus-associated cancers: A systematic review, critical appraisal, and mapping of the epidemiological evidence. J. Glob. Health 2020, 10, 010405. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.J.H.; Schaal, D.L.; Nkadi, E.H.; Scott, R.S. EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses 2022, 14, 2700. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.M.; Cartwright, R.A.; Darwin, C.M.; Richards, I.D.; Roberts, B.; O’Brien, C.; Bird, C.C. Hodgkin’s disease: Case control epidemiological study in Yorkshire. Br. J. Cancer 1987, 55, 85–90. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, W.; Saranath, D. Clinical implications of epigenetic regulation in oral cancer. Oral Oncol. 2015, 51, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H. Progress and Problems in Understanding and Managing Primary Epstein-Barr Virus Infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef] [PubMed]
- Thiruvengadam, R.; Kim, J.H. Therapeutic strategy for oncovirus-mediated oral cancer: A comprehensive review. Biomed. Pharmacother. 2023, 165, 115035. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Sugden, B. Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus. Viruses 2013, 5, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein–Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wiedmer, A.; Martin, K.A.; Wickramasinghe, P.J.M.S.; Kossenkov, A.V.; Lieberman, P.M. Coordinate Regulation of TET2 and EBNA2 Controls the DNA Methylation State of Latent Epstein-Barr Virus. J. Virol. 2017, 91, e00804-17. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16 INK4A and p14 ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein–Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, Y.; Wang, C.; Gan, R. Signaling pathways of EBV-induced oncogenesis. Cancer Cell Int. 2021, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, H.; Chen, J.; Wang, R.; Saleh, A.; Si, H.; Lee, S.; Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; et al. Head and Neck Cancers Promote an Inflammatory Transcriptome through Coactivation of Classic and Alternative NF-κB Pathways. Cancer Immunol. Res. 2019, 7, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Xie, Y.; Tang, J.; Xin, S.; Liu, L.; Zhang, S.; Yan, Q.; Zhu, F.; Lu, J. Targeting Exosomal EBV-LMP1 Transfer and miR-203 Expression via the NF-κB Pathway: The Therapeutic Role of Aspirin in NPC. Mol. Ther. Nucleic Acids 2019, 17, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Jun, S.; Song, M.; Kim, J.H.; Song, Y.J. The extract of Chrysanthemum indicum Linne inhibits EBV LMP1-induced NF-κB activation and the viability of EBV-transformed lymphoblastoid cell lines. Food Chem. Toxicol. 2012, 50, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Uphoff, C.C.; Dirks, W.G.; Pommerenke, C.; Meyer, C.; Drexler, H.G. Epstein-Barr virus (EBV) activates NKL homeobox gene HLX in DLBCL. PLoS ONE 2019, 14, e0216898. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, W.; Feng, L.; Zhang, S.; Zhang, H.; Zhang, X.; Qi, X.; Zhang, Y.; Feng, Q.; Xiang, T.; Zeng, Y.X. Induction of chemokine (C-C motif) ligand 5 by Epstein-Barr virus infection enhances tumor angiogenesis in nasopharyngeal carcinoma. Cancer Sci. 2018, 109, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Lin, Y.X.; Ma, W.; Zhang, H.J.; Chen, K.M.; He, G.P.; Zhang, X.; Xu, M.; Feng, Q.S.; Chen, M.Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Yang, G.D.; Huang, T.J.; Li, R.; Chu, Q.Q.; Xu, L.; Wang, M.S.; Cai, M.D.; Zhong, L.; Wei, H.J.; et al. EB-virus latent membrane protein 1 potentiates the stemness of nasopharyngeal carcinoma via preferential activation of PI3K/AKT pathway by a positive feedback loop. Oncogene 2016, 35, 3419–3431. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.A.; Klingelhutz, A.J.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol. 2003, 77, 12276–12284. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cohen, J.I. Epstein-Barr virus (EBV) tegument protein BGLF2 promotes EBV reactivation through activation of the p38 mitogen-activated protein kinase. J. Virol. 2015, 90, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Poling, B.C.; Price, A.M.; Luftig, M.A.; Cullen, B.R. The Epstein-Barr virus miR-BHRF1 microRNAs regulate viral gene expression in cis. Virology 2017, 512, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Zhou, R.; Zong, J.F.; Lin, W.S.; Tong, S.; Guo, Q.J.; Lin, C.; Lin, S.J.; Chen, Y.X.; Chen, M.R.; et al. Epstein-Barr virus-coded miR-BART13 promotes nasopharyngeal carcinoma cell growth and metastasis via targeting of the NKIRAS2/NF-κB pathway. Cancer Lett. 2019, 447, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, J.; Zhang, X.; Lu, Y.; Wang, J.; Lyu, X.; Chen, Y.; Liu, J.; Cai, H.; Wang, Y.; et al. Gold nano-particles (AuNPs) carrying anti-EBV-miR-BART7-3p inhibit growth of EBV-positive nasopharyngeal carcinoma. Oncotarget 2015, 6, 7838–7850. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Jiang, Q.; Liu, X.; Lin, X.; Tang, Z.; Liu, C.; Zhou, J.; Zhao, M.; Li, X.; Cheng, Z.; et al. Cinobufotalin powerfully reversed EBV-miR-BART22-induced cisplatin resistance via stimulating MAP2K4 to antagonize non-muscle myosin heavy chain IIA/glycogen synthase 3β/β-catenin signaling pathway. EBioMedicine 2019, 48, 386–404. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, J.; Qin, Y.; Yang, C.; Wan, C.; Dai, X.; Sun, Y.; Meng, J.; Lu, Y.; Li, Y.; Zhang, Z.; et al. Downregulation of ABI2 expression by EBV-miR-BART13-3p induces epithelial-mesenchymal transition of nasopharyngeal carcinoma cells through upregulation of c-JUN/SLUG signaling. Aging 2020, 12, 340–358. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [PubMed]
- Blankson, P.K.; Parkins, G.E.; Blankson, H.N.A.; Fasola, A.O.; Pappoe-Ashong, P.J.; Boamah, M.O.; Asmah, R.H. Herpesviruses and human papillomaviruses in saliva and biopsies of patients with orofacial tumors. Clinics 2024, 79, 100477. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.W.; Jayasekara, P.; Amarasinghe, A.A.H.K. Squamous cell carcinoma and precursor lesions of the oral cavity: Epidemiology and aetiology. Periodontol. 2000 2011, 57, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Lautenberger, J.A.; Gao, X.; Sezgin, E.; Hendrickson, S.L.; Troyer, J.L.; David, V.A.; Guan, L.; McIntosh, C.E.; Guo, X.; et al. The Principal Genetic Determinants for Nasopharyngeal Carcinoma in China Involve the HLA Class I Antigen Recognition Groove. PLoS Genet. 2012, 8, e1003103. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, K.V.; Lubenskaya, A.K.; Senyuta, N.B.; Dushenkina, T.E.; Gurtsevitch, V.E. Epstein–Barr virus (Herpesviridae: Lymphocryptovirus) types 1 and 2 and other viral markers in patients with nasopharyngeal carcinoma in two geographically and ethnically distinct regions of Russia. Probl. Virol. 2023, 68, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.Y.; Chung, L.P.; Yuen, S.T.; Ho, C.M.; Wong, M.P.; Chan, S.Y. Lymphoepithelial carcinoma of the salivary gland: In situ detection of Epstein-Barr virus. J. Clin. Pathol. 1995, 48, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Dutoit, S.J.; Therkildsen, M.H.; Nielsen, N.H.; Jensen, H.; Hansen, J.P.H.; Pallesen, G. Undifferentiated carcinoma of the salivary gland in Greenlandic Eskimos: Demonstration of Epstein-Barr virus DNA by in situ nucleic acid hybridization. Hum. Pathol. 1991, 22, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Iezzoni, J.C.; Gaffey, M.J.; Weiss, L.M. The Role of Epstein-Barr Virus in Lymphoepithelioma-like Carcinomas. Am. J. Clin. Pathol. 1995, 103, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Mawardi, H.; Cutler, C.; Treister, N. Medical management update: Non-Hodgkin lymphoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2009, 107, e19–e33. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.D.B.; Tavares Ferreira, C.B.; Boehmer Leite, G.; de Menezes Pontes, J.R.; Antunes, H.S. Oral manifestations of lymphoma: A systematic review. Ecancermedicalscience 2016, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Fornatora, M.; Reich, R.F.; Freedman, P. Extranodal Hodgkin’s lymphoma of the oral soft tissue. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 98, 207–208. [Google Scholar] [CrossRef]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Burkitt’s Lymphoma. N. Engl. J. Med. 2022, 387, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Hämmerl, L.; Colombet, M.; Rochford, R.; Ogwang, D.M.; Parkin, D.M. The burden of Burkitt lymphoma in Africa. Infect. Agent. Cancer 2019, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008, an overview. Pathologica 2010, 102, 83–87. [Google Scholar] [PubMed]
- Lim, S.T.; Karim, R.; Nathwani, B.N.; Tulpule, A.; Espina, B.; Levine, A.M. AIDS-Related Burkitt’s Lymphoma Versus Diffuse Large-Cell Lymphoma in the Pre–Highly Active Antiretroviral Therapy (HAART) and HAART Eras: Significant Differences in Survival With Standard Chemotherapy. J. Clin. Oncol. 2005, 23, 4430–4438. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, A.I.; Pappalardo, V.Y.; Buijs, M.J.; Brandt, B.W.; Mäkitie, A.A.; Meurman, J.H.; Zaura, E. Salivary microbiome profiles of oral cancer patients analyzed before and after treatment. Microbiome 2023, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Di Spirito, F.; Di Palo, M.P.; Folliero, V.; Cannatà, D.; Franci, G.; Martina, S.; Amato, M. Oral Bacteria, Virus and Fungi in Saliva and Tissue Samples from Adult Subjects with Oral Squamous Cell Carcinoma: An Umbrella Review. Cancers 2023, 15, 5540. [Google Scholar] [CrossRef] [PubMed]
- Badwelan, M.; Muaddi, H.; Ahmed, A.; Lee, K.T.; Tran, S.D. Oral Squamous Cell Carcinoma and Concomitant Primary Tumors, What Do We Know? A Review of the Literature. Curr. Oncol. 2023, 30, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Al-Hebshi, N.N.; Speicher, D.J.; Perera, I.; Johnson, N.W. Emerging role of bacteria in oral carcinogenesis: A review with special reference to perio-pathogenic bacteria. J. Oral Microbiol. 2016, 8, 32762. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, Y.; Zhou, X.; Liu, S.; Han, Q.; Cheng, L. Role of Oral Bacteria in the Development of Oral Squamous Cell Carcinoma. Cancers 2020, 12, 2797. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, S.E.; Lamont, R.J. Oral bacteria and cancer. PLoS Pathog. 2014, 10, e1003933. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Shimizu, D.; Hirabayashi, S.; Ueda, S.; Miyabe, S.; Oh-Iwa, I.; Nagao, T.; Shimozato, K.; Nomoto, S. Changes in oral microbial profiles associated with oral squamous cell carcinoma vs leukoplakia. J. Investig. Clin. Dent. 2019, 10, e12445. [Google Scholar] [CrossRef] [PubMed]
- Stasiewicz, M.; Karpiński, T.M. The oral microbiota and its role in carcinogenesis. Semin. Cancer Biol. 2022, 86 Pt 3, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.N.; Deshmukh, R. Oral microbiome and oral cancer—The probable nexus. J. Oral Maxillofac. Pathol. 2020, 24, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Ren, B.; Zhou, X.; Zhang, L.; Xu, X. Cross-kingdom interaction between Candida albicans and oral bacteria. Front. Microbiol. 2022, 13, 911623. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Pallagatti, S.; Sheikh, S.; Aggarwal, A.; Gupta, D.; Singh, R. Candidal species identification in malignant and potentially malignant oral lesions with antifungal resistance patterns. Contemp. Clin. Dent. 2018, 9 (Suppl. 2), S309–S313. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Acurio, D.; Bravo, D.; Aguayo, F. Epstein-Barr virus–oral bacterial link in the development of oral squamous cell carcinoma. Pathogens 2020, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Erira, A.T.; Navarro, A.F.R.; Robayo, D.A.G. Human papillomavirus, Epstein-Barr virus, and Candida albicans co-infection in oral leukoplakia with different degrees of dysplasia. Clin. Exp. Dent. Res. 2021, 7, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Noguchi, Y.; de Rivera, M.W.; Hoshino, M.; Sakashita, H.; Yamada, T.; Inoue, H.; Miyazaki, Y.; Nozaki, T.; González-López, B.S.; et al. Detection of Epstein-Barr virus genome and latent infection gene expression in normal epithelia, epithelial dysplasia, and squamous cell carcinoma of the oral cavity. Tumour Biol. 2016, 37, 3389–3404. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhang, J.-B.; Lu, L.-X.; Jia, Y.-J.; Zheng, M.-Q.; Debelius, J.W.; He, Y.Q.; Wang, T.M.; Deng, C.M.; Tong, X.T.; et al. Oral Microbiota Alteration and Roles in Epstein-Barr Virus Reactivation in Nasopharyngeal Carcinoma. Microbiol. Spectr. 2023, 11, e0344822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Zheng, H.J.; Zhang, C.P. The Oral Microbiota May Have Influence on Oral Cancer. Front. Cell. Infect. Microbiol. 2020, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Yang, L.; Giese, R.A.; Hao, Y.; Nossa, C.W.; Morris, L.G.T.; Rosenthal, M.; Migliacci, J.; Kelly, D.; Tseng, W.; et al. Periodontal pathogens are a risk factor of oral cavity squamous cell carcinoma, independent of tobacco and alcohol and human papillomavirus. Int. J. Cancer 2019, 145, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Drop, B.; Strycharz-Dudziak, M.; Kliszczewska, E.; Polz-Dacewicz, M. Coinfection with Epstein–Barr Virus (EBV), Human Papilloma Virus (HPV) and Polyoma BK Virus (BKPyV) in Laryngeal, Oropharyngeal and Oral Cavity Cancer. Int. J. Mol. Sci. 2017, 18, 2752. [Google Scholar] [CrossRef] [PubMed]
- Sutton, D.N.; Brown, J.S.; Rogers, S.N.; Vaughan, E.D.; Woolgar, J.A. The prognostic implications of the surgical margin in oral squamous cell carcinoma. Int. J. Oral Maxillofac. Surg. 2003, 32, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Makarewicz, J.; Kaźmierczak-Siedlecka, K.; Sobocki, B.K.; Dobrucki, I.T.; Kalinowski, L.; Stachowska, E. Anti-cancer management of head and neck cancers and oral microbiome—What can we clinically obtain? Front. Cell. Infect. Microbiol. 2024, 14, 1329057. [Google Scholar] [CrossRef] [PubMed]
- Vaghela, N. An overview of the oral primary preventive measures at public/community level in India. Int. J. Prev. Clin. Dent. Res. 2022, 9, 48–51. [Google Scholar] [CrossRef]
- Cao, S.-M.; Liu, Z.; Jia, W.-H.; Huang, Q.-H.; Liu, Q.; Guo, X.; Huang, T.B.; Ye, W.; Hong, M.H. Fluctuations of Epstein-Barr Virus Serological Antibodies and Risk for Nasopharyngeal Carcinoma: A Prospective Screening Study with a 20-Year Follow-Up. PLoS ONE 2011, 6, e19100. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Liu, Z.; Yu, K.J.; Coghill, A.E.; Chen, X.X.; Xie, S.H.; Lin, D.F.; Huang, Q.H.; Lu, Y.Q.; Ling, W.; et al. Utility of Epstein–Barr Virus DNA in Nasopharynx Swabs as a Reflex Test to Triage Seropositive Individuals in Nasopharyngeal Carcinoma Screening Programs. Clin. Chem. 2022, 68, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, X.; Tang, C.; Zhang, Y.; Zhou, T.; Yang, X.J.; Liao, Y.; He, Y.Q.; Wang, T.M.; Xue, W.Q.; et al. Detection of Epstein–Barr virus DNA methylation as tumor markers of nasopharyngeal carcinoma patients in saliva, oropharyngeal swab, oral swab, and mouthwash. MedComm 2024, 5, e673. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Vaccine Development for Epstein-Barr Virus. In Epstein Barr Virus Volume 1: EBV Biology, Immunology and Non-Neoplastic Diseases; Münz, C., Ed.; Springer: Singapore, 2018; pp. 477–493. [Google Scholar] [CrossRef] [PubMed]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-Barr Virus gH Is Essential for Penetration of B Cells but Also Plays a Role in Attachment of Virus to Epithelial Cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef] [PubMed]
- Colevas, A.D.D.; Rudek, M.A.; Even, C.; Lee, V.H.-F.; Gillison, M.L.; Khan, S.A.; Rong, L.; Winters, E.; Biedermann, S.; Lai, S.; et al. Phase I/IIa Clinical Trial of a Small Molecule EBNA1 Inhibitor, VK-2019, in Patients with Epstein Barr Virus–Positive Nasopharyngeal Cancer with Pharmacokinetic and Pharmacodynamic Correlative Studies. J. Clin. Oncol. 2023, 41 (Suppl. 16), 6035. [Google Scholar] [CrossRef]
- Keck, K.M.; Moquin, S.A.; He, A.; Fernandez, S.G.; Somberg, J.J.; Liu, S.M.; Martinez, D.M.; Miranda, J.L. Bromodomain and Extraterminal Inhibitors Block the Epstein-Barr Virus Lytic Cycle at Two Distinct Steps. J. Biol. Chem. 2017, 292, 13284–13295. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C. Potential Use of Antiviral l(−)Nucleoside Analogues for the Prevention or Treatment of Viral Associated Cancers. Cancer Lett. 2001, 162 (Suppl. 1), S33–S37. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Pruene, A.; Darguzyte, M.; vom Stein, A.F.; Nguyen, P.-H.; Wagner, D.L.; Kath, J.; Roig-Merino, A.; Heuser, M.; Riehm, L.L.; et al. Non-Viral TRAC-Knocked-In CD19KICAR-T and gp350KICAR-T Cells Tested Against Burkitt Lymphomas with Type 1 or 2 EBV Infection: In Vivo Cellular Dynamics and Potency. Front. Immunol. 2023, 14, 1086433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, X.; Zhang, S.; Wang, N.; Zhang, T.; Zhang, D.; Ao, Q.; Cao, Y.; Huang, L. EBV-Associated Lymphoproliferative Disease Post-CAR-T Cell Therapy. Front. Med. 2024, 18, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of Herpesvirus-Like DNA Sequences in AIDS-Associated Kaposi’s Sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Iftode, N.; Rădulescu, M.A.; Aramă, Ș.S.; Aramă, V. Update on Kaposi Sarcoma-Associated Herpesvirus (KSHV or HHV-8)—Review. Rom. J. Intern. Med. 2020, 58, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Lurain, K.; Yarchoan, R.; Ramaswami, R. Clinical Management of Kaposi Sarcoma Herpesvirus-Associated Diseases: An Update on Disease Manifestations and Treatment Strategies. Expert Rev. Anti-Infect. Ther. 2023, 21, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Said, J.W.; Chien, K.; Tasaka, T.; Koeffler, H.P. Ultrastructural Characterization of Human Herpesvirus 8 (Kaposi’s Sarcoma-Associated Herpesvirus) in Kaposi’s Sarcoma Lesions: Electron Microscopy Permits Distinction from Cytomegalovirus (CMV). J. Pathol. 1997, 182, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Sallah, N.; Palser, A.L.; Watson, S.J.; Labo, N.; Asiki, G.; Marshall, V.; Newton, R.; Whitby, D.; Kellam, P.; Barroso, I. Genome-Wide Sequence Analysis of Kaposi Sarcoma-Associated Herpesvirus Shows Diversification Driven by Recombination. J. Infect. Dis. 2018, 218, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.O.; Marinho, P.N.; Medeiros, L.S.; de Paula, V.S. Human Gammaherpesvirus 8 Oncogenes Associated with Kaposi’s Sarcoma. Int. J. Mol. Sci. 2022, 23, 7203. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi Sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Pauk, J.; Huang, M.-L.; Brodie, S.J.; Wald, A.; Koelle, D.M.; Schacker, T.; Celum, C.; Selke, S.; Corey, L. Mucosal Shedding of Human Herpesvirus 8 in Men. N. Engl. J. Med. 2000, 343, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Atyeo, N.; Rodriguez, M.D.; Papp, B.; Toth, Z. Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections. Viruses 2021, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Krantz, E.; Selke, S.; Kuntz, S.R.; Wang, J.; Huang, M.L.; Pauk, J.S.; Corey, L.; Wald, A. Frequent and Asymptomatic Oropharyngeal Shedding of Human Herpesvirus 8 among Immunocompetent Men. J. Infect. Dis. 2007, 195, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, L.M.; Al-Sulaiman, M.H.; Teo, C.G.; Porter, S.R. Extensive Oral Shedding of Human Herpesvirus 8 in a Renal Allograft Recipient. Oral Microbiol. Immunol. 2009, 24, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Nabiee, R.; Syed, B.; Ramirez Castano, J.; Lalani, R.; Totonchy, J.E. An Update of the Virion Proteome of Kaposi Sarcoma-Associated Herpesvirus. Viruses 2020, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Khammissa, R.A.G.; Lemmer, J.; Feller, L. Oral HIV-Associated Kaposi Sarcoma. J. Oral Pathol. Med. 2013, 42, 201–207. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. Regulation of KSHV Latency and Lytic Reactivation. Viruses 2020, 12, 1034. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; DeFee, M.R.; Cao, Y.; Wen, J.; Wen, X.; Noverr, M.C.; Qin, Z. Lipoteichoic acid (LTA) and lipopolysaccharides (LPS) from periodontal pathogenic bacteria facilitate oncogenic herpesvirus infection within primary oral cells. PLoS ONE 2014, 9, e101326. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Lee, S.H. Understanding Metabolic Pathway Rewiring by Oncogenic Gamma Herpesvirus. J. Microbiol. Biotechnol. 2024, 34, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, M.; Zhang, T.; Marshall, V.; Gonçalves, P.; Ramaswami, R.; Labo, N.; Whitby, D.; Uldrick, T.S.; Yarchoan, R.; Huang, Y.; et al. Signatures of oral microbiome in HIV-infected individuals with oral Kaposi’s sarcoma and cell-associated KSHV DNA. PLoS Pathog. 2020, 16, e1008114. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qiao, J.; Yin, J.; Goldstein, A.; Lin, H.-Y.; Post, S.R.; Qin, Z. Kaposi Sarcoma–Associated Herpesvirus and Staphylococcus aureus Coinfection in Oral Cavities of HIV-Positive Patients: A Unique Niche for Oncogenic Virus Lytic Reactivation. J. Infect. Dis. 2019, 221, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Etemad, S.A.; Dewan, A.K. Kaposi Sarcoma Updates. Dermatol. Clin. 2019, 37, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Ertürk Yılmaz, T.; Akay, B.N.; Okçu Heper, A. Dermoscopic Findings of Kaposi Sarcoma and Dermatopathological Correlations. Australas. J. Dermatol. 2020, 61, e309–e313. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Website. Available online: https://www.who.int/ (accessed on 17 December 2024).
- Denis, D.; Seta, V.; Regnier-Rosencher, E.; Kramkimel, N.; Chanal, J.; Avril, M.-F.; Dupin, N. A Fifth Subtype of Kaposi’s Sarcoma, Classic Kaposi’s Sarcoma in Men Who Have Sex with Men: A Cohort Study in Paris. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Bender Ignacio, R.A.; Goldman, J.D.; Magaret, A.S.; Selke, S.; Huang, M.-L.; Gantt, S.; Johnston, C.; Phipps, W.T.; Schiffer, J.T.; Zuckerman, R.A.; et al. Patterns of Human Herpesvirus-8 Oral Shedding among Diverse Cohorts of Human Herpesvirus-8 Seropositive Persons. Infect. Agents Cancer 2016, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.; Orem, J.; Okuku, F.; Kalinaki, M.; Saracino, M.; Katongole-Mbidde, E.; Sande, M.; Ronald, A.; McAdam, K.; Huang, M.L.; et al. Impact of HIV Infection and Kaposi Sarcoma on Human Herpesvirus-8 Mucosal Replication and Dissemination in Uganda. PLoS ONE 2009, 4, e4222. [Google Scholar] [CrossRef] [PubMed]
- Phipps, W.; Saracino, M.; Selke, S.; Huang, M.-L.; Jaoko, W.; Mandaliya, K.; Wald, A.; Casper, C.; McClelland, R.S. Oral HHV-8 Replication among Women in Mombasa, Kenya. J. Med. Virol. 2014, 86, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Coogan, M.M.; Greenspan, J.; Challacombe, S.J. Oral Lesions in Infection with Human Immunodeficiency Virus. Bull. World Health Organ. 2005, 83, 700–706. [Google Scholar] [PubMed]
- Little, R.F.; Uldrick, T.S. Are There Clues to Oral Kaposi Sarcoma–Associated Herpesvirus Shedding and Kaposi Sarcoma Oncogenesis in the Oral Microbiome? J. Infect. Dis. 2019, 221, 1226–1228. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.S.; Maronian, N.; Vieira, J. Activation of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Gene Expression during Epithelial Differentiation. J. Virol. 2005, 79, 13769–13777. [Google Scholar] [CrossRef] [PubMed]
- The Lancet HIV. Putting People First in Communication about HIV. Lancet HIV 2023, 10, e623. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shahir, A.-M.; Sha, J.; Feng, Z.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-Chain Fatty Acids from Periodontal Pathogens Suppress Histone Deacetylases, EZH2, and SUV39H1 To Promote Kaposi’s Sarcoma-Associated Herpesvirus Replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Barrett, L.; Plaisance-Bonstaff, K.; Post, S.R.; Qin, Z. Porphyromonas gingivalis Coinfects with KSHV in Oral Cavities of HIV+ Patients and Induces Viral Lytic Reactivation. J. Med. Virol. 2020, 92, 3862–3867. [Google Scholar] [CrossRef] [PubMed]
- Markazi, A.; Bracci, P.M.; McGrath, M.; Gao, S.J. Pseudomonas aeruginosa stimulates inflammation and enhances Kaposi’s sarcoma herpesvirus-induced cell proliferation and cellular transformation through both lipopolysaccharide and flagellin. mBio 2020, 11, e02843-20. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fu, W.; Li, S.; Yeh, W.; She, R.; Brenner, C.; Chen, C.; Feng, P. Oral Neisseria gonorrhoeae promotes KSHV lytic replication. J. Med. Virol. 2025, 97, e70304. [Google Scholar] [CrossRef] [PubMed]
- Nachega, J.B.; Scarsi, K.K.; Gandhi, M.; Scott, R.K.; Mofenson, L.M.; Archary, M.; Nachman, S.; Decloedt, E.; Geng, E.H.; Wilson, L.; et al. Long-acting antiretrovirals and HIV treatment adherence. Lancet HIV 2023, 10, e332–e342. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, R.; Lurain, K.; Yarchoan, R. Oncologic treatment of HIV-associated Kaposi sarcoma 40 years on. J. Clin. Oncol. 2022, 40, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.A.; Blazkova, J.; Kardava, L.; Justement, J.S.; Shi, V.; Manning, M.R.; Shahid, A.; Dong, W.; Kennedy, B.D.; Sewack, A.B.; et al. Sustained virologic suppression of multidrug-resistant HIV in an individual treated with anti-CD4 domain 1 antibody and lenacapavir. Nat. Med. 2025, 31, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Leidner, R.S.; Aboulafia, D.M. Recrudescent Kaposi’s Sarcoma After Initiation of HAART: A Manifestation of Immune Reconstitution Syndrome. AIDS Patient Care STDs 2005, 19, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Granato, M. Nanotechnology Frontiers in γ-Herpesviruses Treatments. Int. J. Mol. Sci. 2021, 22, 11407. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Lee, J.J.; Park, A.; Jung, K.L.; Lee, S.A.; Choi, Y.J.; Lee, H.R.; Lai, C.J.; Eoh, H.; Jung, J.U. Herpesvirus-induced spermidine synthesis and eIF5A hypusination for viral episomal maintenance. Cell Rep. 2022, 40, 111234. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, F.O. Early Studies of Viral Hepatitis. Br. Med. Bull. 1972, 28, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Asandem, D.A.; Segbefia, S.P.; Kusi, K.A.; Bonney, J.H.K. Hepatitis B Virus Infection: A Mini Review. Viruses 2024, 16, 724. [Google Scholar] [CrossRef] [PubMed]
- Magnius, L.; Mason, W.S.; Taylor, J.; Kann, M.; Glebe, D.; Dény, P.; Sureau, C.; Norder, H.; Ictv Report Consortium. ICTV Virus Taxonomy Profile: Hepadnaviridae. J. Gen. Virol. 2020, 101, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xu, M.; Li, X.; Zhang, Z. Distribution of Hepatitis B Virus Genotypes and Subgenotypes: A Meta-Analysis. Medicine 2021, 100, e27941. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B Virus Infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, E.J.; Hawkins, G.; Fraser, A.; Hutchinson, S.J.; Goldberg, D. Hepatitis B Prevention, Diagnosis, Treatment and Care: A Review. Occup. Med. 2011, 61, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.H.; Li, C.L.; Lin, Y.Y.; Ho, M.C.; Wang, Y.C.; Tseng, S.T.; Chen, P.J. Hepatitis B Virus DNA Integration Drives Carcinogenesis and Provides a New Biomarker for HBV-Related HCC. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, D.N.; Bennardo, F.; Popescu, D.M.; Nicolae, F.M.; Ionele, C.M.; Boldeanu, M.V.; Camen, A.; Rogoveanu, I.; Surlin, P. Oral and Periodontal Implications of Hepatitis Type B and D. Current State of Knowledge and Future Perspectives. J. Pers. Med. 2022, 12, 1580. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Cheng, Y.; Jiang, X.; Jiang, H.; Wang, Y.; Li, L. Decreased Diversity of the Oral Microbiota of Patients with Hepatitis B Virus-Induced Chronic Liver Disease: A Pilot Project. Sci. Rep. 2015, 5, 17098. [Google Scholar] [CrossRef] [PubMed]
- Neag, M.A.; Mitre, A.O.; Catinean, A.; Buzoianu, A.D. Overview of the microbiota in the gut-liver axis in viral B and C hepatitis. World J. Gastroenterol. 2021, 27, 7446–7461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, Q.; Lu, F.; Chen, K.; Ni, Z.; Wang, H.; Zhou, C.; Zhang, Y.; Chen, B.; Bo, Z.; et al. distinct microbiota signature precedes the clinical diagnosis of hepatocellular carcinoma. Gut Microbes 2023, 15, 2201159. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Min, Y.; Wei, X.; Xia, X.; Wei, Z.; Li, R.; Jin, J.; Liu, Z.; Hu, X.; Peng, X. Hepatitis B Virus Infection: An Insight into the Clinical Connection and Molecular Interaction Between Hepatitis B Virus and Host Extrahepatic Cancer Risk. Front. Immunol. 2023, 14, 1141956. [Google Scholar] [CrossRef] [PubMed]
- Komori, M.F.; Kimura, T.; Kariya, S.; Onoda, T.; Takeda, S.; Mizukawa, N.; Iida, S.; Kimata, Y.; Nishizaki, K. Epidemiological Correlations Between Head and Neck Cancer and Hepatitis B Core Antibody Positivity. Anticancer Res. 2020, 40, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Lin, C.; Su, Y.; Shih, Y.; Wang, C.; Teng, C.J.; Chou, C.W. Survival Outcomes of Patients with Head and Neck Squamous Cell Cancer with Hepatitis B Virus Infection: An Analysis from an Endemic Tertiary Center. Cancer Med. 2023, 12, 6802–6810. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Zhu, X.; Sun, Y.; Yang, S.; Peng, C.; Feng, X.; Chen, Z.; Yimamu, Y.; Liao, G.; Yang, L. The Association of HBV Infection and Head and Neck Cancer: A Systematic Review and Meta-Analysis. BMC Cancer 2024, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Donà, S.; Borsetto, D.; Fussey, J.; Biscaro, V.; Vian, E.; Spinato, G.; Menegaldo, A.; Da Mosto, M.C.; Rigoli, R.; Polesel, J.; et al. Association Between Hepatitis C and B Viruses and Head and Neck Squamous Cell Carcinoma. J. Clin. Virol. 2019, 121, 104209. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.H.; Yang, T.H.; Cheng, Y.F.; Chen, C.S.; Lin, H.C. Associations of Head and Neck Cancer with Hepatitis B Virus and Hepatitis C Virus Infection. Cancers 2023, 15, 4510. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Takahashi, T.; Fukuda, J. Prevalence of Hepatitis Virus Infection in Association with Oral Diseases Requiring Surgery. Oral Dis. 2002, 8, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Lv, J.; Liu, Y.; Chen, J.G.; Ge, Z.; Zhu, J.; Dai, J.; Du, L.B.; Yu, C.; Guo, Y.; et al. Associations Between Hepatitis B Virus Infection and Risk of All Cancer Types. JAMA Netw. Open 2019, 2, e195718. [Google Scholar] [CrossRef] [PubMed]
- Kamiza, A.B.; Fatumo, S.; Singini, M.G.; Yeh, C.C.; Chikowore, T. Hepatitis B Infection Is Causally Associated with Extrahepatic Cancers: A Mendelian Randomization Study. EBioMedicine 2022, 79, 104003. [Google Scholar] [CrossRef] [PubMed]
- Dejean, A.; Lugassy, C.; Zafrani, S.; Tiollais, P.; Brechot, C. Detection of Hepatitis B Virus DNA in Pancreas, Kidney and Skin of Two Human Carriers of the Virus. J. Gen. Virol. 1984, 65, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Wick, M.; White, H.; Perrillo, R. Hepatitis B Virus Replication in Diverse Cell Types During Chronic Hepatitis B Virus Infection. Hepatology 1993, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Barreiro, P.; Cachay, E.; Kottilil, S.; Fernandez-Montero, J.V.; de Mendoza, C. Advances in Hepatitis B Therapeutics. Ther. Adv. Infect. Dis. 2020, 7, 2049936120965027. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.F. Toward a Functional Cure for Hepatitis B. Gut Liver 2024, 18, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-Associated Hepatitis Not Due to Viral Hepatitis Type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Preciado, M.V.; Valva, P.; Escobar-Gutierrez, A.; Rahal, P.; Ruiz-Tovar, K.; Yamasaki, L.; Vazquez-Chacon, C.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Fonseca-Coronado, S.; et al. Hepatitis C Virus Molecular Evolution: Transmission, Disease Progression and Antiviral Therapy. World J. Gastroenterol. 2014, 20, 15992–16013. [Google Scholar] [CrossRef] [PubMed]
- Vo-Quang, E.; Pawlotsky, J.M. ‘Unusual’ HCV Genotype Subtypes: Origin, Distribution, Sensitivity to Direct-Acting Antiviral Drugs and Behaviour on Antiviral Treatment and Retreatment. Gut 2024, 73, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and Natural History of HCV Infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Butt, M.B.; Alfayad, M.; Saqib, S.; Khan, M.A.; Ahmad, M.; Khan, M.A.; Elmitwally, N.S. Diagnosing the Stage of Hepatitis C Using Machine Learning. J. Healthc. Eng. 2021, 2021, 8062410. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, F.; Beisel, C.; Binder, M. Hepatitis C Virus and Hepatocellular Carcinoma: Carcinogenesis in the Era of Direct-Acting Antivirals. Curr. Opin. Virol. 2024, 67, 101423. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular Mechanisms of Hepatitis C Virus–Induced Hepatocellular Carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, S.; Bacchi-Reggiani, L.; de Biase, D.; Fornelli, A.; Masetti, M.; Tura, A.; Grizzi, F.; Zanello, M.; Mastrangelo, L.; Lombardi, R.; et al. Possible association between hepatitis C virus and malignancies different from hepatocellular carcinoma: A systematic review. World J. Gastroenterol. 2015, 21, 12896–12953. [Google Scholar] [CrossRef] [PubMed]
- Darvishian, M.; Tang, T.; Wong, S.; Binka, M.; Yu, A.; Alvarez, M.; Alexander Velásquez García, H.; Adu, P.A.; Jeong, D.; Bartlett, S.; et al. Chronic hepatitis C infection is associated with higher incidence of extrahepatic cancers in a Canadian population based cohort. Front. Oncol. 2022, 12, 983238. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, D.N.; Foia, L.; Toma, V.; Surdu, A.; Herascu, E.; Popescu, D.M.; Surlin, P.; Vere, C.C.; Rogoveanu, I. Hepatitis C Infection and Periodontal Disease: Is There a Common Immunological Link? J. Immunol. Res. 2018, 2018, 8720101. [Google Scholar] [CrossRef] [PubMed]
- Carrozzo, M. Oral manifestations of hepatitis C virus infection. World J. Gastroenterol. 2014, 20, 7534. [Google Scholar] [CrossRef] [PubMed]
- Manfrè, V.; Chatzis, L.G.; Cafaro, G.; Fonzetti, S.; Calvacchi, S.; Fulvio, G.; Navarro Garcia, I.C.; La Rocca, G.; Ferro, F.; Perricone, C.; et al. Sjögren’s syndrome: One year in review 2022. Clin. Exp. Rheumatol. 2022, 40, 2211–2224. [Google Scholar] [CrossRef] [PubMed]
- Mahale, P.; Sturgis, E.M.; Tweardy, D.J.; Ariza-Heredia, E.J.; Torres, H.A. Association between hepatitis C virus and head and neck cancers. J. Natl. Cancer Inst. 2016, 108, djw035. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Sata, M.; Noguchi, S.; Seno’o, T.; Kinoshita, M.; Kameyama, T.; Ueno, T. Detection of hepatitis C virus RNA in oral lichen planus and oral cancer tissues. J. Oral Pathol. Med. 2000, 29, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Borsetto, D.; Fussey, J.; Fabris, L.; Bandolin, L.; Gaudioso, P.; Phillips, V.; Polesel, J.; Boscolo-Rizzo, P. HCV infection and the risk of head and neck cancer: A meta-analysis. Oral Oncol. 2020, 109, 104869. [Google Scholar] [CrossRef] [PubMed]
- de Brito Rangel, J.; Thuler, L.C.S.; da Cunha Pinto, J.F. Prevalence of hepatitis C virus infection and its impact on the prognosis of head and neck cancer patients. Oral Oncol. 2018, 87, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Maasoumy, B. Breakthroughs in hepatitis C research: From discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Adugna, A. Therapeutic strategies and promising vaccine for hepatitis C virus infection. Immun. Inflamm. Dis. 2023, 11, e977. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and burden of human papillomavirus and related diseases, molecular pathogenesis, and vaccine evaluation. Front. Public Health 2021, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Becker, G.L.; Jackson, J.B.; Rysavy, M.B. Human papillomavirus and associated cancers: A review. Viruses 2024, 16, 680. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Hahn, E.; Salunkhe, R.; Barcelona, M.V.N.; O’Sullivan, B. Are we ready for deintensification in human papillomavirus-positive oropharyngeal carcinomas? Curr. Opin. Otolaryngol. Head Neck Surg. 2023, 31, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.E.; Chiocca, S. Human papillomavirus as a driver of head and neck cancers. Br. J. Cancer 2020, 122, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Oyouni, A.A.A. Human papillomavirus in cancer: Infection, disease transmission, and progress in vaccines. J. Infect. Public Health 2023, 16, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Petca, A.; Borislavschi, A.; Zvanca, M.; Petca, R.C.; Sandru, F.; Dumitrascu, M. Non-sexual HPV transmission and role of vaccination for a better future (Review). Exp. Ther. Med. 2020, 20, 186. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.; Santonocito, S.; Bruno, M.T.; Polizzi, A.; Mastroianni, A.; Chaurasia, A.; Isola, G. Oral and periodontal manifestation related during human papilloma virus infections: Update on early prognostic factors. Heliyon 2024, 10, e31061. [Google Scholar] [CrossRef] [PubMed]
- Katirachi, S.K.; Grønlund, M.P.; Jakobsen, K.K.; Grønhøj, C.; von Buchwald, C. The prevalence of HPV in oral cavity squamous cell carcinoma. Viruses 2023, 15, 451. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Narwal, M.; Min, S.Y.; Keam, B.; Kang, H. Squamous cell carcinoma of head and neck: What internists should know. Korean J. Intern. Med. 2020, 35, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Meites, E.; Wilkin, T.J.; Markowitz, L.E. Review of human papillomavirus (HPV) burden and HPV vaccination for gay, bisexual, and other men who have sex with men and transgender women in the United States. Hum. Vaccines Immunother. 2022, 18, 2016007. [Google Scholar] [CrossRef] [PubMed]
- Bruni, L.; Albero, G.; Rowley, J.; Alemany, L.; Arbyn, M.; Giuliano, A.R.; Markowitz, L.E.; Broutet, N.; Taylor, M. Global and regional estimates of genital human papillomavirus prevalence among men: A systematic review and meta-analysis. Lancet Glob. Health 2023, 11, e1345–e1362. [Google Scholar] [CrossRef] [PubMed]
- Mesia, R.; Iglesias, L.; Lambea, J.; Martínez-Trufero, J.; Soria, A.; Taberna, M.; Trigo, J.; Chaves, M.; García-Castaño, A.; Cruz, J. SEOM clinical guidelines for the treatment of head and neck cancer (2020). Clin. Transl. Oncol. 2021, 23, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Parmar, A.; Macluskey, M.; Mc Goldrick, N.; Conway, D.I.; Glenny, A.M.; Clarkson, J.E.; Worthington, H.V.; Chan, K.K. Interventions for the treatment of oral cavity and oropharyngeal cancer: Chemotherapy. Cochrane Database Syst. Rev. 2021, 12, CD006386. [Google Scholar] [CrossRef] [PubMed]
- Irani, S. New insights into oral cancer—Risk factors and prevention: A review of literature. Int. J. Prev. Med. 2020, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka, M.; Klussmann, J.P.; San Giorgi, M.R.; Wuerdemann, N.; Dikkers, F.G. Oral and laryngeal HPV infection: Incidence, prevalence and risk factors, with special regard to concurrent infection in head, neck and genitals. Vaccine 2021, 39, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Brenner, N.; Mentzer, A.J.; Hill, M.; Almond, R.; Allen, N.; Pawlita, M.; Waterboer, T. Characterization of human papillomavirus (HPV) 16 E6 seropositive individuals without HPV-associated malignancies after 10 years of follow-up in the UK Biobank. EBioMedicine 2020, 62, 103123. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Grønhøj, C.; Jensen, D.H.; Carlander, A.F.; Agander, T.; Kiss, K.; Olsen, C.; Baandrup, L.; Nielsen, F.C.; Andersen, E.; et al. The current epidemic of HPV-associated oropharyngeal cancer: An 18-year Danish population-based study with 2169 patients. Eur. J. Cancer 2020, 134, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Sichero, L.; Gonçalves, M.G.; Bettoni, F.; Coser, E.M.; Mota, G.; Nunes, R.A.L.; Mercante, A.M.D.C.; Natalino, R.; Uno, M.; Ferreira Alves, M.J.; et al. Detection of serum biomarkers of HPV-16 driven oropharynx and oral cavity cancer in Brazil. Oral Oncol. 2024, 149, 106676. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; Rollo, F.; Donà, M.G.; Garbuglia, A.R. Human Papillomavirus Oral Infection: Review of Methodological Aspects and Epidemiology. Pathogens 2021, 10, 1411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palmieri, A.; Lauritano, D.; Pellati, A.; Scapoli, L.; Arcuri, C.; Baggi, L.; Gatto, R.; Carinci, F. Prevalence of Human Papillomavirus in the Oropharynx of Healthy Individuals in an Italian Population. J. Clin. Med. 2022, 11, 1935. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mello, J.B.; Carvalho, B.F.S.; Dacio, S.F.; Silva, K.C.; Monteiro, M.J.F. Associação do HPV com lesões orais e orofaringe: Revisão sistemática. Res. Soc. Dev. 2024, 13, e6213345255. [Google Scholar] [CrossRef]
- Burger-Calderon, R.; Webster-Cyriaque, J. Human BK Polyomavirus—The Potential for Head and Neck Malignancy and Disease. Cancers 2015, 7, 1244–1270. [Google Scholar] [CrossRef] [PubMed]
- Prado, J.C.M.; Monezi, T.A.; Amorim, A.T.; Lino, V.; Paladino, A.; Boccardo, E. Human polyomaviruses and cancer: An overview. Clinics 2018, 73, e558s. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Koralnik, I.J. JC, BK, and Other Polyomaviruses. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1807–1814.e3. [Google Scholar] [CrossRef]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.N.; Cortinhas, J.M.; Dos Santos, M.B.; Queiroz, M.A.F.; da Silva, A.N.M.R.; Cayres-Vallinoto, I.M.V.; Vallinoto, A.C.R. Detection of Human polyomavirus 2 (HPyV2) in oyster samples in northern Brazil. Virol. J. 2020, 17, 85. [Google Scholar] [CrossRef] [PubMed]
- Hämetoja, H.; Hagström, J.; Haglund, C.; Bäck, L.; Mäkitie, A.; Syrjänen, S. Polyomavirus JCPyV infrequently detectable in adenoid cystic carcinoma of the oral cavity and the airways. Virchows Arch. 2019, 475, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Polz, D.; Morshed, K.; Stec, A.; Podsiadło, Ł.; Polz-Dacewicz, M. Do polyomavirus hominis strains BK and JC play a role in oral squamous cell carcinoma? Ann. Agric. Environ. Med. 2015, 22, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Passerini, S.; Messina, S.; Moens, U.; Pietropaolo, V. Merkel Cell Polyomavirus (MCPyV) and Its Possible Role in Head and Neck Cancers. Biomedicines 2025, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Grimm, J.; Willmes, C.; Weinkam, R.; Becker, J.C.; Schrama, D. Merkel cell carcinoma and Merkel cell polyomavirus: Evidence for hit-and-run oncogenesis. J. Investig. Dermatol. 2012, 132, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Vilkin, A.; Ronen, Z.; Levi, Z.; Morgenstern, S.; Halpern, M.; Niv, Y. Presence of JC Virus DNA in the Tumor Tissue and Normal Mucosa of Patients with Sporadic Colorectal Cancer (CRC) or with Positive Family History and Bethesda Criteria. Dig. Dis. Sci. 2012, 57, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C.; Xue, H.; Zhang, C.Y. The oncogenic roles of JC polyomavirus in cancer. Front. Oncol. 2022, 12, 976577. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; White, M.K.; Khalili, K. Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J. Neuropathol. Exp. Neurol. 2008, 67, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Levican, J.; Acevedo, M.; León, O.; Gaggero, A.; Aguayo, F. Role of BK human polyomavirus in cancer. Infect. Agents Cancer 2018, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Passerini, S.; Babini, G.; Merenda, E.; Carletti, R.; Scribano, D.; Rosa, L.; Conte, A.L.; Moens, U.; Ottolenghi, L.; Romeo, U.; et al. Merkel Cell Polyomavirus in the Context of Oral Squamous Cell Carcinoma and Oral Potentially Malignant Disorders. Biomedicines 2024, 12, 709. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.T.; Sobrinho, J.S.; Pimentel, D.R.N.; López, R.V.M.; Uno, M.; Silva, A.C.P.R.E.; Santos-Silva, A.R.; Sichero, L.; Tomimori, J. Prevalence of Human Polyomaviruses in Oral and Oropharyngeal Squamous Cell Carcinoma From Patients Treated at a Cancer Center in São Paulo, Brazil. J. Med. Virol. 2024, 96, e70041. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; You, J. Molecular mechanisms of Merkel cell polyomavirus transformation and replication. Annu. Rev. Virol. 2020, 7, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Robaina, T.F.; Mendes, G.S.; Benati, F.J.; Pena, G.A.; Silva, R.C.; Montes, M.A.; Otero, R.; Castro, G.F.; Câmara, F.P.; Santos, N. Polyomavirus in saliva of HIV-infected children, Brazil. Emerg. Infect. Dis. 2013, 19, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Martelli, F.; Mencarini, J.; Rocca, A.; Malva, N.D.; Bartolozzi, D.; Giannecchini, S. Polyomavirus microRNA in saliva reveals persistent infectious status in the oral cavity. Virus Res. 2018, 249, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Hashida, Y.; Higuchi, T.; Ohno, S.; Sento, S.; Sasabe, E.; Murakami, I.; Yamamoto, T.; Daibata, M. Detection of Merkel cell polyomavirus in multiple primary oral squamous cell carcinomas. Odontology 2023, 111, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Al-Talib, M.; Welberry-Smith, M.; Macdonald, A.; Griffin, S. BK Polyomavirus-associated nephropathy—Diagnostic and treatment standard. Nephrol. Dial. Transplant. 2025, 40, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Robaina, T.F.; Mendes, G.S.; Benati, F.J.; Pena, G.A.; Silva, R.C.; Montes, M.A.; Janini, M.E.; Câmara, F.P.; Santos, N. Shedding of polyomavirus in the saliva of immunocompetent individuals. J. Med. Virol. 2013, 85, 144–148. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C.; Wu, X.; Wang, M.; Guo, Y.; Zhang, W.; Sun, Y.; Stha, A. Expression and methylation of Dickkopf-1 in the pathogenesis and malignant transformation of oral submucous fibrosis. J. Oral Pathol. Med. 2020, 49, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, X.; Peng, X.; Li, M.; Ren, B.; Cheng, G.; Cheng, L. Porphyromonas gingivalis promotes immunoevasion of oral cancer by protecting cancer from macrophage attack. J. Immunol. 2020, 205, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Roider, J.; Ngoepe, A.; Muenchhoff, M.; Adland, E.; Groll, A.; Ndung’u, T.; Kløverpris, H.; Goulder, P.; Leslie, A. Increased regulatory T-cell activity and enhanced T-cell homeostatic signaling in slow progressing HIV-infected children. Front. Immunol. 2019, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Alkan, B.; Tuncer, M.A.; İnkaya, A.Ç. Advances in virus-specific T-cell therapy for polyomavirus infections: A comprehensive review. Int. J. Antimicrob. Agents 2024, 64, 107333. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.S.; Peh, C.A.; Nolan, J.; Bannister, K.M.; Clarkson, A.R.; Faull, R.J. BK transplant nephropathy successfully treated with cidofovir. Nephrol. Dial. Transplant. 2003, 18, 1013–1014. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, D.R.; Vandooren, A.K.; Lerut, E.; Evenepoel, P.; Claes, K.; Snoeck, R.; Naesens, L.; Vanrenterghem, Y. Adjuvant low-dose cidofovir therapy for BK polyomavirus interstitial nephritis in renal transplant recipients. Am. J. Transplant. 2005, 5, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Vats, A.; Shapiro, R.; Singh Randhawa, P.; Scantlebury, V.; Tuzuner, A.; Saxena, M.; Moritz, M.L.; Beattie, T.J.; Gonwa, T.; Green, M.D.; et al. Quantitative viral load monitoring and cidofovir therapy for the management of BK virus-associated nephropathy in children and adults. Transplantation 2003, 75, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Faguer, S.; Hirsch, H.H.; Kamar, N.; Guilbeau-Frugier, C.; Ribes, D.; Guitard, J.; Esposito, L.; Cointault, O.; Modesto, A.; Lavit, M.; et al. Leflunomide treatment for polyomavirus BK-associated nephropathy after kidney transplantation. Transpl. Int. 2007, 20, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Josephson, M.A.; Gillen, D.; Javaid, B.; Kadambi, P.; Meehan, S.; Foster, P.; Harland, R.; Thistlethwaite, R.J.; Garfinkel, M.; Atwood, W.; et al. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation 2006, 81, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Krisl, J.C.; Taber, D.J.; Pilch, N.; Chavin, K.; Bratton, C.; Thomas, B.; McGillicuddy, J.; Baliga, P. Leflunomide efficacy and pharmacodynamics for the treatment of BK viral infection. Clin. J. Am. Soc. Nephrol. 2012, 7, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Gabardi, S.; Waikar, S.S.; Martin, S.; Roberts, K.; Chen, J.; Borgi, L.; Sheashaa, H.; Dyer, C.; Malek, S.K.; Tullius, S.G.; et al. Evaluation of fluoroquinolones for the prevention of BK viremia after renal transplantation. Clin. J. Am. Soc. Nephrol. 2010, 5, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.Y.; Chan, M.T.; Yuen, K.Y.; Cheng, V.C.; Chan, K.H.; Wong, C.L.; Liang, R.; Lie, A.K.; Kwong, Y.L. Ciprofloxacin decreased polyoma BK virus load in patients who underwent allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 2005, 40, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Peretti, A.; Scorpio, D.G.; Kong, W.P.; Pang, Y.S.; McCarthy, M.P.; Ren, K.; Jackson, M.; Graham, B.S.; Buck, C.B.; McTamney, P.M.; et al. A multivalent polyomavirus vaccine elicits durable neutralizing antibody responses in macaques. Vaccine 2023, 41, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Kanse, S.; Khandelwal, M.; Pandey, R.K.; Khokhar, M.; Desai, N.; Kumbhar, B.V. Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus. Vaccines 2023, 11, 1182. [Google Scholar] [CrossRef] [PubMed]
- Millen, S.; Thoma-Kress, A.K. Milk Transmission of HTLV-1 and the Need for Innovative Prevention Strategies. Front. Med. 2022, 9, 867147. [Google Scholar] [CrossRef] [PubMed]
- Carod-Artal, F.J. Immunopathogenesis and treatment of the myelopathy associated to the HTLV-I virus. Rev. Neurol. 2009, 48, 147–155. [Google Scholar]
- Forlani, G.; Shallak, M.; Accolla, R.S.; Romanelli, M.G. HTLV-1 Infection and Pathogenesis: New Insights from Cellular and Animal Models. Int. J. Mol. Sci. 2021, 22, 8001. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, S.; Rezaee, S.A.; Farjami, Z.; Ebrahimi, N.; Abdullabass, H.K.; Ibrahim Jebur, M.I.; Rafatpanah, H.; Akbarin, M.M. HTLV, a multi organ oncovirus. Microb. Pathog. 2022, 169, 105622. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, S.; Enriquez-Vera, D.; Jahan, M.I.; Sugata, K.; Satou, Y. Understanding the Immunopathology of HTLV-1-Associated Adult T-Cell Leukemia/Lymphoma: A Comprehensive Review. Biomolecules 2023, 13, 1543. [Google Scholar] [CrossRef] [PubMed]
- Kamoi, K.; Uchimaru, K.; Tojo, A.; Watanabe, T.; Ohno-Matsui, K. HTLV-1 uveitis and Graves’ disease presenting with sudden onset of blurred vision. Lancet 2022, 399, 60. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Harhaj, E.W. Mechanisms of oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S. The road to HTLV-1-induced leukemia by following the subcellular localization of HTLV-1-encoded HBZ protein. Front. Immunol. 2022, 13, 940131. [Google Scholar] [CrossRef] [PubMed]
- Eusebio-Ponce, E.; Anguita, E.; Paulino-Ramirez, R.; Candel, F.J. HTLV-1 infection: An emerging risk. Pathogenesis, epidemiology, diagnosis and associated diseases. Rev. Esp. Quimioter. 2019, 32, 485–496. [Google Scholar] [PubMed]
- Chen, I.S.Y.; Slamon, D.J.; Rosenblatt, J.D.; Shah, N.P.; Quan, S.G.; Wachsman, W. The x gene is essential for HTLV replication. Science 1985, 229, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Sodroski, J.G.; Rosen, C.A.; Haseltine, W.A. Trans-acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science 1984, 225, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Ernzen, K.J.; Panfil, A.R. Regulation of HTLV-1 transformation. Biosci. Rep. 2022, 42, BSR20211921. [Google Scholar] [CrossRef] [PubMed]
- Mahieux, R.; Pise-Masison, C.A.; Nicot, C.; Green, P.; Hall, W.W.; Brady, J.N. Inactivation of p53 by HTLV type 1 and HTLV type 2 Tax trans-activators. AIDS Res. Hum. Retroviruses 2000, 16, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Tavakolian, S.; Goudarzi, H.; Faghihloo, E. Cyclin-dependent kinases and CDK inhibitors in virus-associated cancers. Infect. Agent. Cancer 2020, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Kang, X.; Niu, Y.; Zhao, T.; Wang, H. PCBP1 interacts with the HTLV-1 Tax oncoprotein to potentiate NF-κB activation. Front. Immunol. 2024, 15, 1375168. [Google Scholar] [CrossRef] [PubMed]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV Tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takahashi, C.; Yamaoka, S.; Nosaka, T.; Maki, M.; Hatanaka, M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Daenthanasanmak, A.; Bamford, R.N.; Yoshioka, M.; Yang, S.M.; Homan, P.; Karim, B.; Bryant, B.R.; Petrus, M.N.; Thomas, C.J.; Green, P.L.; et al. Triple combination of BET plus PI3K and NF-κB inhibitors exhibit synergistic activity in adult T-cell leukemia/lymphoma. Blood Adv. 2022, 6, 2346–2360. [Google Scholar] [CrossRef] [PubMed]
- Kannagi, M.; Harada, S.; Maruyama, I.; Inoko, H.; Igarashi, H.; Kuwashima, G.; Sato, S.; Morita, M.; Kidokoro, M.; Sugimoto, M. Predominant recognition of human T-cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int. Immunol. 1991, 3, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T. The role of HBZ in HTLV-1-induced oncogenesis. Viruses 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Yasunaga, J.-I.; Taniguchi, Y.; Tamiya, S.; Nakahata, T.; Matsuoka, M. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J. Virol. 2007, 81, 5714–5723. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kubota, R.; Tara, M.; Izumo, S.; Osame, M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood 2001, 97, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Yeh, C.; Bai, X.T.; Nicot, C. The HTLV-I oncoprotein Tax inactivates the tumor suppressor FBXW7. J. Virol. 2024, 98, e0040524. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Nosaka, K.; Yasunaga, J.; Maeda, M.; Mueller, N.; Okayama, A.; Matsuoka, M. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2005, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Domingues, W.; Folgosi, V.Â.; Sanabani, S.S.; Leite Junior, P.D.; Assone, T.; Casseb, J. Novel approaches for HTLV-1 therapy: Innovative applications of CRISPR-Cas9. Rev. Inst. Med. Trop. Sao Paulo 2024, 66, e48. [Google Scholar] [CrossRef] [PubMed]
- Gaudray, G.; Gachon, F.; Basbous, J.; Biard-Piechaczyk, M.; Devaux, C.; Mesnard, J.-M. The complementary strand of the Human T-cell Leukemia Virus Type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J. Virol. 2002, 76, 12813–12822. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Satou, Y.; Sugata, K.; Miyazato, P.; Green, P.L.; Imamura, T.; Matsuoka, M. HTLV-1 bZIP factor enhances TGF-β signaling through p300 coactivator. Blood 2011, 118, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kannian, P.; Dissinger, N.; Haines, R.; Niewiesk, S.; Green, P.L. Human T-cell leukemia virus type 2 antisense viral protein 2 is dispensable for in vitro immortalization but functions to repress early virus replication in vivo. J. Virol. 2012, 86, 8412–8421. [Google Scholar] [CrossRef] [PubMed]
- Polakowski, N.; Gregory, H.; Mesnard, J.-M.; Lemasson, I. Expression of a protein involved in bone resorption, Dkk1, is activated by HTLV-1 bZIP factor through its activation domain. Retrovirology 2010, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Takahashi, M.; Fukunaga, Y.; Takahashi, H. HTLV-I-infected breast milk macrophages inhibit monocyte differentiation to dendritic cells. Viral Immunol. 2012, 25, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Kemeter, L.M.; Birzer, A.; Heym, S.; Thoma-Kress, A.K. Milk transmission of mammalian retroviruses. Microorganisms 2023, 11, 1777. [Google Scholar] [CrossRef] [PubMed]
- Alais, S.; Pasquier, A.; Jegado, B.; Journo, C.; Rua, R.; Gessain, A.; Tobaly-Tapiero, J.; Lacoste, R.; Turpin, J.; Mahieux, R. STLV-1 co-infection is correlated with an increased SFV proviral load in the peripheral blood of SFV/STLV-1 naturally infected non-human primates. PLoS Negl. Trop. Dis. 2018, 12, e0006812. [Google Scholar] [CrossRef] [PubMed]
- Woo, T.; Rosadas, C.; Ijaz, S.; Dicks, S.; Tosswill, J.H.C.; Tedder, R.S.; Taylor, G.P. Noninvasive detection of antibodies to Human T-cell Lymphotropic Virus types 1 and 2 by use of oral fluid. J. Clin. Microbiol. 2019, 57, e01179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chu, A. Sjögren’s syndrome and viral infections. Rheumatol. Ther. 2021, 8, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Falcão, G.G.V.S.C.; Sarmento, V.A.; Dutra, B.S.; Russoni, B.; de Oliveira, L.S.; Costa, D.A.; Brites, C.; Bouqout, J.E.; Lins-Kusterer, L. Oral health and quality of life of people living with human T-cell leukemia virus-1 in Salvador, Brazil: A cross-sectional study. Clin. Oral Investig. 2022, 26, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Casseb, J.; Penalva-de-Oliveira, A.; de Paiva, M.; Watanuki, F.; Ortega, K. Oral manifestations of human T-cell lymphotropic virus infection in adult patients from Brazil. Oral Dis. 2010, 16, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Lins, L.; de Carvalho, V.J.; de Almeida Rego, F.F.; Azevedo, R.; Kashima, S.; Gallazi, V.N.; Xavier, M.T.; Galvão-Castro, B.; Alcantara, L.C., Jr. Oral health profile in patients infected with HTLV-1: Clinical findings, proviral load, and molecular analysis from HTLV-1 in saliva. J. Med. Virol. 2012, 84, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Plemons, J.M.; Al-Hashimi, I.; Marek, C.L. Managing xerostomia and salivary gland hypofunction. J. Am. Dent. Assoc. 2014, 145, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Rafii, S.; Wu, M.H.; Wijelath, E.S.; Yu, C.; Ishida, A.; Fujita, Y.; Kothari, S.; Mohle, R.; Sauvage, L.R.; et al. Evidence for circulating bone marrow-derived endothelial cells. Blood 1998, 92, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yamanashi, H.; Kitamura, M.; Furugen, R.; Iwasaki, T.; Fukuda, H.; Hayashida, H.; Kawasaki, K.; Kiyoura, K.; Kawashiri, S.Y.; et al. Association between human T cell leukemia virus 1 (HTLV-1) infection and advanced periodontitis in relation to hematopoietic activity among elderly participants: A cross-sectional study. Environ. Health Prev. Med. 2019, 24, 42. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shrivastava, S.; Ray, R.M.; Holguin, L.; Echavarria, L.; Grepo, N.; Scott, T.A.; Burnett, J.; Morris, K.V. Exosome-mediated stable epigenetic repression of HIV-1. Nat. Commun. 2021, 12, 5541. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Hirons, A.; Doerflinger, M.; Morris, K.V.; Ledger, S.; Purcell, D.F.J.; Kelleher, A.D.; Ahlenstiel, C.L. Current state of therapeutics for HTLV-1. Viruses 2024, 16, 1616. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Utsunomiya, A.; Fukuda, H.; Shibata, T.; Fukushima, T.; Takatsuka, Y.; Ikeda, S.; Masuda, M.; Nagoshi, H.; Ueda, R.; et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J. Clin. Oncol. 2007, 25, 5458–5464. [Google Scholar] [CrossRef] [PubMed]
- Stuver, R.; Horwitz, S.M.; Epstein-Peterson, Z.D. Treatment of adult T-cell leukemia/lymphoma: Established paradigms and emerging directions. Curr. Treat. Options Oncol. 2023, 24, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.A. Advances in the treatment of HTLV-1-associated adult T-cell leukemia lymphoma. Curr. Opin. Virol. 2023, 58, 101289. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, G.; Rodríguez, S.M.; de Brogniez, A.; Gillet, N.; Golime, R.; Burny, A.; Jaworski, J.P.; Alvarez, I.; Vagnoni, L.; Trono, K.; et al. Vaccination against δ-retroviruses: The bovine leukemia virus paradigm. Viruses 2014, 6, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Brasil. Mistério da Saúde, Secretaria de Vigilância em Saúde, Departamento de Doenças de Condições Crônicas e Infecções Sexualmente Transmissíveis. Protocolo Clínico e Diretrizes Terapêuticas para Atenção Integral às Pessoas com Infecções Sexualmente Transmissíveis—IST. 2022. Available online: https://www.gov.br/aids/pt-br/central-de-conteudo/pcdts/2022/ist/pcdt-ist-2022_isbn-1.pdf (accessed on 10 July 2025).
- Nahmias, A.J.; Dowdle, W.R. Antigenic and biologic differences in herpesvirus hominis. Prog. Med. Virol. 1968, 10, 110–159. [Google Scholar] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cell. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Norberg, P.; Bergström, T.; Rekabdar, E.; Lindh, M.; Liljeqvist, J.-A. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J. Virol. 2004, 78, 10755–10764. [Google Scholar] [CrossRef] [PubMed]
- Norberg, P.; Bergström, T.; Liljeqvist, J.-A. Genotyping of clinical herpes simplex virus type 1 isolates by use of restriction enzymes. J. Clin. Microbiol. 2006, 44, 4511–4514. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef] [PubMed]
- de Souza Carneiro, V.C.; Pereira, J.G.; de Paula, V.S. Family Herpesviridae and neuroinfections: Current status and research in progress. Mem. Inst. Oswaldo Cruz 2022, 117, e220200. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.G.; Leon, L.A.A.; de Almeida, N.A.A.; Raposo-Vedovi, J.V.; Fontes-Dantas, F.L.; Farinhas, J.G.D.; Pereira, V.C.S.R.; Alves-Leon, S.V.; de Paula, V.S. Higher frequency of Human herpesvirus-6 (HHV-6) viral DNA simultaneously with low frequency of Epstein-Barr virus (EBV) viral DNA in a cohort of multiple sclerosis patients from Rio de Janeiro, Brazil. Mult. Scler. Relat. Disord. 2023, 76, 104747. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5, e22766. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights Into Proposed Interrelationships With Neurodegenerative Disorders. Front. Cell. Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- von Stebut, J.; Heiland, M.; Preissner, R.; Rendenbach, C.; Preissner, S. Association of Herpes simplex infection with significantly increased risk of head and neck cancer: Real-world evidence of about 500,000 patients. Int. J. Dermatol. 2024, 63, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Jalouli, J.; Ibrahim, S.O.; Sapkota, D.; Jalouli, M.M.; Vasstrand, E.N.; Hirsch, J.M.; Larsson, P.A. Presence of human papilloma virus, herpes simplex virus and Epstein-Barr virus DNA in oral biopsies from Sudanese patients with regard to toombak use. J. Oral Pathol. Med. 2010, 39, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wang, D.; Zhao, X.; Feng, Z.; Chen, Q.; Shen, Y. The Dilemma of HSV-1 Oncolytic Virus Delivery: The Method Choice and Hurdles. Int. J. Mol. Sci. 2023, 24, 3681. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kumar, S.P.; Chandy, M.L.; Shanavas, M.; Khan, S.; Suresh, K.V. Pathogenesis and life cycle of herpes simplex virus infection-stages of primary, latency and recurrence. J. Oral Maxillofac. Surg. Med. Pathol. 2016, 28, 350–353. [Google Scholar] [CrossRef]
- Saha, D.; Wakimoto, H.; Rabkin, S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016, 21, 26–34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cox, M.; Maitlan, N.; Scully, C. Human herpes simplex-1 and papillomavirus type 16 homologous DNA sequences in normal, potentially malignant and malignant oral mucosa. Eur. J. Cancer B Oral Oncol. 1993, 29, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Starr, J.R.; Daling, J.R.; Fitzgibbons, E.D.; Madeleine, M.M.; Ashley, R.; Galloway, D.A.; Schwartz, S.M. Serologic evidence of herpes simplex virus 1 infection and oropharyngeal cancer risk. Cancer Res. 2001, 61, 8459–8464, Erratum in Cancer Res. 2002, 62, 2445. [Google Scholar] [PubMed]
- Slavkin, H.C. The human genome, implications for oral health and diseases, and dental education. J. Dent. Educ. 2001, 65, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Kassim, K.H.; Daley, T.D. Herpes simplex virus type 1 proteins in human oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. 1988, 65, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Eglin, R.P.; Scully, C.; Lehner, T.; Ward-Booth, P.; McGregor, I.A. Detection of RNA complementary to herpes simplex virus in human oral squamous cell carcinoma. Lancet 1983, 2, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; McDougall, J.K. The oncogenic potential of herpes simplex viruses: Evidence for a ‘hit-and-run’ mechanism. Nature 1983, 302, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, L. Transformation and Mutagenic Effects Induced by Herpes Simplex Virus Types 1 and 2, In DNA Tumor Viruses: Oncogenic Mechanisms; Barbanti-Brodano, G., Bendinelli, M., Friedman, H., Eds.; Plenum Press: New York, NY, USA, 1995; pp. 253–280. [Google Scholar] [CrossRef]
- Jain, M. Assesment of Correlation of Herpes Simplex Virus-1 with Oral Cancer and Precancer- A Comparative Study. J. Clin. Diagn. Res. 2016, 10, ZC14–ZC17. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Koh, L.W.; Tsai, J.H.; Tsai, C.H.; Wong, E.F.; Lin, S.J.; Yang, C.C. Involvement of viral and chemical factors with oral cancer in Taiwan. Jpn. J. Clin. Oncol. 2004, 34, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.; Johansson, S.L.; Vahine, A.; Hirsch, J.M. Snuff tumorigenesis: Effects of long-term snuff administration after initiation with 4-nitroquinoline-N-oxide and herpes simplex virus type 1. J. Oral Pathol. Med. 1989, 18 (Suppl. 1), 187–192. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, J.C.; Martin Kast, W.; Flexman, J.P.; McCallum, D.; French, M.A.; Phillips, M. Syndemic synergy of HPV and other sexually transmitted pathogens in the development of high-grade anal squamous intraepithelial lesions. Papillomavirus Res. 2017, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M. Keratin, a dual role in herpes simplex virus pathogenesis. J. Clin. Virol. 2006, 35, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Fabbri, C.; D’Accolti, M.; Soffritti, I.; Bassi, C.; Mazzacane, S.; Franchi, M. Defining the oral microbiome by whole-genome sequencing and resistome analysis: The complexity of the healthy picture. BMC Microbiol. 2020, 20, 120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kilian, M. The oral microbiome—Friend or foe? Eur. J. Oral Sci. 2018, 126 (Suppl. 1), 5–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, X.; Yu, H.; Zhou, H.; Xu, S. Oral microbiota as promising diagnostic biomarkers for gastrointestinal cancer: A systematic review. OncoTargets Ther. 2019, 12, 11131–11144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irfan, M.; Delgado, R.Z.R.; Frias-Lopez, J. The oral microbiome and cancer. Front. Immunol. 2020, 11, 591088. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shillitoe, E.J.; Silverman, S. Oral cancer and herpes simplex virus—A review. Oral Surg. Oral Med. Oral Pathol. 1979, 48, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- The Clinical Evaluation of Antiviral Agents. Report of a Working Party of the British Society for Antimicrobial Chemotherapy and the Society of Pharmaceutical Medicine. J. Antimicrob. Chemother. 1997, 40, 1–67. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Selective anti-herpesvirus agents. Antivir. Chem. Chemother. 2013, 23, 93–101. [Google Scholar] [CrossRef] [PubMed]
- James, S.H.; Prichard, M.N. Current and future therapies for herpes simplex virus infections: Mechanism of action and drug resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Han, L.; Shi, C.; Li, Y.; Qian, S.; Feng, Z.; Yu, L. An updated review of HSV-1 infection-associated diseases and treatment, vaccine development, and vector therapy application. Virulence 2024, 15, 2425744. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Russell, L.; Peng, K.-W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Ghouse, S.M.; Martuza, R.L.; Rabkin, S.D. In Situ Cancer Vaccination and Immunovirotherapy Using Oncolytic HSV. Viruses 2021, 13, 1740. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef] [PubMed]
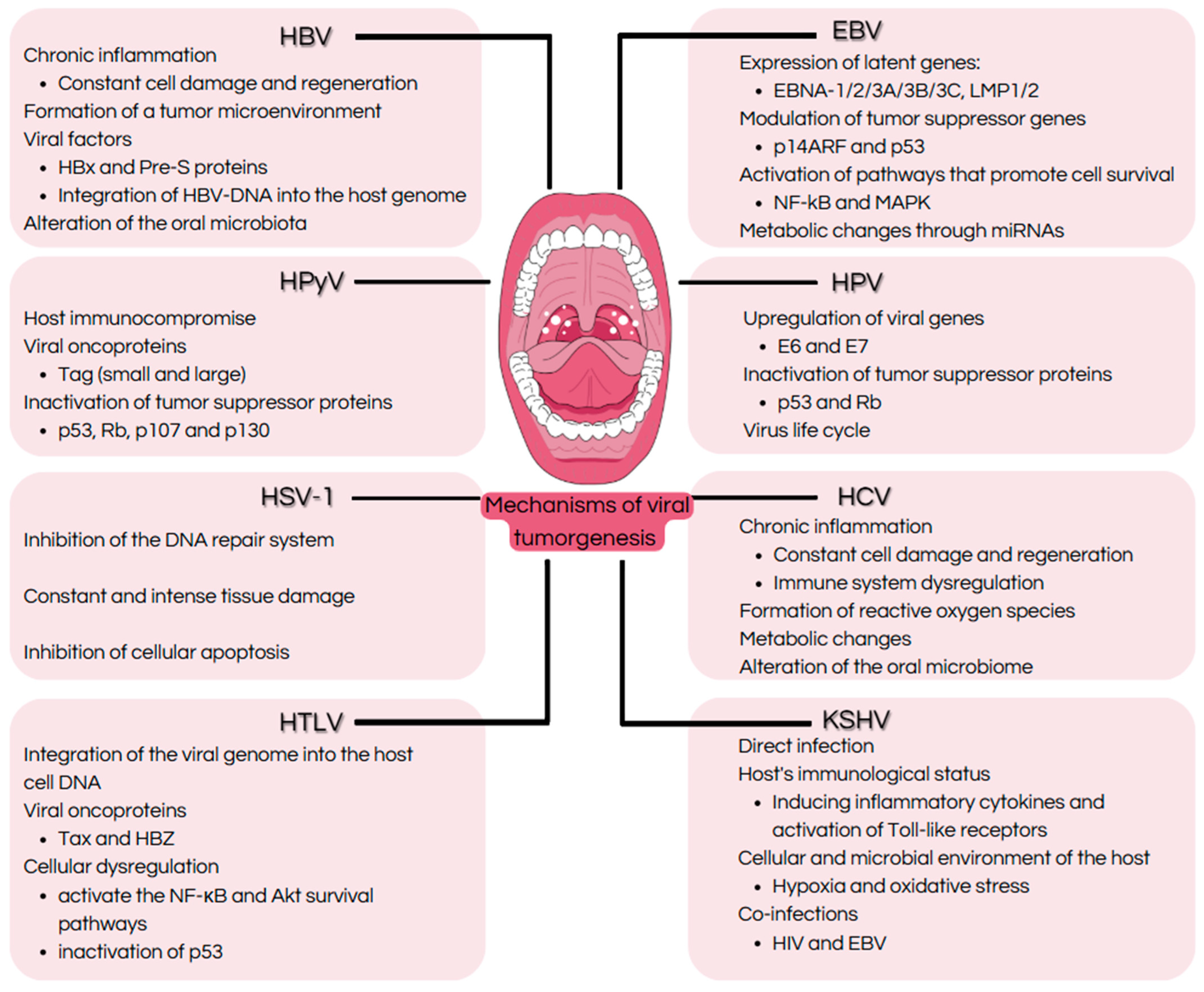
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, L.B.; Guimarães, A.C.S.; Pereira, J.G.; Silva, C.S.d.; de Almeida, N.A.A.; Marinho, P.d.N.; de Sousa, R.M.P.; Duś-Ilnicka, I.; de Paula, V.S. Oncoviruses in the Oral Cavity: Recent Advances in Understanding Viral Infections and Tumorigenesis. Int. J. Mol. Sci. 2025, 26, 6721. https://doi.org/10.3390/ijms26146721
Campos LB, Guimarães ACS, Pereira JG, Silva CSd, de Almeida NAA, Marinho PdN, de Sousa RMP, Duś-Ilnicka I, de Paula VS. Oncoviruses in the Oral Cavity: Recent Advances in Understanding Viral Infections and Tumorigenesis. International Journal of Molecular Sciences. 2025; 26(14):6721. https://doi.org/10.3390/ijms26146721
Chicago/Turabian StyleCampos, Letícia Bomfim, Ana Carolina Silva Guimarães, Jéssica Gonçalves Pereira, Carla Sousa da Silva, Nathália Alves Araújo de Almeida, Pedro do Nascimento Marinho, Rafaela Moraes Pereira de Sousa, Irena Duś-Ilnicka, and Vanessa Salete de Paula. 2025. "Oncoviruses in the Oral Cavity: Recent Advances in Understanding Viral Infections and Tumorigenesis" International Journal of Molecular Sciences 26, no. 14: 6721. https://doi.org/10.3390/ijms26146721
APA StyleCampos, L. B., Guimarães, A. C. S., Pereira, J. G., Silva, C. S. d., de Almeida, N. A. A., Marinho, P. d. N., de Sousa, R. M. P., Duś-Ilnicka, I., & de Paula, V. S. (2025). Oncoviruses in the Oral Cavity: Recent Advances in Understanding Viral Infections and Tumorigenesis. International Journal of Molecular Sciences, 26(14), 6721. https://doi.org/10.3390/ijms26146721