Abstract
As a crucial aspect of epigenetic research, DNA methylation is fundamental to genomic stability, gene transcription regulation, and chromatin remodeling. Rice is a staple food source for roughly half of the world’s population. Therefore, optimizing rice yield and stress tolerance is vital for global food security. With the continuous advancement of DNA methylation detection technologies, studies have shown that DNA methylation regulates various rice growth and development processes, including root differentiation and grain development, through the dynamic equilibrium of de novo methylation, maintenance methylation, and demethylation. Furthermore, DNA methylation is crucial in the plant’s response to environmental stressors like high or low temperature, drought and salinity. The patterns of DNA methylation modifications are also closely linked to rice domestication and heterosis formation. Therefore, a comprehensive investigation of the DNA methylation regulatory network holds significant theoretical value for rice genetic improvement and molecular breeding. This review offers a systematic analysis of the molecular mechanisms and detection technologies of DNA methylation, as well as its regulatory roles in rice growth and development, stress responses, and other biological processes, aiming to provide a theoretical foundation for rice genetic improvement research.
1. Introduction
Classical genetics primarily focuses on the study of DNA sequence variations, emphasizing their regulatory impact on phenotypic variation traits. However, it encounters challenges in genetic phenomena, such as significant phenotypic variations despite consistent genotypes and the transgenerational inheritance of environmentally induced traits. In response to these limitations, epigenetics has emerged as a critical area of investigation aimed at addressing these complexities [1]. Epigenetics primarily explores heritable regulatory mechanisms that modulate gene expression without altering the DNA sequence, encompassing key aspects such as DNA methylation, histone modification, chromatin conformation remodeling, and non-coding RNAs [2].
DNA methylation represents a fundamental area of investigation within plant epigenetics. Currently, the scientific community has comprehensively elucidated the dynamic regulatory network of DNA methylation in the model plant Arabidopsis, with subsequent research progressively extending to various crop species [3,4,5]. Mechanistically, DNA methylation influences these processes by modulating DNA methylation patterns within gene and transposon regions, thereby affecting chromatin accessibility and gene transcriptional activity, ultimately regulating plant growth processes and stress response [6,7,8,9,10]. The mddcc mutant of Arabidopsis, a line engineered to be completely devoid of DNA methylation, not only exhibited extreme developmental defects, including dwarfism and failure to flower, but also a high frequency of active transposition events [11]. Meanwhile, exposure to cold stress induces the demethylase ROS1 to remove DNA methylation of promoter regions from genes associated with the CBF pathway, thereby activating the expression of downstream antifreeze genes [12].
Rice, as a staple crop, is critical for global food security [13]. Faced with the dual challenges of population growth and climate change, the crop scientific community has conducted extensive research on key biological processes, including yield formation, quality improvement, and disease resistance, leading to significant advances in genetic breeding and molecular regulation [14]. Relying on the established theoretical framework of DNA methylation in Arabidopsis and the ongoing refinement of the molecular regulatory network in rice, the scientific community has elucidated the critical roles of rice DNA methylation in growth, agronomic trait development, and stress response, thereby establishing a robust theoretical basis for epigenetic breeding in rice [15,16,17]. Concurrently, advancements in DNA methylation detection technologies and epigenomic techniques have provided crucial technical support for epigenetic breeding in rice. Specifically, in the field of epigenetic editing engineering, researchers have recently addressed challenges such as plant cell wall structures and low transformation efficiency, significantly improving editing efficiency and specificity to bolster the technical foundation for epigenetic breeding in rice [18,19]. Driven by both the elucidation of theoretical mechanisms and technological innovation, epigenetic breeding in rice has progressed from fundamental research to applied exploration, resulting in significant breakthroughs. The recent systematic investigation of rice cold stress by Song et al. represents a landmark achievement, proposing a novel “stress acclimation–epigenetic editing–stable trait inheritance” paradigm for epigenetic breeding [20]. Therefore, this review provides a comprehensive research progress of rice DNA methylation, aiming to inform strategies for genetic improvement of yield and quality, molecular breeding of stress-resistant varieties, and germplasm innovation in modern agriculture.
2. Molecular Mechanism of DNA Methylation
DNA methylation represents an epigenetic modification, catalyzed by methyltransferases, where SAM (S-adenosylmethionine) donates methyl groups to cytosine (C), generating 5mC (5-methylcytosine) [21]. The distribution of 5mC is typically enriched in transposable elements (TEs) and repetitive sequences within the genome. In mammals, DNA methylation is predominantly observed at symmetrical CG sites, while non-CG methylation (CHG and CHH, where H represents A, T, or C) is specifically distributed in certain cell types, including pluripotent stem cells, the brain, and oocytes [22,23,24,25]. In contrast, plants exhibit DNA methylation at all contents (CG, CHG, and CHH) [26]. Significant interspecies variations exist in DNA methylation and distribution patterns. In higher animal cells, 60-90% of CG sites are methylated [27], while the C methylation proportion in higher plant genomes can reach up to 43% [28]. Notably, DNA methylation in rice genome displays a fragmented distribution pattern, with an overall methylation level four times higher than that of Arabidopsis [29]. Plant DNA methylation is a dynamic regulatory process, with its modification status undergoing alterations in response to environmental changes and developmental stages [30]. The dynamic equilibrium of DNA methylation in plants is primarily regulated by three processes: de novo methylation, maintenance methylation, and demethylation [3].
2.1. De Novo Methylation
De novo methylation is defined as the process of establishing novel methylation patterns through the addition of methyl groups at specific genomic sites by DNA methyltransferases, facilitated by RdDM (RNA-directed DNA methylation) pathway in regions lacking prior methylation modifications [31].
Studies have demonstrated that the RdDM de novo methylation pathway can be categorized into canonical (Figure 1A) and non-canonical types. The canonical pathway involves the histone-binding protein SHH1 (Suppressor of hairy-wing 1), which specifically recognizes histone H3K9me2, and the ZMP (Zinc finger protein), which specifically recognizes H3K4me0. These proteins cooperatively regulate the plant-specific RNA Pol IV (Polymerase IV) across various chromatin environments [32,33,34]. Following binding to these histone modification recognition proteins, specific POL IV subunits, such as the largest subunit NRPD1 (RNA polymerase IV subunit 1), transcribe ssRNA (single-stranded RNA) using specific chromatin loci as templates [35,36,37]. Simultaneously, other Pol IV subunits interact with RDR2 (RNA dependent RNA polymerase 2), employing ssRNA as a template to synthesize complementary strands, thereby forming dsRNA (double stranded RNA) [38,39,40,41]. Subsequently, dsRNA is cleaved by DCL3 (Dicer-like 3) into 24-nucleotide siRNA (small interfering RNA) [42,43]. The non-canonical pathway initiates with the cleavage of POL II transcripts mediated by miRNA (microRNA). After stabilization by SGS3 (Suppressor of gene silencing 3), the cleavage products recruit RDR6 to synthesize dsRNA, which DCL4 then processes to generate 21-nt siRNA [44,45,46,47]. siRNAs generated by both pathways undergo 3’ terminal methylation by the small RNA methyltransferase HEN1 (Hua enhancer 1) to maintain structural stability [48]. The modified siRNAs bind to AGO4 (Argonaute 4) or AGO6 to form the RISC (RNA-induced silencing complex), which targets DNA sequences based on complementary base pairing [49]. Furthermore, Pol V, after being recruited by DNA methylation, transcribes a 200-nt scaffold RNA. This scaffold RNA interacts with the RISC, and the cytosine residues of the target sequence are methylated by DRM2 (Domains rearranged methyltransferase 2) [50,51].
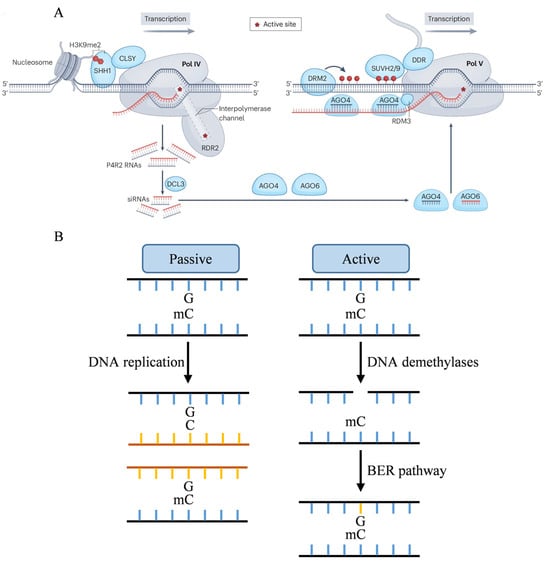
Figure 1.
Mechanism of DNA de novo methylation and demethylation. (A) SHH1 exhibits preferential binds to demethylated H3K9me2, thereby facilitating the recruitment of DNA-dependent RNA Pol IV to a subset of RdDM loci through direct interaction with CLSY1 or CLSY2 chromatin remodeling factors. Pol IV and RDR2 form a holoenzyme that produces double-stranded precursor RNAs (P4R2 RNAs). These P4R2 RNAs undergo cleavage by DCL3 to produce 24-nt siRNA. One strand of the siRNA is subsequently bound and stabilized by AGO4 and AGO6 proteins. The siRNA-AGO complex is then recruited by Pol V transcripts through base pairing. SUVH2 and SUVH9, acting as methylcytosine binding proteins, bind to pre-existing methylcytosines, along with the DRD1-DMS3-RDM1 (DDR) complex. The siRNA-AGO complex, potentially in conjunction with other proteins, then recruits DRM2 to affect DNA methylation [52]. Reproduced with permission from Heng Zhang and Jian-Kang Zhu, Nature; published by Springer Nature, 2025. (B) DNA demethylation can occur passively during DNA replication. Active DNA demethylation is mediated by plant-specific DNA demethylases. The single-nucleotide gap will be filled with a unmethylated cytosine through BER pathway.
In Arabidopsis, DRM1/2 function via the RdDM pathway, primarily establishing methylation in CG and CHH contexts [53,54]. Meanwhile, CMT3 (Chromomethylase 3) localizes methylation sites via histone modification marks, predominantly responsible for methylation establishment in CHG context [55,56]. In rice, OsDRM2, the homologous of Arabidopsis DRM2, participates in the de novo methylation of CG and non-CG sites (Table 1) [57]. Rice has two copies of CMT3 with distinct functions (Table 1). OsCMT3a is responsible for CHG methylation of TEs and centromeric region repetitive sequences, regulating rice development by suppressing TE transposition [58]. It also functions as the primary CHG methyltransferase in spermatocytes [59]. OsCMT3b, a rice-specific non-CG DNA methyltransferase, maintains non-CG methylation in specific GC-rich regions and regulates CHG methylation in microspores [59].

Table 1.
Some cloned DNA methylation-related genes in rice.
2.2. Maintenance Methylation
Methylation maintenance is the process of maintaining a stable methylation status during semi-conservative DNA replication, guided by the methylation pattern of parental strand. This is achieved through DNA methyltransferase, which add methylation modifications to the corresponding cytosine sites of the daughter strand [67].
In Arabidopsis, various DNA methyltransferases are responsible for maintaining the DNA methylation status of different cytosine sequences. CG methylation maintenance primarily relies on MET1 (Methyltransferase 1) [55,56,68]; CHG methylation maintenance is mainly dependent on CMT3; CHH methylation maintenance is primarily reliant on CMT2 and DRM1/2. Specifically, CMT2 maintains CHH methylation in heterochromatin, while DRM1/2 maintains CHH methylation in euchromatin or at the boundaries of long TEs [31,69,70]. DRM1/2 also maintains DNA methylation through the RdDM pathway. CMT3 forms a positive feedback regulatory loop with the histone methyltransferase SUVH4 (Suppressor of variegation 3-9 homolog 4), reinforcing CHG methylation maintenance. The catalytic activity of SUVH4 depends on the CHG methylation established by CMT3, while CMT3 recognizes the H3K9me2 modification catalyzed by SUVH4 through its BAH (Bromo adjacent homology) and chromo domains, thereby forming a positive feedback regulatory loop between DNA methylation and histone modification [71]. Similarly, CMT2 also maintains CHH methylation by recognizing H3K9me2-modified nucleosomes and collaborating with histone methyltransferases [72]. In the CG methylation maintenance pathway, the VIM1 (Variant in methylation 1) binds to MET1 via its SRA (Set and ring-finger-associated) domain, which enhances MET1 protein stability and precisely anchors MET1 to heterochromatin regions by binding to hypomethylated DNA and H3K9me2 modifications, thus establishing CG methylation [73].
In the rice genome, two homologous genes of MET1 (OsMET1a and OsMET1b) have been cloned, exhibiting functional differentiation (Table 1) [60]. Both OsMET1a and OsMET1b are expressed in dividing cells-containing tissues, with OsMET1b showing significantly higher expression levels than OsMET1a [60]. In addition to OsCMT3, rice also has two unique OsDDM1 (OsDDM1a and OsDDM1b) genes, which play a crucial role in CHG methylation and partial CG methylation of heterochromatic regions, and also participate in CHH methylation of gene regions, performing some functions of OsCMT2 and OsCMT3 (Table 1) [67,74].
2.3. DNA Demethylation
DNA demethylation can activate specific genes or reset the epigenetic status of the genome during development, encompassing passive and active demethylation (Figure 1B). Passive DNA demethylation arises when newly formed DNA strands fail to maintain their original methylation status post-replication, resulting in an unmethylated state. This process is often driven by factors such as the inhibition or loss of methyltransferase activity, methylation donor deficiency, and alterations in chromatin structure [75,76]. Active demethylation, on the other hand, is mediated by DNA demethylases, which excise methylated cytosine, followed by the synthesis of unmethylated cytosine through the BER (Base excision repair) pathway, ultimately resulting in DNA demethylation [77,78].
The active DNA demethylation process in plants can be delineated into three stages: substrate recognition, glycosidic bond hydrolysis, and DNA repair. Initially, DNA demethylases recognize 5mC sites and catalyze glycosidic bond hydrolysis, cleaving the bond between the base and deoxyribose, thereby removing 5mC and producing a base-free site. Subsequently, the BER pathway is activated, with the AP (Apurinic/apyrimidinic) endonuclease identifying the base site and cleaving the adjacent DNA strand. DNA polymerase then fills the gap with unmethylated cytosine, and finally, DNA ligase ligates the repaired DNA strand [79].
Unlike in plants, the active DNA demethylation process in animals is facilitated by the TET (Ten-eleven translocation) family of oxidases [80]. These enzymes catalyze the sequential oxidation of 5mC to 5hmC (5-hydroxymethylcytosine) [81], followed by 5fC (5-formylcytosine), and ultimately 5caC (5-carboxylcytosine) [82]. Subsequently, these oxidized derivatives are excised and replaced with unmethylated cytosine via the BER pathway [83,84]. Although plant genomes lack dioxygenases homologous to the mammalian TET family, the detection of 5mC oxidation products in species such as Arabidopsis [85], rice [86], and rye [87] suggests the presence of an oxidative demethylation mechanism in plants. Moreover, the heterologous expression of the catalytic domain of human TET3 in Arabidopsis induces the accumulation of 5hmC and 5fC, resulting in alterations in DNA methylation patterns [88]. Notably, these methylation modifications are stably inherited even after the transgene removal, demonstrating that exogenous TET enzymes retain oxidative functionality within plant systems.
Currently, researchers have identified eight DNA demethylase genes in rice, including four OsROS1 (Repressor of silencing 1) genes (OsROS1a, OsROS1b, OsROS1c, and OsROS1d), two Demeter-Like 3 (OsDML3) genes (OsDML3a and OsDML3b), and one each of OsDML4 and OsDML5 (Table 1) [89]. Studies have indicated that OsROS1a is involved in the demethylation process of CG and CHG in rice endosperm [62], while OsROS1a/b/c exert DNA demethylation effects in various genomic regions of gametes and zygotes [63]. Additionally, OsDML4 plays a crucial role in cytosine demethylation. In the osdml4 mutant, the global methylation levels of CG, CHG, and CHH in seeds are significantly increased, impacting endosperm formation [65].
3. DNA Methylation Detection Methods
Epigenetics researchers have developed several DNA methylation sequencing methods to identify DNA methylation sites. The advent of BS (Bisulfite sequencing) represented the initial technology for DNA methylation sequencing [90]. Advances in sequencing technologies, particularly NGS (Next-generation sequencing), have enabled the development of various approaches like RRBS (Reduced representation bisulfite sequencing) [91], MeDIP-seq (Methylated DNA immunoprecipitation sequencing) [92], and WGBS (Whole-genome bisulfite sequencing) [93]. These technologies have found increasing application in plant science, from fundamental epigenetic mechanism studies to applications in crop breeding, stress response, and developmental regulation [94,95,96]. DNA methylation detection technologies are classified into two main categories based on their detection targets: targeted methylation detection technologies, which focus on specific genomic regions, and genome-wide methylation detection technologies, which analyze the entire genome.
Targeted methylation detection technologies encompass a range of techniques, including MSP (Methylation-specific PCR), BSP (Bisulfite sequencing PCR), methylation pyrosequencing, TBS (Targeted bisulfite sequencing), MSRE-PCR (Methylation-sensitive restriction enzyme-PCR), MALDI-TOF (Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry), oxBS-seq (oxidative Bisulfite sequencing), and TAB-seq (TET-assisted bisulfite sequencing) [97]. Among these, BS-PCR represents an early approach, utilizing bisulfite treatment to distinguish between unmethylated cytosine, which is converted to uracil (C→U), and 5mC, which remains unchanged. Following PCR amplification, U is read as thymine (T), whereas C that persist correspond to 5mC (Figure 2) [90]. Bisulfite treatment is a critical step in many other methylation detection methods. Pyrosequencing, after bisulfite treatment, enables real-time sequencing by detecing pyrophosphate release, directly quantifying the C/T ratio, which corresponds to 5mC/C [98]. TBS combines BS-PCR principles with high-throughput sequencing, allowing for the simultaneous analysis of multiple targeted loci [99]. Additionally, MALDI-TOF mass spectrometry, following bisulfite treatment and enzymatic digestion, quantifies methylation levels by detecting mass differences in DNA fragments [100]. To specifically detect 5hmC in plant genomes, oxBS-seq and TAB-seq were developed. oxBS-seq involves oxidizing 5hmC to 5fC, which is subsequently converted to U during bisulfite treatment, leaving only 5mC as C [91]. TAB-seq employs βGT (β-glucosyltransferase) to protect 5hmC by converting it to β-glucosylhydroxymethylcytosine (5gmC), while the TET enzyme oxidizes 5mC to 5caC, which is subsequently converted to U during bisulfite treatment, thus preserving 5gmC [101]. Additional targeted detection techniques including MSP, which bypass bisulfite treatment and determines methylation status through PCR amplification using primers specific to methylated or unmethylated sequences [102], and MSRE-PCR, which utilizes restriction enzymes that selectively cleave unmethylated sites, enabling the detection of 5mC via PCR amplification of uncleaved fragments containing methylated cytosines [103].
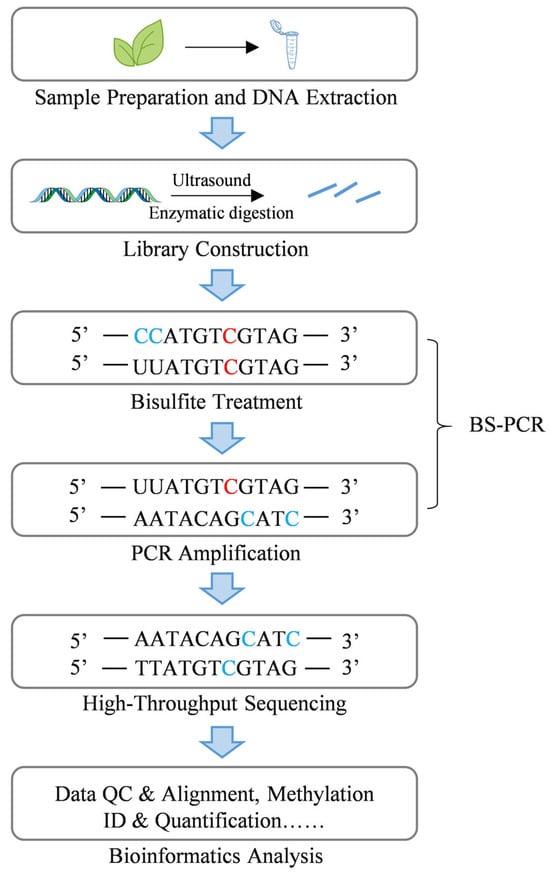
Figure 2.
Principles and procedures of BS-PCR and WGBS. The select of appropriate samples and the extraction of high-quality DNA. The extracted DNA is then fragmented into smaller segments via ultrasound or enzymatic digestion, followed by adapter ligation for library construction. The library undergoes bisulfite treatment, which converts unmethylated cytosine (C) to uracil (U), while methylated cytosine (5mC) remains unchanged. PCR amplification is performed using the treated DNA as a template, where unmethylated C is replaced by thymine (T), and 5mC remains as C. the amplified products are then sequenced via high-throughput sequencing. Finally, bioinformatics analysis conducted, including data quality control, sequence alignment, and methylation qualitative and quantitative analysis. BS-PCR utilizes the bisulfite treatment and PCR amplification steps of WGBS to determines the DNA methylation status of specific loci through sequencing the amplified products. Unmethylated cytosine is denoted by the blue “C”, while methylated cytosine is denoted by the red “C”.
Genome-wide methylation detection technologies include WGBS, RRBS, XRBS (Extended representation bisulfite sequencing), methylation microarrays, MeDIP-seq, and hMeDIP-seq (Hydroxymethylated DNA immunoprecipitation sequencing) [97]. These methodologies enable the creation of plant epigenetic maps and facilitate large-scale screening. RRBS, originally developed with Sanger sequencing, employs restriction enzymes to digest genomic DNA, selectively enriches CpG-dense regions, followed by bisulfite conversion and sequencing for cost-effective genome-wide methylation analysis [91]. XRBS improves library preparation and sequencing protocols compared to RRBS, expanding CpG site coverage and enabling methylation status assessment across more gene regulatory elements [104]. WGBS involves high-throughput sequencing of the entire genomic DNA post-bisulfite treatment, providing comprehensive coverage of almost all C sites (Figure 2) [105]. It is considered the "gold standard" for methylation detection due to its genome-wide scope and single-base resolution. However, it has limitations, including high cost, data complexity, and the need for significant input DNA [106]. Besides bisulfite-dependent technologies, other genome-wide methylation detection techniques include methylation microarrays, MeDIP-seq and hMeDIP-seq. Methylation microarrays detect predetermined CpG sites (covering promoters, gene bodies, etc.) via probe hybridization, quantifying methylation levels based on fluorescent signal intensities. These arrays offer advantages such as high throughput, affordability, and straightforward data analysis, but are limited by probe dependency and potential hybridization biases [107]. MeDIP-seq uses 5mC-specific antibodies to immunoprecipitate DNA fragments containing 5mC, followed by high-throughput sequencing to map 5mC distribution [108]. hMeDIP-seq, based on MeDIP-seq, uses 5hmC-specific antibodies for genome-wide specific sequencing analysis of 5hmC, addressing the need to differentiate between 5hmC and 5mC modifications [109].
Various DNA methylation detection technologies previously documented have been implemented in rice research. The selection of detection methodologies in rice DNA methylation studies is contingent upon aligning research objectives (genome-wide analysis, specific gene validation, large-scale population screening) with technical attributes (coverage, resolution, cost). Among genome-wide methylation detection approaches, WGBS is frequently utilized [110,111]. WGBS serves as a fundamental tool for elucidating genome-wide methylation patterns, such as tissue-specific methylation variations and epigenetic modifications associated with domestication in rice [59,112]. Despite its associated costs, WGBS remains essential for in-depth mechanistic investigations [106]. Furthermore, rice, as a model species within the Poaceae family, characterized by a comapct and well-assembled genome, provides a crucial framework for methodologies that depend on high-quality genome alignment for WGBS [113,114]. In research focused on specific genes or genomic regions, the commonly employed targeted methylation detection technologies in rice are BSP and MSRE-PCR. BSP is widely used for ensure DNA methylation results due to its simplicity, low cost, and ability to accurately verify methylation dynamics of target genes [110,115,116]. MSRE-PCR, which obviates the need for bisulfite treatment, enables rapid qualitative preliminary screening of methylation differences in target genes, thereby rendering it advantageous for initial sample screening [117].
In rice research, genome-wide and targeted DNA methylation detection methodologies are commonly used complementarily [20,118]. The strategic integration of these techniques has significantly enhanced the understanding of epigenetic regulatory mechanisms involved in rice development, domestication, and stress responses, thereby providing essential epigenetic theoretical foundations for molecular breeding in rice.
4. Role of DNA Methylation in Rice Growth and Development
The rice life cycle, encompassing seed germination to the maturity of new seeds, is bifurcated into vegetative and reproductive phases. The vegetative growth stage initiates with seed germination, progressing through the seedling and tillering stage, culminating in the jointing stage. The reproductive growth stage commences with the booting stage, advancing through the heading and flowering stages, and concluding with seed maturity, which includes the milk, dough, and full maturity stages [119]. DNA methylation is a critical regulator throughout rice entire growth and development stages. The osdrm2 mutant exhibits reduced genome-wide DNA methylation, it results in phenotypes, such as dwarfism, reduced tillering, and aberrant leave morphology in vegetative stage. While in the reproductive stage, it shows delayed heading, abnormal panicles and spikelets morphology, and complete sterility [57].
4.1. Vegetative Growth Stage
Rice vegetative organs, including root, stem, leaf, and tiller, function synergistically to support vegetative growth. The root system, consisting of primary, postembryonic crown, and lateral roots, facilitates water and mineral uptake. Primary roots are ephemeral, while the fibrous root system, composed of postembryonic crown and lateral roots, forms the primary absorptive network. Research indicates that CHH methylation influences the development of these root types, affecting transcriptome regulation. The activation of OsROS1a and the inhibition of OsDRM2, resulting in reduced DNA methylation, are essential for the expression of key functional genes (Figure 3) [120].
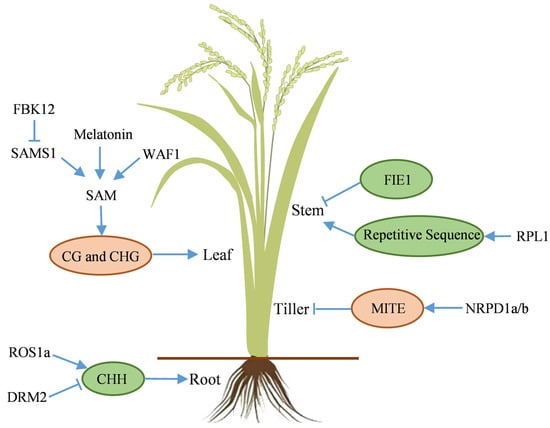
Figure 3.
Mechanism of DNA methylation in the vegetative growth stage of rice. Blue arrows and T-bars indicate promotion and inhibition, respectively. Pink and green ovals represent increased and decreased DNA methylation, respectively. DRM2: Domains rearranged methyltransferase 2, FBK12: F-box protein containing a Kelch repeat motif, FIE1: Fertilization-independent endosperm 1, MITE: Miniature inverted-repeat transposable element, NRPD1a/b: Nuclear RNA polymerase D1a/b, ROS1a: Repressor of silencing 1, RPL1: Ribosomal protein l1, SAMS1: S-adenosylmethionine synthase 1, SAM: S-adenosylmethionine, WAF1: Wavy leaf1.
The stem provides structural support and serves as a conduit for nutrient transport. Stem development, consequently plant height, is regulated by DNA methylation via epigenetic mechanisms (Figure 3). The epigenetic allele epi-DF (Epigenetic-dwarf) of the FIE1 (Fertilization-independent endosperm 1) gene, despite lacking DNA sequence alterations, exhibits hypomethylation in the promoter and 5’ region, accompanied with histone modification, leading to ectopic gene expression in stem internodes, ultimately causing plant dwarfing [116]. Additionally, mutation of the rpl1 (ribosomal protein l1) gene increases methylation of repetitive sequences, disrupting hormone signaling and causing dwarf phenotype [121].
Leaf, the primary site of photosynthesis, converts light energy into chemical energy, supplying carbon sources and energy for plant growth. Rice leaf development is closely related to SAM levels. Melatonin deficiency induces premature leaf senescence, characterized by accelerated chlorophyll degradation, increased ROS (Reactive oxygen species) accumulation, and aberrant activation of senescence-related genes. Melatonin maintains DNA methyltransferase activity by stabilizing SAM levels (Figure 3). Melatonin deficiency impacts SAM synthesis, leading to a shortage of methyl donors and significant hypomethylation, particularly in TEs and promoters of genes involved in carbon metabolism and redox, ultimately resulting in elevated gene expression [122]. The F-box protein OsFBK12 (F-box protein containing a Kelch repeat motif 12) reduces SAM content by targeting and degrading OsSAMS1 (S-adenosylmethionine synthase 1), affecting the ethylene synthesis pathway and delaying leaf senescence [123]. Although direct DNA methylation data are lacking, the reduction in SAM content suggests that DNA methylation may regulate leaf senescence. Furthermore, the rice waf1 (wavy leaf 1) (HEN1 orthologous in Arabidopsis) mutant exhibits SAM deficiency, resulting in seedlings mortality and developmental abnormalities, such as wavy leaves and increased leaf angle [124].
Tillering, a unique form of vegetative reproduction in rice, originating from axillary buds at the stem base, and the subsequent panicle formation rate are critical determinants of effective panicle number per unit area, thereby influencing yield composition. The disruption of OsNRPD1a/b, the largest subunit genes of Pol IV, leads to a significant reduction in 24-nt siRNA levels. This alteration affects DNA methylation status of MITEs (Miniature inverted-repeat transposable elements), subsequently impacting the expression of nearby genes involved in tiller development, and resulting in increased rice tiller number (Figure 3) [125]. Furthermore, the rpl1 mutation also exhibits a phenotype of significantly increased tiller number [121].
4.2. Reproductive Growth Stage
4.2.1. Heading and Flowering
The heading stage, a critical transition from vegetative to reproductive growth in rice, is a key factor in determining plant development and yield formation. DNA methylation is also involved in regulating the heading process. Knockdown the rice SAMS gene, which encodes S-adenosylmethionine synthetase, leads to a decrease in SAM content, resulting in reduced methylation levels at non-CG sites and histone H3K4me3. These epigenetic changes further suppress the expression of key flowering genes, such as Ehd1 (Early heading date 1), Hd3a (Heading date 3a), and RFT1 (Rice Flowering Locus 1), ultimately causing delayed heading in rice (Figure 4) [126].
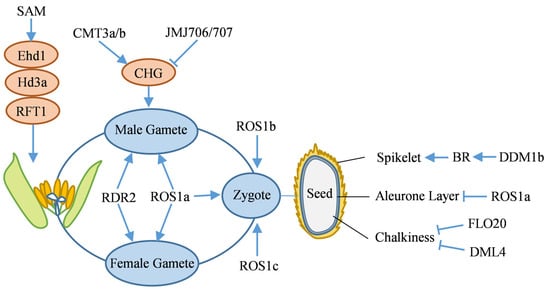
Figure 4.
Mechanism of DNA methylation in the reproductive growth stage of rice. Blue arrows and T-bars indicate promotion and inhibition, respectively. Pink ovals represent the increase in DNA methylation levels. BR: Brassinosteroid, CMT3a/b: Chromomethylase 3a/b, DML4: Demeter-like 4, Ehd1: Early heading date 1, FLO20: Floral organ number 20, Hd3a: Heading date 3a, JMJ706/707: Jumonji 706/707, RDR2: RNA dependent RNA polymerase 2, RFT1: Rice flowering locus 1, ROS1a/b/c: Repressor of silencing 1 a/b/c, SAM: S-adenosylmethionine.
4.2.2. Gamete and Zygote Development
The formation of male and female gametes, along with zygotes development, are central to plant sexual reproduction, directly determining the transmission of genetic material and the development of new individuals. During male gamete development, pollen mother cells undergo meiosis to form microspores, which then generate mature pollen grains containing vegetative cells and sperm cells through mitosis. Female gamete development begins with meiosis of the megaspore mother cell, where the surviving megaspore undergoes three mitotic divisions to form an eight-nucleate embryo sac containing an egg cell. During fertilization, sperm cells enter the embryo sac via the pollen tube: one fuses with the egg cell to form a zygote, while the other fuses with polar nuclei to form an endosperm nucleus [127,128].
A loss-of-function mutation in the OsROS1a DNA demethylase gene leads to elevated CG and CHG methylation within the promoter regions of key gametophyte development genes, consequently repressing their expression. The osros1a mutant exhibits smaller pollen grains, starch accumulation defects, and iodine-stained sterility, alongside aberrant proliferation of antipodal cells, morphological abnormalities in egg and synergid cells, and partial ovary degeneration post-pollination, resulting in seed abortion (Figure 4) [129]. Additionally, OsDRM2 maintains elevated CHH methylation via the RdDM pathway to ensure proper sexual reproduction [67]. The osrdr2 mutant impairs 24-nt siRNAs production, leading to diminished CHH methylation, TE activation, and interference with reproductive development gene expression, ultimately causing male and female gamete development abnormalities and sterility (Figure 4) [130].
In rice male gametogenesis, the regulation of DNA methylation is crucial (Figure 4). During the microspore stage, CMT3a/b synergistically enhances CHG methylation, silences TE, and suppresses excessive genome transcription to maintain male gamete genomic stability [59]. In the vegetative cell stage, ROS1a targets promoter and TE regions for local demethylation, and DNA demethylation in vegetative cells can indirectly promote non-CG methylation in sperm [64]. In the sperm stage, histone demethylases JMJ706/707 mediate reduced CHG methylation, activate sperm functional genes, and complete the epigenetic regulatory conversion [59].
In rice, DNA methylation orchestrates zygotic gene expression via dynamic demethylation remodeling, thereby establishing a critical epigenetic foundation for typical zygote development and reproductive functions (Figure 4). Research indicates that regional methylation remodeling is initiated within the zygotic genome 6.5 h post-fertilization in rice. Genetic and multi-omics investigations demonstrate that DNA demethylases DNG702 (ROS1a), DNG701 (ROS1b), and DNG704 (ROS1c) facilitate DNA demethylation across diverse genomic regions in gametes and zygotic cells. These demethylases are essential for activating zygotic gene expression and maintaining developmental processes [63].
4.2.3. Seed Development
The primary attributes of rice seeds encompass grain shape (affecting yield) and quality (determining edibility). Grain shape is characterized by metrics such as grain length, width, and thickness, while quality is assessed through appearance, milling properties, taste, and nutritional content [131].
Previous studies have demonstrated that dynamic DNA methylation regulation is critical in the developmental process governing rice grain shape (Figure 4). The rice chromatin remodeling factor OsDDM1b (Decrease in DNA methylation 1b) maintains heterochromatin DNA methylation via ATP-dependent nucleosome remodeling, which subsequently regulates cell cycle genes to promote glume cell division. Additionally, it may modulate brassinosteroid (BR) homeostasis and signal transduction by maintaining methylation. The loss function of OsDDM1b results in cell cycle arrest, reduced BR content, and significantly smaller grains [61]. Furthermore, DNA methylation plays a crucial role in regulating rice seed quality by influencing the epigenetic modification status of genes involved in storage proteins and starch synthesis, as well as affecting nutrient accumulation and distribution during endosperm development (Figure 4). The FLO20 (Floral organ number 20), encoding SHMT4 protein, interacts with SAMS2 to modulate SAM synthesis. This interaction maintains the SAM/SAH (S-adenosyl-l-homocysteine) balance, which is crucial for DNA methylation, and subsequently regulate the expression of starch and storage protein genes. In the flo20 mutant, a reduction in SAM concentration induces elevated genome-wide DNA methylation, thereby repressing the expression of key transcription factors (TFs) involved in starch and storage protein synthesis. This ultimately results in endosperm chalkiness, disrupted starch granules, and aberrant protein bodies (PBs) [132]. OsDML4 regulates the expression of storage protein genes via DNA methylation. Functional deficiency of OsDML4 results in elevated CG, CHG, and CHH methylation levels in the endosperm under high temperature conditions, leading to hypermethylation and downregulation of storage protein-related genes. This, in turn, causes aberrant PBs formation and disrupted starch granule arrangement, ultimately increasing grain chalkiness [66].
A dominant negative mutation in the DNA demethylase gene OsROS1 leads to an expansion of the aleurone layer in the wild type to multiple layers in the mutant (Figure 4). This specific mutation causes aberrant splicing, leading to increased CG and CHG methylation in the endosperm. Two aleurone layer differentiation-related TFs are hypermethylated and exhibit reduced expression, promoting an increase in aleurone layer cell numbers and enhancing the content of non-starch nutritional components [62].
5. Role of DNA Methylation in Rice Stress Response
5.1. Abiotic Stress
Rice is frequently exposed to abiotic stresses during its growth and development, which can significantly affect its physiological metabolism, growth status, and yield quality. Common abiotic stresses include high/low temperature, drought, salinity, heavy metal, and nutrient stresses [14].
Temperature is a critical environmental determinant affecting rice production. Analyzing the response mechanisms of rice to both high and low temperature stresses can provide genetic resources for breeding temperature-tolerant rice varieties. Guo et al. observed that in the cold-tolerant rice variety P427, the number of genes exhibiting DNA methylation alterations under low-temperature stress was significantly higher compared to other varieties. Specifically, the expression of certain genes is significantly upregulated due to reduced DNA methylation, potentially playing a key role in the cold tolerance of P427 [133]. Additionally, the cold tolerance conferred by DNA methylation in rice demonstrates stable genetic characteristics. Song et al. indicated that low-temperature stress can suppress the expression of the DNA methyltransferase gene MET1b, resulting in DNA demethylation within the promoter region of the ACT1 (Acquired cold tolerance 1) gene (Figure 5). This demethylation facilitates the binding of the upstream Dof1 (DNA binding with one finger 1) transcription factor, which subsequently activates ACT1 expression and enhances rice cold tolerance. The hypomethylated region in the ACT1 promoter represents a key domestication site for rice cold adaptation, and cold-tolerant lines carrying this epigenetic modification can be stably inherited for at least five generations under normal temperature conditions (Figure 5) [20]. Conversely, high temperature stress during rice grain development negatively impacts the synthesis of various substances, leading to increased chalkiness. Research has confirmed that under high-temperature stress conditions, the genome-wide DNA methylation is significantly increased in the osdml4 mutant, and the content of gluten and alcohol-soluble proteins decreases, ultimately resulting in a chalky endosperm phenotype [65].
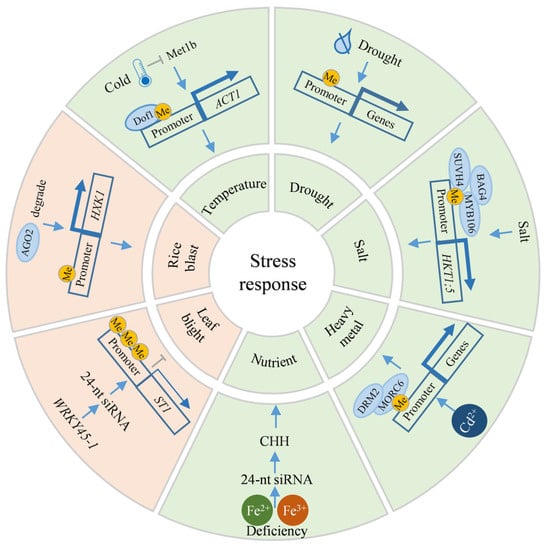
Figure 5.
Mechanism of DNA methylation in stress resistance processes. Blue arrows and gray T-bars indicate promotion and inhibition, respectively. Pink and green regions correspond to biotic and abiotic stresses, respectively. A single yellow “Me” marker represents a low methylation level, while three yellow “Me” markers represent a high methylation level. Thick blue arrows indicate high expression, and thin blue arrows indicate low expression. AGO2: Argonaute 2, BAG4: Bcl-2-associated athanogene protein 4, Dof1: DNA binding with one finger 1, DRM2: Domains rearranged methyltransferase 2, HKT1;5: High-affinity potassium transporter 1;5, HXK1: Hexokinase 1, Me: Methylation, Met1b: Methyltransferase 1b, MORC6: Minichromosome maintenance 1-related protein 6, ST1: Susceptibility to blast 1.
Drought stress poses a major abiotic constraint on rice production, with roughly half of the world’s rice-growing regions experiencing seasonal drought, resulting in an average yield reduction of 30–50% [134]. Drought treatment experiments on drought-tolerant and drought-sensitive rice varieties have shown that drought-tolerant varieties display a hypomethylated status under drought stress, which correlates with the high expression of some abiotic stress-responsive genes [110]. Differentially methylated regions (DMRs) linked to rice drought stress memory exhibit dynamic and unique patterns of change [135]. Similarly, epigenetic mutations induced by drought treatment in rice can perpetuate the altered DNA methylation status in subsequent generations, with genes associated with transgenerational epigenetic mutations directly involved in drought stress responses (Figure 5) [136].
Soil salinity poses a substantial constraint on global agricultural practices, especially in rice cultivation, where salinization can impede rice development and significantly diminish yield. Unlike drought stress, salt stress conditions induce elevated DNA methylation in salt-tolerant varieties compared to sensitive ones. This observation correlates with the upregulated expression of specific genes responsive to abiotic stressors [110]. Wang et al. elucidated a mechanism wherein a complex comprising a DNA methylation recognition enzyme, a chaperone regulatory protein, and a TF modulates the expression of OsHKT1;5 (High-affinity potassium transporter 1;5) to mediate salt stress response: the DNA methylation recognition enzyme OsSUVH7 recognizes CHG and CHH methylation of the MITE element upstream of OsHKT1;5. Simultaneously, OsBAG4 (Bcl-2-associated athanogene protein 4) facilitates the interaction between OsSUVH7 and OsMYB106, thereby prompting the binding of OsMYB106 to the upstream region of OsHKT1;5 and activating OsHKT1;5 expression in response to salt stress (Figure 5) [137]. OsDML4 has been identified as a participant in the rice salt stress response. The osdml4 mutant exhibits increased sensitivity to salt stress, characterized by aberrant ROS accumulation and an elevated Na⁺/K⁺ ratio, potentially influencing ROS homeostasis and the jasmonic acid (JA) signaling pathway [65].
The cultivation of rice in heavy metal-contaminated soil presents multifaceted challenges, impacting not only plant growth, yield, and quality, but also posing risks to human health via the food chain. Tan et al. indicates that the rice Microrchidia family protein OsMORC6 (Minichromosome maintenance 1-related protein 6) recruits OsDRM2 through the RdDM pathway to facilitate DNA methylation, thereby positively influencing rice’s chromium tolerance (Figure 5) [138]. Furthermore, heavy metal stress-induced alterations in DNA methylation patterns can be stably inherited in rice, enhancing heavy metal tolerance in subsequent generations [139].
Nutrient stress, encompassing both macro- and micronutrient imbalances, significantly affects rice’s physiological metabolism, yield formation, and quality. Sun et al. observed that under iron-deficient conditions, the abundance of 24-nt siRNA increases, accompanied by elevated genome-wide CHH methylation, particularly in TE regions in rice (Figure 5). This methylation change suppresses TE activity and activates adjacent iron-deficiency response genes, thereby improving rice’s adaptability to iron-deficient conditions [140].
The aforementioned environmental stressors, including cold, drought, and heavy metals, have been demonstrated to trigger transgenerational epigenetic memory in rice. In fact, plants frequently exhibit transgenerational epigenetic memory under abiotic stress [141]. Current research indicates that the preservation and inheritance of epigenetic modifications underpin this phenomenon at the molecular level. During germ cell formation and zygotic development, the genome undergoes DNA methylation reprogramming, whereas DNA methylation status of specific loci can resist this reprogramming and remain stable, acting as transgenerational memory carriers [118]. Histone modifications can prevent DNA methylation resetting via the RdDM pathway. For example, H3K4me3 directly inhibits the recruitment of core components in the RdDM pathway, while H3K18ac actively removes DNA methylation by recruiting DNA demethylases. This dual mechanism of inhibiting RdDM activity preserves the transgenerational stability of the initial epigenetic states [118]. Small RNAs can also influence DNA methylation inheritance. In maize, 24-nt siRNAs can be transmitted transgenerationally and induce CHH methylation via the RdDM pathway, which can then be converted into a stable and inheritable state [142].
5.2. Biotic Stress
Current investigations into biotic stress in rice primarily emphasize the fungal disease rice blast and the bacterial disease bacterial leaf blight. Rice blast, incited by Magnaporthe oryzae, can infect rice throughout its entire development stages, from the spindle-shaped lesions of leaf blast during the seedling phase to white panicles caused by panicle neck blast, frequently leading to a 40-50% yield reduction [143]. Early studies indicated that the expression of the rice blast resistance gene Pib is significantly upregulated upon exposure to the rice blast fungus, with its core promoter region exhibiting a highly CG-methylated state. Notably, promoter region demethylation leads to reduced rice blast resistance [144]. Another study revealed that OsAGO2, a critical component of the silencing complex in the rice RdDM pathway, participates in the rice blast resistance response. In the absence of rice blast fungus infection, OsAGO2 binds to miR1875 and suppresses the expression of the target gene OsHXK1 (Hexokinase 1) by methylating its promoter region via DNA methylation (Figure 5). Upon infection by the rice blast fungus, OsAGO2 is degraded, releasing the inhibition of OsHXK1 expression, thereby enhancing the plant’s resistance to the disease [145]. Additionally, a novel miR812w originating from TEs in rice, is crucial for immunity against rice blast disease. miR812w modulates host defense genes via both cis and trans DNA methylation. These regulations induce CHH hypermethylation of target genes, thereby inhibiting their expression, which subsequently suppresses pathogen infection and positively regulates rice resistance to the rice blast fungus [146]. Deng et al. discovered that the Pigm locus harbors two antagonistic NBS-LRR-type receptor proteins, PigmR and PigmS. These proteins regulate the CHH methylation of their promoter regions via RdDM pathway, ensuring that PigmR is expressed primarily in tissues like leaf, while PigmS is highly expressed in pollen. This epigenetic regulatory mechanism dynamically balances the relationship between disease resistance and yield. Upon pathogens invasion, PigmR governs the disease resistance response in leaves, during reproductive development, PigmS in pollen maintains yield by enhancing fertility [147].
Bacterial leaf blight, incited by Xanthomonas oryzae, manifests as characteristic yellow-white lesions that progress along leaf margins. Zhang et al. identified two WRKY45 alleles, WRKY45-1 and WRKY45-2. A TE insertion within the WRKY45-1 intron leads to the generation of multiple, partially overlapping 24-nt siRNAs from its transcript. These siRNAs trigger CHH methylation in the promoter region of ST1 (Susceptibility to blast 1) gene, a key element in the downstream disease resistance signaling pathway, via the RdDM pathway, consequently suppressing ST1 expression and ultimately compromising rice’s resistance to blast disease (Figure 5). In contrast, WRKY45-2, lacking TE insertion, permits normal ST1 expression, thereby conferring resistance to bacterial leaf blight in rice [148].
6. Role of DNA Methylation in Other Biological Process
In rice, DNA methylation is implicated in the regulation of growth and development processes, and stress responses, and also plays a crucial role in significant biological processes, including domestication and heterosis formation, through dynamic epigenetic modifications.
Rice domestication is characterized by the anthropogenic conversion of wild rice into cultivated rice through sustained human selection. Conversely, de-domestication denotes the phenomenon where specific rice varieties gradually revert to some phenotypic traits of wild rice, evolving into weedy rice [149]. A comprehensive methylome analysis of wild, cultivated, and weedy rice revealed a significant decrease in genome-wide DNA methylation during rice domestication, contrasting with a marked increase during de-domestication. The methylation variation regions associated with domestication and de-domestication do not overlap, suggesting that de-domestication is not a simple reversal of domestication. Furthermore, distinct DNA methylation patterns have been observed among various cultivated rice subtypes [112]. A separate investigation into the genome-wide DNA methylation, transcriptome, and metabolome of japonica and indica rice germplasms revealed that the genome-wide DNA methylation of indica rice is notably lower than that of japonica rice [150]. Peng et al. observed that under natural conditions, an active retrotransposon HUO is prevalent in wild rice, whereas it is absent in cultivated rice varieties. HUO influences functional genes at the genomic level via the RdDM pathway, thereby activating the genome’s defense mechanisms. This activation facilitates wild rice’s adaption to intricate and dynamic natural environments; however, it is not conducive to high and stable yields in cultivated rice. Therefore, it has been gradually and selectively eliminated during the domestication and breeding of rice [151].
The manifestation of heterosis in rice is intricately linked to DNA methylation. Zhou et al. revealed that the overall DNA methylation in hybrid rice seeds is reduced compared to their parents, a factor critical for the onset and persistence of hybrid vigor [152]. Ma et al. demonstrated an inverse relationship between CHG methylation and allelic-specific expression (ASE), with the CMT3 enzyme implicated in this regulatory mechanism. In addition, non-additive CHG methylation regions are abundant in genes associated with hormone signaling pathways, potentially contributing to the enhanced environmental adaptability observed in hybrid rice cultivars [153].
7. Summary and Prospects
DNA methylation is widely recognized as a pivotal regulatory mechanism influencing rice growth and development, stress response, domestication, and heterosis. Nevertheless, current studies present several limitations. Firstly, despite the known involvement of various DNA methylation-related enzymes in regulation, the precise molecular mechanisms by which these enzymes specifically regulate methylation sites and levels across diverse biological processes remain incompletely elucidated. For instance, in response to intricate and fluctuating natural environmental conditions, the adaptive strategies of rice, mediated by dynamic alterations in DNA methylation status through the regulation of related enzyme activity, require further investigation into the signaling pathways and key regulatory nodes involved. Secondly, although certain DMRs and genes associated with significant traits have been identified, the causal relationships between these DMRs and gene expression, as well as their synergistic regulation of rice phenotypes, necessitate extensive functional verification experiments. Finally, the interplay among different epigenetic regulatory mechanisms, including DNA methylation, histone modification, and non-coding RNA regulation, along with their integrated regulatory networks in complex rice biological processes, remains poorly understood.
Future advancements in technology will facilitate more precise and comprehensive analysis of the rice DNA methylation landscape and its dynamic alterations. This will involve investigating key regulatory genes and molecular modules governing DNA methylation in rice growth, development and stress response. Furthermore, employing gene editing and related technologies to precisely modulate rice DNA methylation status will enable the breeding of novel rice varieties with enhanced yields, quality and stress tolerance. To expedite the attainment of the aforementioned objectives, several technical hurdles in DNA methylation research necessitate immediate resolution. Current methodologies for DNA methylation detection continue to grapple with the inherent "triple trade-off" encompassing coverage, resolution, and cost, with no single technology universally applicable across all experimental contexts [154,155]. Additionally, functional validation of differentially methylated sites heavily relies on targeted editing technologies, which present notable limitations. Specifically, the editing efficiencies in plants are relatively low, ranging from 10% to 30% [156,157,158,159,160]. Both issues impede large-scale functional verification of candidate sites. Moreover, these technologies are currently unable to disentangle the specific contributions of DNA methylation alterations from other epigenetic modifications, such as histone modifications, thereby complicating the attribution of functional outcomes [161,162].
With the ongoing progress in DNA methylation research, breeders are increasingly exploring the application of DNA methylation-based regulatory strategies in epigenetic breeding. Utilizing epigenetic editing technologies, researchers can precisely modulate methylation levels in the promoter regions of specific DNA methylation sites, thereby enabling the development of stress-tolerant rice varieties. A targeted DNA demethylation system based on SunTag-dCas9-TETcd was developed in rice, enabling precise regulation of methylation within the promoter region of the OsFIE1 gene with stable inheritance across generations [163]. Furthermore, targeted editing of the methylation status of the ACT1 gene has enabled the directed modulation of cold tolerance in rice [20]. By leveraging epigenetic marker-assisted selection (EMAS) technology, researchers can screen for materials or loci associated with target traits. Methylation quantitative trait loci (meQTL), which serve as the foundation for screening valid markers in EMAS, are currently a significant research focus. meQTLs, defined as genetic variants exhibiting statistically significant associations with specific DNA methylation patterns, can be identified through large-scale population DNA methylation analyses combined with linkage mapping of genetic variants [94]. Similar meQTL analyses have been performed in plants such as tomato, pear, and cotton [94,164,165]. Despite the existence of population-level DNA methylation studies in rice, linkage analyses correlating DNA methylation with genetic variants remain unexplored, which could provide guidance for future applications of DNA methylation in rice breeding [150,151,152,153].
Theoretically, a comprehensive understanding of DNA methylation mechanisms in rice clarifies how such epigenetic modifications dynamically regulate genes associated with yield, quality, and stress tolerance. Practically, unraveling these mechanisms establishes a foundational basis for innovative breeding technologies. In conclusion, in-depth investigation of the mechanism of DNA methylation in rice is of significant theoretical and practical importance for advancing rice genetic improvement and ensuring global food security.
Author Contributions
T.L. conceived the article. T.L. and W.-J.L. drafted the manuscript. J.-H.X. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jablonka, E.; Lamb, M.J. Précis of evolution in four dimensions. Behav. Brain Sci. 2007, 30, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Niu, Q.; Xu, Y.; Huang, H.; Li, L.; Tang, D.; Wu, S.; Liu, P.; Liu, R.; Ma, Y.; Zhang, B.; et al. Two transcription factors play critical roles in mediating epigenetic regulation of fruit ripening in tomato. Proc. Natl. Acad. Sci. 2025, 122, e2422798122. [Google Scholar] [CrossRef]
- Miao, L.; Xu, W.; Liu, Y.; Huang, X.; Chen, Z.; Wang, H.; Wang, Z.; Chen, Y.; Song, Q.; Zhang, J.; et al. Reshaped DNA methylation cooperating with homoeolog-divergent expression promotes improved root traits in synthesized tetraploid wheat. New Phytol. 2024, 242, 507–523. [Google Scholar] [CrossRef]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [PubMed]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; Richard McCombie, W.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman Derr, D.; Eshed Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Lyons, D.B.; Zilberman, D. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife 2017, 6, e30674. [Google Scholar] [CrossRef]
- He, L.; Huang, H.; Bradai, M.; Zhao, C.; You, Y.; Ma, J.; Zhao, L.; Lozano-Durán, R.; Zhu, J.K. DNA methylation-free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat. Commun. 2022, 13, 1335. [Google Scholar] [CrossRef]
- Yang, L.; Lang, C.; Wu, Y.; Meng, D.; Yang, T.; Li, D.; Jin, T.; Zhou, X. ROS1-mediated decrease in DNA methylation and increase in expression of defense genes and stress response genes in Arabidopsis thaliana due to abiotic stresses. BMC Plant Biol. 2022, 22, 104. [Google Scholar] [CrossRef]
- Mohidem, N.A.; Hashim, N.; Shamsudin, R.; Che Man, H. Rice for food security: Revisiting its rroduction, diversity, rice milling process and nutrient content. Agriculture 2022, 12, 741. [Google Scholar] [CrossRef]
- Chen, R.; Deng, Y.; Ding, Y.; Guo, J.; Qiu, J.; Wang, B.; Wang, C.; Xie, Y.; Zhang, Z.; Chen, J.; et al. Rice functional genomics: Decades’ efforts and roads ahead. Sci. China Life Sci. 2022, 65, 33–92. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Batley, J. DNA methylation: Toward crop disease resistance improvement. Trends Plant Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Song, X.; Wei, L.; Liu, C.; Cao, X. Epigenetic regulation and epigenomic landscape in rice. Natl. Sci. Rev. 2016, 3, 309–327. [Google Scholar] [CrossRef]
- Xue, Y.; Cao, X.; Chen, X.; Deng, X.; Deng, X.W.; Ding, Y.; Dong, A.; Duan, C.G.; Fang, X.; Gong, L.; et al. Epigenetics in the modern era of crop improvements. Sci. China Life Sci. 2025, 68, 1570–1609. [Google Scholar] [CrossRef]
- Liu, S.; Sretenovic, S.; Fan, T.; Cheng, Y.; Li, G.; Qi, A.; Tang, X.; Xu, Y.; Guo, W.; Zhong, Z.; et al. Hypercompact CRISPR–Cas12j2 (CasΦ) enables genome editing, gene activation, and epigenome editing in plants. Plant Commun. 2022, 3, 100453. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Alariqi, M.; Li, B.; Hussain, A.; Zhou, H.; Wang, Q.; Wang, F.; Wang, G.; Zhu, X.; Hui, F.; et al. CRISPR/dCas13(Rx) derived RNA N6-methyladenosine (m6A) dynamic modification in plant. Adv. Sci. 2024, 11, 2401118. [Google Scholar] [CrossRef]
- Song, X.; Tang, S.; Liu, H.; Meng, Y.; Luo, H.; Wang, B.; Hou, X.L.; Yan, B.; Yang, C.; Guo, Z.; et al. Inheritance of acquired adaptive cold tolerance in rice through DNA methylation. Cell 2025, 188, 4213–4224.E12. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Shirane, K.; Toh, H.; Kobayashi, H.; Miura, F.; Chiba, H.; Ito, T.; Kono, T.; Sasaki, H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013, 9, e1003439. [Google Scholar] [CrossRef]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Covering your bases: Inheritance of DNA methylation in plant genomes. Mol. Plant 2014, 7, 472–480. [Google Scholar] [CrossRef]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Vidalis, A.; Živković, D.; Wardenaar, R.; Roquis, D.; Tellier, A.; Johannes, F. Methylome evolution in plants. Genome Biol. 2016, 17, 264. [Google Scholar] [CrossRef]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef]
- Lloyd, J.P.B.; Lister, R. Epigenome plasticity in plants. Nat. Rev. Genet. 2022, 23, 55–68. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Z.Y.; Zeng, L.; Tanaka, K.; Zhang, C.J.; Ma, J.; Bai, G.; Wang, P.; Zhang, S.W.; Liu, Z.W.; et al. DTF1 is a core component of RNA-directed DNA methylation and may assist in the recruitment of Pol IV. Proc. Natl. Acad. Sci. USA 2013, 110, 8290–8295. [Google Scholar] [CrossRef]
- Law, J.A.; Du, J.; Hale, C.J.; Feng, S.; Krajewski, K.; Palanca, A.M.S.; Strahl, B.D.; Patel, D.J.; Jacobsen, S.E. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature 2013, 498, 385–389. [Google Scholar] [CrossRef]
- Wang, Y.; Le, B.H.; Wang, J.; You, C.; Zhao, Y.; Galli, M.; Xu, Y.; Gallavotti, A.; Eulgem, T.; Mo, B.; et al. ZMP recruits and excludes Pol IV–mediated DNA methylation in a site-specific manner. Sci. Adv. 2022, 8, eadc9454. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.J.; Jensen, M.B.; Dalmay, T.; Baulcombe, D.C. RNA polymerase IV directs silencing of endogenous DNA. Science 2005, 308, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Huettel, B.; Mette, M.F.; Aufsatz, W.; Jaligot, E.; Daxinger, L.; Kreil, D.P.; Matzke, M.; Matzke, A.J.M. Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat. Genet. 2005, 37, 761–765. [Google Scholar] [CrossRef]
- Onodera, Y.; Haag, J.R.; Ream, T.; Nunes, P.C.; Pontes, O.; Pikaard, C.S. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 2005, 120, 613–622. [Google Scholar] [CrossRef]
- Huang, K.; Wu, X.X.; Fang, C.L.; Xu, Z.G.; Zhang, H.W.; Gao, J.; Zhou, C.M.; You, L.L.; Gu, Z.X.; Mu, W.H.; et al. Pol IV and RDR2: A two-RNA-polymerase machine that produces double-stranded RNA. Science 2021, 374, 1579–1586. [Google Scholar] [CrossRef]
- Zhai, J.; Bischof, S.; Wang, H.; Feng, S.; Lee, T.; Teng, C.; Chen, X.; Park, S.Y.; Liu, L.; Gallego-Bartolome, J.; et al. One precursor one siRNA model for Pol IV-dependent siRNAs biogenesis. Cell 2015, 163, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 2015, 4, e09591. [Google Scholar] [CrossRef]
- Li, S.; Vandivier, L.E.; Tu, B.; Gao, L.; Won, S.Y.; Li, S.; Zheng, B.; Gregory, B.D.; Chen, X. Detection of Pol IV/RDR2-dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res. 2015, 25, 235–245. [Google Scholar] [CrossRef]
- Wang, Q.; Xue, Y.; Zhang, L.; Zhong, Z.; Feng, S.; Wang, C.; Xiao, L.; Yang, Z.; Harris, C.J.; Wu, Z.; et al. Mechanism of siRNA production by a plant Dicer-RNA complex in dicing-competent conformation. Science 2021, 374, 1152–1157. [Google Scholar] [CrossRef]
- Loffer, A.; Singh, J.; Fukudome, A.; Mishra, V.; Wang, F.; Pikaard, C.S. A DCL3 dicing code within Pol IV-RDR2 transcripts diversifies the siRNA pool guiding RNA-directed DNA methylation. eLife 2022, 11, e73260. [Google Scholar] [CrossRef] [PubMed]
- Rajeswaran, R.; Pooggin, M.M. RDR6-mediated synthesis of complementary RNA is terminated by miRNA stably bound to template RNA. Nucleic Acids Res. 2012, 40, 594–599. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Peragine, A.; Park, M.Y.; Poethig, R.S. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005, 19, 2164–2175. [Google Scholar] [CrossRef]
- Xie, Z.; Allen, E.; Wilken, A.; Carrington, J.C. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2005, 102, 12984–12989. [Google Scholar] [CrossRef] [PubMed]
- Elmayan, T.; Adenot, X.; Gissot, L.; Lauressergues, D.; Gy, I.; Vaucheret, H. A neomorphic sgs3 allele stabilizing miRNA cleavage products reveals that SGS3 acts as a homodimer. FEBS J. 2009, 276, 835–844. [Google Scholar] [CrossRef]
- Yang, Z.; Ebright, Y.W.; Yu, B.; Chen, X. HEN1 recognizes 21–24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucleic Acids Res. 2006, 34, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Sigman, M.J.; Panda, K.; Kirchner, R.; McLain, L.L.; Payne, H.; Peasari, J.R.; Husbands, A.Y.; Slotkin, R.K.; McCue, A.D. An siRNA-guided ARGONAUTE protein directs RNA polymerase V to initiate DNA methylation. Nat. Plants 2021, 7, 1461–1474. [Google Scholar] [CrossRef]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA Polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.K. Epigenetic gene regulation in plants and its potential applications in crop improvement. Nat. Rev. Mol. Cell Biol. 2025, 26, 51–67. [Google Scholar] [CrossRef]
- Naumann, U.; Daxinger, L.; Kanno, T.; Eun, C.; Long, Q.; Lorkovic, Z.J.; Matzke, M.; Matzke, A.J.M. Genetic evidence that DNA methyltransferase DRM2 has a direct catalytic role in RNA-directed DNA methylation in Arabidopsis thaliana. Genetics 2011, 187, 977–979. [Google Scholar] [CrossRef]
- Cao, X.; Jacobsen, S.E. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef]
- Moritoh, S.; Eun, C.H.; Ono, A.; Asao, H.; Okano, Y.; Yamaguchi, K.; Shimatani, Z.; Koizumi, A.; Terada, R. Targeted disruption of an orthologue of DOMAINS REARRANGED METHYLASE 2, OsDRM2, impairs the growth of rice plants by abnormal DNA methylation. Plant J. 2012, 71, 85–98. [Google Scholar] [CrossRef]
- Cheng, C.; Tarutani, Y.; Miyao, A.; Ito, T.; Yamazaki, M.; Sakai, H.; Fukai, E.; Hirochika, H. Loss of function mutations in the rice chromomethylase OsCMT3a cause a burst of transposition. Plant J. 2015, 83, 1069–1081. [Google Scholar] [CrossRef]
- Li, X.; Zhu, B.; Lu, Y.; Zhao, F.; Liu, Q.; Wang, J.; Ye, M.; Chen, S.; Nie, J.; Xiong, L.; et al. DNA methylation remodeling and the functional implication during male gametogenesis in rice. Genome Biol. 2024, 25, 84, https://doi.org/10.1186/s13059-024-03222-w. Correction in Genome Biol. 2024, 25, 196. [Google Scholar] [CrossRef]
- Yamauchi, T.; Moritoh, S.; Johzuka-Hisatomi, Y.; Ono, A.; Terada, R.; Nakamura, I.; Iida, S. Alternative splicing of the rice OsMET1 genes encoding maintenance DNA methyltransferase. J. Plant Physiol. 2008, 165, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhang, W.; Mohammadi, M.A.; He, Z.; She, Z.; Yan, M.; Shi, C.; Lin, L.; Wang, A.; Liu, J.; et al. OsDDM1b controls grain size by influencing cell cycling and regulating homeostasis and signaling of brassinosteroid in rice. Front. Plant Sci. 2022, 873993. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, X.; Yao, X.; Yu, R.; Larkin, P.J.; Liu, C.M. Mutations in the DNA demethylase OsROS1 result in a thickened aleurone and improved nutritional value in rice grains. Proc. Natl. Acad. Sci. USA 2018, 115, 11327–11332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, X.; Liu, Q.; Zhao, Y.; Jiang, W.; Wu, A.; Zhou, D.X. DNA demethylases remodel DNA methylation in rice gametes and zygote and are required for reproduction. Mol. Plant 2021, 14, 1569–1583. [Google Scholar] [CrossRef]
- Kim, M.Y.; Ono, A.; Scholten, S.; Kinoshita, T.; Zilberman, D.; Okamoto, T.; Fischer, R.L. DNA demethylation by ROS1a in rice vegetative cells promotes methylation in sperm. Proc. Natl. Acad. Sci. USA 2019, 116, 9652–9657. [Google Scholar] [CrossRef]
- Li, C.; Kong, J.R.; Yu, J.; He, Y.Q.; Yang, Z.K.; Zhuang, J.J.; Ruan, C.C.; Yan, Y.; Xu, J.H. DNA demethylase gene OsDML4 controls salt tolerance by regulating the ROS homeostasis and the JA signaling in rice. Environ. Exp. Bot. 2023, 209, 105276. [Google Scholar] [CrossRef]
- Yan, Y.; Li, C.; Liu, Z.; Zhuang, J.J.; Kong, J.R.; Yang, Z.K.; Yu, J.; Shah Alam, M.; Ruan, C.C.; Zhang, H.M.; et al. A new demethylase gene, OsDML4, is involved in high temperature-increased grain chalkiness in rice. J. Exp. Bot. 2022, 73, 7273–7284. [Google Scholar] [CrossRef]
- Tan, F.; Zhou, C.; Zhou, Q.; Zhou, S.; Yang, W.; Zhao, Y.; Li, G.; Zhou, D.X. Analysis of chromatin regulators reveals specific features of rice DNA methylation pathways. Plant Physiol. 2016, 171, 2041–2054. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Haag, J.R.; Pikaard, C.S. Multisubunit RNA polymerases IV and V: Purveyors of non-coding RNA for plant gene silencing. Nat. Rev. Mol. Cell Biol. 2011, 12, 483–492. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhao, C.; Zhang, Q.; Zinta, G.; Wang, D.; Lozano-Durán, R.; Zhu, J.K. Pathway conversion enables a double-lock mechanism to maintain DNA methylation and genome stability. Proc. Natl. Acad. Sci. USA 2021, 118, e2107320118. [Google Scholar] [CrossRef]
- Yadav, N.S.; Khadka, J.; Domb, K.; Zemach, A.; Grafi, G. CMT3 and SUVH4/KYP silence the exonic Evelknievel retroelement to allow for reconstitution of CMT1 mRNA. Epigenetics Chromatin 2018, 11, 69. [Google Scholar] [CrossRef]
- Sasaki, E.; Kawakatsu, T.; Ecker, J.R.; Nordborg, M. Common alleles of CMT2 and NRPE1 are major determinants of CHH methylation variation in Arabidopsis thaliana. PLoS Genet. 2019, 15, e1008492. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.H.; Richards, E.J.; Chung, K.M.; Woo, H.R. Arabidopsis VIM proteins regulate epigenetic silencing by modulating DNA methylation and histone modification in cooperation with MET1. Mol. Plant 2014, 7, 1470–1485. [Google Scholar] [CrossRef]
- Higo, H.; Tahir, M.; Takashima, K.; Miura, A.; Watanabe, K.; Tagiri, A.; Ugaki, M.; Ishikawa, R.; Eiguchi, M.; Kurata, N.; et al. DDM1 (Decrease in DNA Methylation) genes in rice (Oryza sativa). Mol. Genet. Genom. 2012, 287, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Groth, M.; Moissiard, G.; Wirtz, M.; Wang, H.; Garcia-Salinas, C.; Ramos-Parra, P.A.; Bischof, S.; Feng, S.; Cokus, S.J.; John, A.; et al. MTHFD1 controls DNA methylation in Arabidopsis. Nat. Commun. 2016, 7, 11640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Zhang, F.F.; Ma, Z.Y.; Huang, H.W.; Jiang, L.; Cai, T.; Zhu, J.K.; Zhang, C.; He, X.J. Folate polyglutamylation is involved in chromatin silencing by maintaining global DNA methylation and histone H3K9 dimethylation in Arabidopsis. Plant Cell 2013, 25, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32, 33–42. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.R.; Krawczyk, C.; Robertson, A.B.; Kuśnierczyk, A.; Vågbø, C.B.; Schuermann, D.; Klungland, A.; Schär, P. Biochemical reconstitution of TET1–TDG–BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 2016, 7, 10806. [Google Scholar] [CrossRef]
- Liu, S.; Dunwell, T.L.; Pfeifer, G.P.; Dunwell, J.M.; Ullah, I.; Wang, Y. Detection of oxidation products of 5-methyl-2′-deoxycytidine in Arabidopsis DNA. PLoS ONE 2013, 8, e84620. [Google Scholar] [CrossRef]
- Tang, Y.; Xiong, J.; Jiang, H.P.; Zheng, S.J.; Feng, Y.Q.; Yuan, B.F. Determination of oxidation products of 5-methylcytosine in plants by chemical derivatization coupled with liquid chromatography/tandem mass spectrometry analysis. Anal. Chem. 2014, 86, 7764–7772. [Google Scholar] [CrossRef]
- Kalinka, A.; Starczak, M.; Gackowski, D.; Stępień, E.; Achrem, M. Global DNA 5-hydroxymethylcytosine level and its chromosomal distribution in four rye species. J. Exp. Bot. 2023, 74, 3488–3502. [Google Scholar] [CrossRef]
- Hollwey, E.; Watson, M.; Meyer, P. Expression of the c-terminal domain of mammalian TET3 DNA dioxygenase in Arabidopsis thaliana induces heritable methylation changes at rDNA loci. Adv. Biosci. Biotechnol. 2016, 7, 243–250. [Google Scholar] [CrossRef][Green Version]
- Liu, Q.; Xue, Q.; Xu, J. Phylogenetic analysis of DNA demethylase genes in angiosperm. Hereditas 2014, 36, 276–285. [Google Scholar]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef]
- Stroud, H.; Feng, S.; Morey Kinney, S.; Pradhan, S.; Jacobsen, S.E. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011, 12, R54. [Google Scholar] [CrossRef] [PubMed]
- Urich, M.A.; Nery, J.R.; Lister, R.; Schmitz, R.J.; Ecker, J.R. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat. Protoc. 2015, 10, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Guan, X.; Hu, Y.; Zhang, Z.; Yang, H.; Shi, X.; Han, J.; Mei, H.; Wang, L.; Shao, L.; et al. Population-wide DNA methylation polymorphisms at single-nucleotide resolution in 207 cotton accessions reveal epigenomic contributions to complex traits. Cell Res. 2024, 34, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Saini, R.; Upadhyay, R.; Tiwari, K.N. Revealing the Epigenetic Mechanisms Underlying the Stress Response in Medicinal Plants. In Stress-Responsive Factors and Molecular Farming in Medicinal Plants; Singh, D., Mishra, A.K., Srivastava, A.K., Eds.; Springer Nature: Singapore, 2023; pp. 107–122. ISBN 978-981-9944-80-4. [Google Scholar]
- Simko, I. Differentially methylated genomic regions of lettuce seeds relate to divergence across morphologically distinct horticultural types. AoB Plants 2023, 15, plad060. [Google Scholar] [CrossRef]
- Tost, J. Current and Emerging Technologies for the Analysis of the Genome-Wide and Locus-Specific DNA Methylation Patterns. In DNA Methyltransferases-Role and Function; Jeltsch, A., Jurkowska, R.Z., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 343–430. ISBN 978-3-319-43624-1. [Google Scholar]
- Full Article: Serial Pyrosequencing for Quantitative DNA Methylation Analysis. Available online: https://www.tandfonline.com/doi/10.2144/000112190?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed (accessed on 16 August 2025).
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Park, B.; Min, J.H.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- Melnikov, A.A.; Gartenhaus, R.B.; Levenson, A.S.; Motchoulskaia, N.A.; Levenson, V.V. MSRE-PCR for analysis of gene-specific DNA methylation. Nucleic Acids Res. 2005, 33, e93. [Google Scholar] [CrossRef]
- Shareef, S.J.; Bevill, S.M.; Raman, A.T.; Aryee, M.J.; van Galen, P.; Hovestadt, V.; Bernstein, B.E. Extended representation bisulfite sequencing of gene regulatory elements in multiplexed samples and single cells. Nat. Biotechnol. 2021, 39, 1086–1094. [Google Scholar] [CrossRef]
- Kawakatsu, T. Whole-Genome Bisulfite Sequencing and Epigenetic Variation in Cereal Methylomes. In Cereal Genomics: Methods and Protocols; Vaschetto, L.M., Ed.; Springer: New York, NY, USA, 2020; pp. 119–128. ISBN 978-1-4939-9865-4. [Google Scholar]
- Lentini, A.; Nestor, C.E. Mapping DNA Methylation in Mammals: The State of the Art. In DNA Modifications: Methods and Protocols; Ruzov, A., Gering, M., Eds.; Springer: New York, NY, USA, 2021; pp. 37–50. ISBN 978-1-07-160876-0. [Google Scholar]
- Gitan, R.S.; Shi, H.; Chen, C.M.; Yan, P.S.; Huang, T.H.M. Methylation-specific oligonucleotide microarray: A new potential for high-throughput methylation analysis. Genome Res. 2002, 12, 158–164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Tan, L.; Xiong, L.; Xu, W.; Wu, F.; Huang, N.; Xu, Y.; Kong, L.; Zheng, L.; Schwartz, L.; Shi, Y.; et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP-seq method. Nucleic Acids Res. 2013, 41, e84. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Cui, N.; Chen, X.; Shi, Y.; Chi, M.; Hu, J.; Lai, K.; Wang, Z.; Wang, H. Changes in the epigenome and transcriptome of rice in response to Magnaporthe oryzae infection. Crop J. 2021, 9, 843–853. [Google Scholar] [CrossRef]
- Cao, S.; Chen, K.; Lu, K.; Chen, S.; Zhang, X.; Shen, C.; Zhu, S.; Niu, Y.; Fan, L.; Chen, Z.J.; et al. Asymmetric variation in DNA methylation during domestication and de-domestication of rice. Plant Cell 2023, 35, 3429–3443. [Google Scholar] [CrossRef]
- Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Li, C.; Buell, C.R. The rice genome annotation project: An updated database for mining the rice genome. Nucleic Acids Res. 2025, 53, D1614–D1622. [Google Scholar] [CrossRef]
- Hu, J.; Chen, X.; Zhang, H.; Ding, Y. Genome-wide analysis of DNA methylation in photoperiod- and thermo-sensitive male sterile rice Peiai 64S. BMC Genom. 2015, 16, 102. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Z.; Qin, R.; Qiu, Y.; Wang, J.L.; Cui, X.; Gu, L.; Zhang, X.; Guo, X.; Wang, D.; et al. Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 2012, 24, 4407–4421. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Chaudhuri, S. Analysis of DNA Methylation Profile in Plants by Chop-PCR. In Plant Innate Immunity: Methods and Protocols; Gassmann, W., Ed.; Springer: New York, NY, USA, 2019; pp. 79–90. ISBN 978-1-4939-9458-8. [Google Scholar]
- Li, J.; Yang, D.L.; Huang, H.; Zhang, G.; He, L.; Pang, J.; Lozano-Durán, R.; Lang, Z.; Zhu, J.K. Epigenetic memory marks determine epiallele stability at loci targeted by de novo DNA methylation. Nat. Plants 2020, 6, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Knowledge of Rice Growth Stages Important. Available online: https://www.lsuagcenter.com/portals/our_offices/research_stations/rice/features/publications/knowledge-of-rice-growth-stages-important (accessed on 18 June 2025).
- Jiang, W.; Zhou, Z.; Li, X.; Zhao, Y.; Zhou, S. DNA methylation dynamics play crucial roles in shaping the distinct transcriptomic profiles for different root-type initiation in rice. Genome Biol. 2025, 26, 99. [Google Scholar] [CrossRef]
- Zhang, C.C.; Yuan, W.Y.; Zhang, Q.F. RPL1, a gene involved in epigenetic processes regulates phenotypic plasticity in rice. Mol. Plant 2012, 5, 482–493. [Google Scholar] [CrossRef][Green Version]
- Lu, Y.; Gharib, A.; Chen, R.; Wang, H.; Tao, T.; Zuo, Z.; Bu, Q.; Su, Y.; Li, Y.; Luo, Y.; et al. Rice melatonin deficiency causes premature leaf senescence via DNA methylation regulation. Crop J. 2024, 12, 721–731. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Y.; Luo, W.; Li, W.; Chen, N.; Zhang, D.; Chong, K. The F-box protein OsFBK12 targets OsSAMS1 for degradation and affects pleiotropic phenotypes, including leaf senescence, in rice. Plant Physiol. 2013, 163, 1673–1685. [Google Scholar] [CrossRef]
- Abe, M.; Yoshikawa, T.; Nosaka, M.; Sakakibara, H.; Sato, Y.; Nagato, Y.; Itoh, J. WAVY LEAF1, an ortholog of Arabidopsis HEN1, regulates shoot development by maintaining microRNA and trans-acting small interfering RNA accumulation in rice. Plant Physiol. 2010, 154, 1335–1346. [Google Scholar] [CrossRef]
- Xu, L.; Yuan, K.; Yuan, M.; Meng, X.; Chen, M.; Wu, J.; Li, J.; Qi, Y. Regulation of rice tillering by RNA-directed DNA methylation at miniature inverted-repeat transposable elements. Mol. Plant 2020, 13, 851–863. [Google Scholar] [CrossRef]
- Li, W.; Han, Y.; Tao, F.; Chong, K. Knockdown of SAMS genes encoding S-adenosyl-l-methionine synthetases causes methylation alterations of DNAs and histones and leads to late flowering in rice. J. Plant Physiol. 2011, 168, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Shi, D.Q.; Chen, Y.H. Female gametophyte development in flowering plants. Annu. Rev. Plant Biol. 2010, 61, 89–108. [Google Scholar] [CrossRef]
- Hafidh, S.; Honys, D. Reproduction multitasking: The male gametophyte. Annu. Rev. Plant Biol. 2021, 72, 581–614. [Google Scholar] [CrossRef]
- Ono, A.; Yamaguchi, K.; Fukada-Tanaka, S.; Terada, R.; Mitsui, T.; Iida, S. A null mutation of ROS1a for DNA demethylation in rice is not transmittable to progeny. Plant J. 2012, 71, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, K.; Zeng, L.; Xu, D.; Zhu, T.; Yin, Y.; Zhan, H.; Wu, Y.; Yang, D.-L. Reinforcement of CHH methylation through RNA-directed DNA methylation ensures sexual reproduction in rice. Plant Physiol. 2021, 188, 1189–1209. [Google Scholar] [CrossRef]
- Ren, D.; Ding, C.; Qian, Q. Molecular bases of rice grain size and quality for optimized productivity. Sci. Bull. 2023, 68, 314–350. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Pan, T.; Zhu, Y.; Jiang, X.; Yu, M.; Wang, R.; Zhang, F.; Luo, S.; Bao, X.; Chen, Y.; et al. FLOURY ENDOSPERM20 encoding SHMT4 is required for rice endosperm development. Plant Biotechnol. J. 2022, 20, 1438–1440. [Google Scholar] [CrossRef]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W.; Gan, Y.; Sun, P.; Xiang, G.; Zhang, H.; et al. The methylation patterns and transcriptional responses to chilling stress at the seedling stage in rice. Int. J. Mol. Sci. 2019, 20, 5089. [Google Scholar] [CrossRef]
- Kim, W.; Iizumi, T.; Nishimori, M. Global patterns of crop production losses associated with droughts from 1983 to 2009. J. Appl. Meteorol. Climatol. 2019, 58, 1233–1244. [Google Scholar] [CrossRef]
- Kou, S.; Gu, Q.; Duan, L.; Liu, G.; Yuan, P.; Li, H.; Wu, Z.; Liu, W.; Huang, P.; Liu, L. Genome-wide bisulphite sequencing uncovered the contribution of DNA methylation to rice short-term drought memory formation. J. Plant Growth Regul. 2022, 41, 2903–2917. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef]
- Wang, J.; Nan, N.; Li, N.; Liu, Y.; Wang, T.J.; Hwang, I.; Liu, B.; Xu, Z.Y. A DNA methylation reader–chaperone regulator–transcription factor complex activates OsHKT1;5 expression during salinity stress. Plant Cell 2020, 32, 3535–3558. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Fahad, M.; Zhang, L.; Wu, L.; Wu, X. Microrchidia OsMORC6 positively regulates cadmium tolerance and uptake by mediating DNA methylation in rice. Rice 2025, 18, 25. [Google Scholar] [CrossRef]
- Cong, W.; Miao, Y.; Xu, L.; Zhang, Y.; Yuan, C.; Wang, J.; Zhuang, T.; Lin, X.; Jiang, L.; Wang, N.; et al. Transgenerational memory of gene expression changes induced by heavy metal stress in rice (Oryza sativa L.). BMC Plant Biol. 2019, 19, 282. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhu, J.; Guo, R.; Whelan, J.; Shou, H. DNA methylation is involved in acclimation to iron-deficiency in rice (Oryza sativa). Plant J. 2021, 107, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xing, L.; Li, Z.; Jiang, D. Epigenetic control of plant abiotic stress responses. J. Genet. Genom. 2025, 52, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wang, L.; Han, T.; Ye, W.; Liu, Y.; Sun, Y.; Moose, S.P.; Song, Q.; Chen, Z.J. Small RNAs mediate transgenerational inheritance of genome-wide trans-acting epialleles in maize. Genome Biol. 2022, 23, 53. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, I.; Coughlin, M.W.; Perley, D.A.; Yao, Y.; Lu, W.; Cenko, S.B.; Kumar, H.; Anand, S.; Ho, A.Y.Q.; Kasliwal, M.M.; et al. A very luminous jet from the disruption of a star by a massive black hole. Nature 2022, 612, 430–434. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Q.; Kou, H.; Wang, D.; Lin, X.; Wu, Y.; Xu, C.; Xing, S.; Liu, B. Induced Pib expression and resistance to Magnaporthe grisea are compromised by cytosine demethylation at critical promoter regions in rice. J. Integr. Plant Biol. 2011, 53, 814–823. [Google Scholar] [CrossRef]
- Sheng, C.; Li, X.; Xia, S.; Zhang, Y.; Yu, Z.; Tang, C.; Xu, L.; Wang, Z.; Zhang, X.; Zhou, T.; et al. An OsPRMT5-OsAGO2/miR1875-OsHXK1 module regulates rice immunity to blast disease. J. Integr. Plant Biol. 2023, 65, 1077–1095. [Google Scholar] [CrossRef]
- Campo, S.; Sánchez-Sanuy, F.; Camargo-Ramírez, R.; Gómez-Ariza, J.; Baldrich, P.; Campos-Soriano, L.; Soto-Suárez, M.; San Segundo, B. A novel Transposable element-derived microRNA participates in plant immunity to rice blast disease. Plant Biotechnol. J. 2021, 19, 1798–1811. [Google Scholar] [CrossRef]
- Deng, Y.; Zhai, K.; Xie, Z.; Yang, D.; Zhu, X.; Liu, J.; Wang, X.; Qin, P.; Yang, Y.; Zhang, G.; et al. Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 2017, 355, 962–965. [Google Scholar] [CrossRef]
- Zhang, H.; Tao, Z.; Hong, H.; Chen, Z.; Wu, C.; Li, X.; Xiao, J.; Wang, S. Transposon-derived small RNA is responsible for modified function of WRKY45 locus. Nat. Plants 2016, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhou, Y.; Mao, L.; Ye, C.; Wang, W.; Zhang, J.; Yu, Y.; Fu, F.; Wang, Y.; Qian, F.; et al. Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 2017, 8, 15323. [Google Scholar] [CrossRef]
- Yan, T.; Kuang, L.; Gao, F.; Chen, J.; Li, L.; Wu, D. Differentiation of genome-wide DNA methylation between japonica and indica rice. Plant J. 2025, 121, e17218. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Y.; Gui, Y.; An, D.; Liu, J.; Xu, X.; Li, Q.; Wang, J.; Wang, W.; Shi, C.; et al. Elimination of a retrotransposon for quenching genome instability in modern rice. Mol. Plant 2019, 12, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xing, M.; Zhao, Z.; Gu, Y.; Xiao, Y.; Liu, Q.; Xue, H. DNA methylation modification in heterosis initiation through analyzing rice hybrid contemporary seeds. Crop J. 2021, 9, 1179–1190. [Google Scholar] [CrossRef]
- Ma, X.; Xing, F.; Jia, Q.; Zhang, Q.; Hu, T.; Wu, B.; Shao, L.; Zhao, Y.; Zhang, Q.; Zhou, D.X. Parental variation in CHG methylation is associated with allelic-specific expression in elite hybrid rice. Plant Physiol. 2021, 186, 1025–1041. [Google Scholar] [CrossRef]
- Sigurpalsdottir, B.D.; Stefansson, O.A.; Holley, G.; Beyter, D.; Zink, F.; Hardarson, M.Þ.; Sverrisson, S.Þ.; Kristinsdottir, N.; Magnusdottir, D.N.; Magnusson, O.Þ.; et al. A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes. Genome Biol. 2024, 25, 69. [Google Scholar] [CrossRef]
- Liu, J.; Zhong, X. Population epigenetics: DNA methylation in the plant omics era. Plant Physiol. 2024, 194, 2039–2048. [Google Scholar] [CrossRef]
- Rozov, S.M.; Permyakova, N.V.; Deineko, E.V. The problem of the low rates of CRISPR/Cas9-mediated knock-ins in plants: Approaches and solutions. Int. J. Mol. Sci. 2019, 20, 3371. [Google Scholar] [CrossRef]
- Yan, N.; Feng, H.; Sun, Y.; Xin, Y.; Zhang, H.; Lu, H.; Zheng, J.; He, C.; Zuo, Z.; Yuan, T.; et al. Cytosine base editors induce off-target mutations and adverse phenotypic effects in transgenic mice. Nat. Commun. 2023, 14, 1784. [Google Scholar] [CrossRef]
- Shankar, S.; Sreekumar, A.; Prasad, D.; Das, A.V.; Pillai, M.R. Genome editing of oncogenes with ZFNs and TALENs: Caveats in nuclease design. Cancer Cell Int. 2018, 18, 169. [Google Scholar] [CrossRef]
- Gong, Z.; Previtera, D.A.; Wang, Y.; Botella, J.R. Geminiviral-induced genome editing using miniature CRISPR/Cas12j (CasΦ) and Cas12f variants in plants. Plant Cell Rep. 2024, 43, 71. [Google Scholar] [CrossRef]
- Tang, X.; Ren, Q.; Yan, X.; Zhang, R.; Liu, L.; Han, Q.; Zheng, X.; Qi, Y.; Song, H.; Zhang, Y. Boosting genome editing in plants with single transcript unit surrogate reporter systems. Plant Commun. 2024, 5, 100921. [Google Scholar] [CrossRef]
- Přibylová, A.; Fischer, L.; Pyott, D.; Bassett, A.; Molnar, A. DNA methylation can alter CRISPR/Cas9 editing frequency and DNA repair outcome in a target-specific manner. New Phytol. 2022, 235, 2285–2299. [Google Scholar] [CrossRef]
- Aussel, C.; Cathomen, T.; Fuster-García, C. The hidden risks of CRISPR/Cas: Structural variations and genome integrity. Nat. Commun. 2025, 16, 7208. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Yang, C.; Wang, D.; Deng, X.; Cao, X.; Song, X. Targeted DNA demethylation produces heritable epialleles in rice. Sci. China Life Sci. 2022, 65, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cao, P.; Wang, C.; Lai, J.; Deng, Y.; Li, C.; Hao, Y.; Wu, Z.; Chen, R.; Qiang, Q.; et al. Population analysis reveals the roles of DNA methylation in tomato domestication and metabolic diversity. Sci. China Life Sci. 2023, 66, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Yu, J.; Li, X.; Li, J.; Fan, J.; Liu, H.; Wei, W.; Zhang, L.; Gu, K.; Liu, D.; et al. Increased DNA methylation contributes to the early ripening of pear fruits during domestication and improvement. Genome Biol. 2024, 25, 87. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).