
Identification of Four New Mutations in the GLA Gene Associated with Anderson–Fabry Disease


Abstract
:1. Introduction
2. Results
2.1. CASE 1: Mutation p.G261C
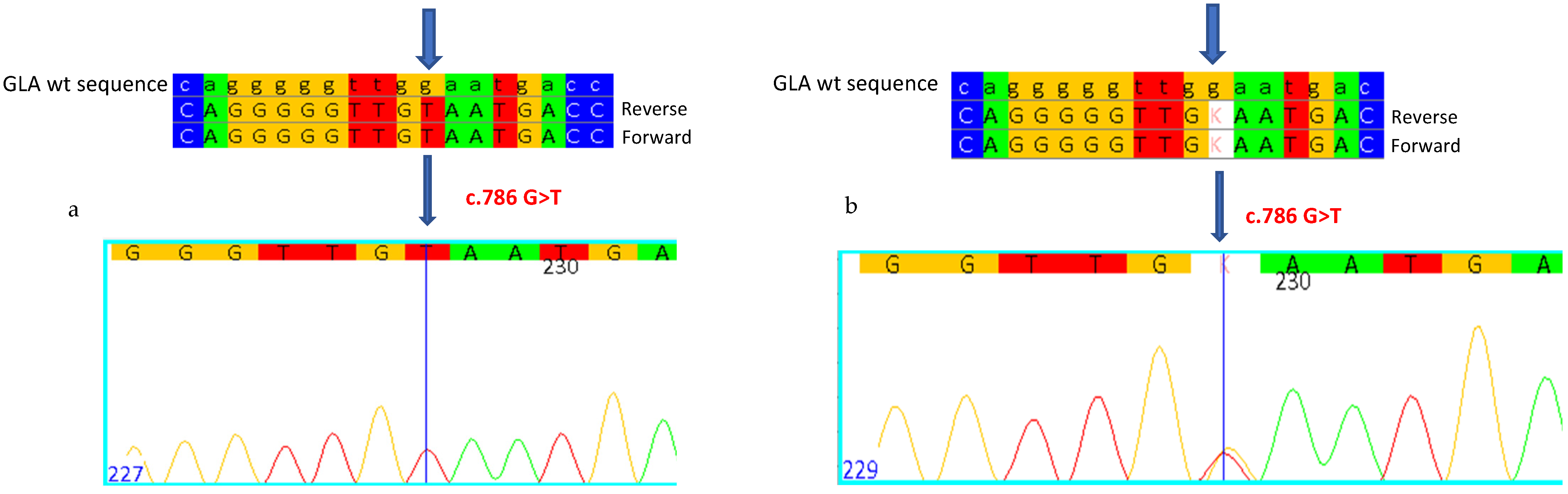
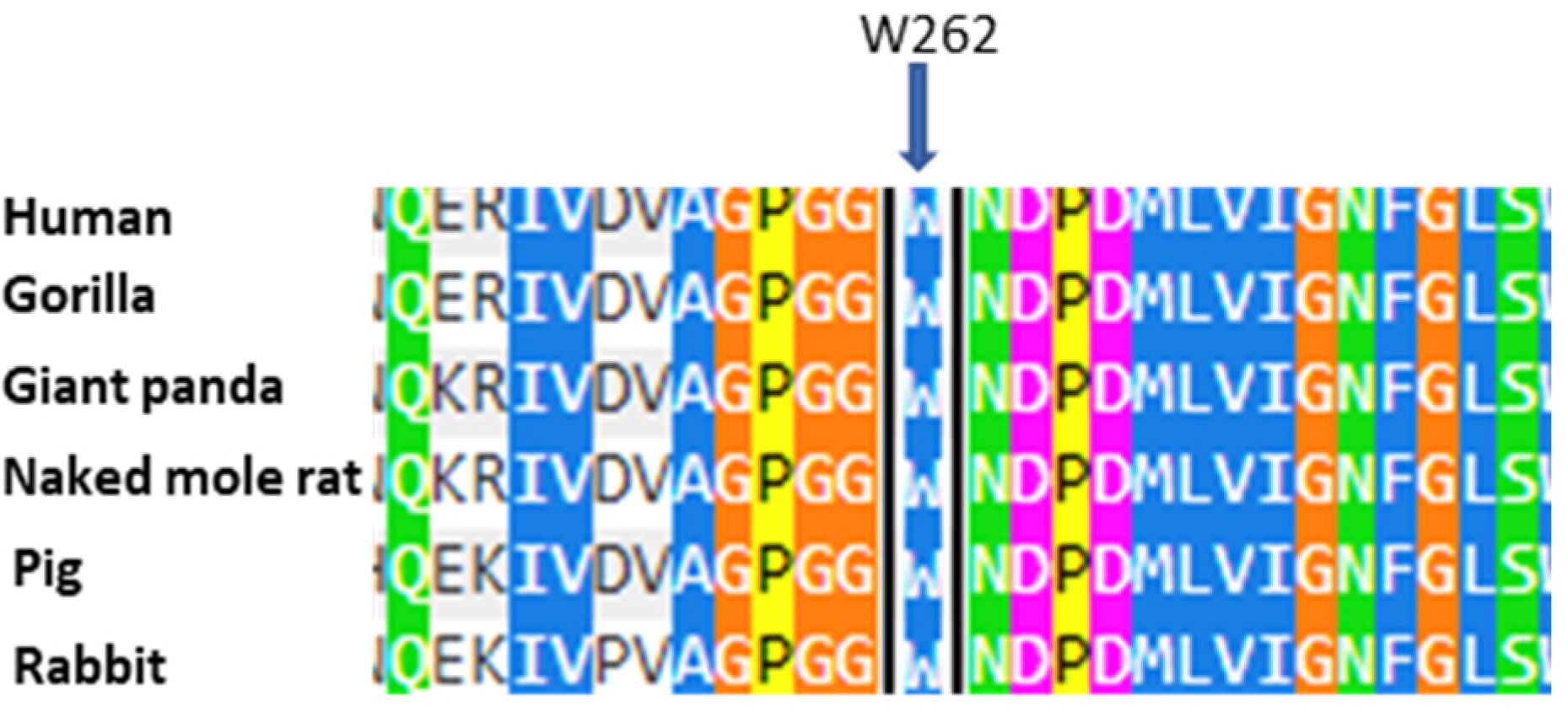
2.2. CASE 2: Mutation c.786G>T, Which Determines p.W262C
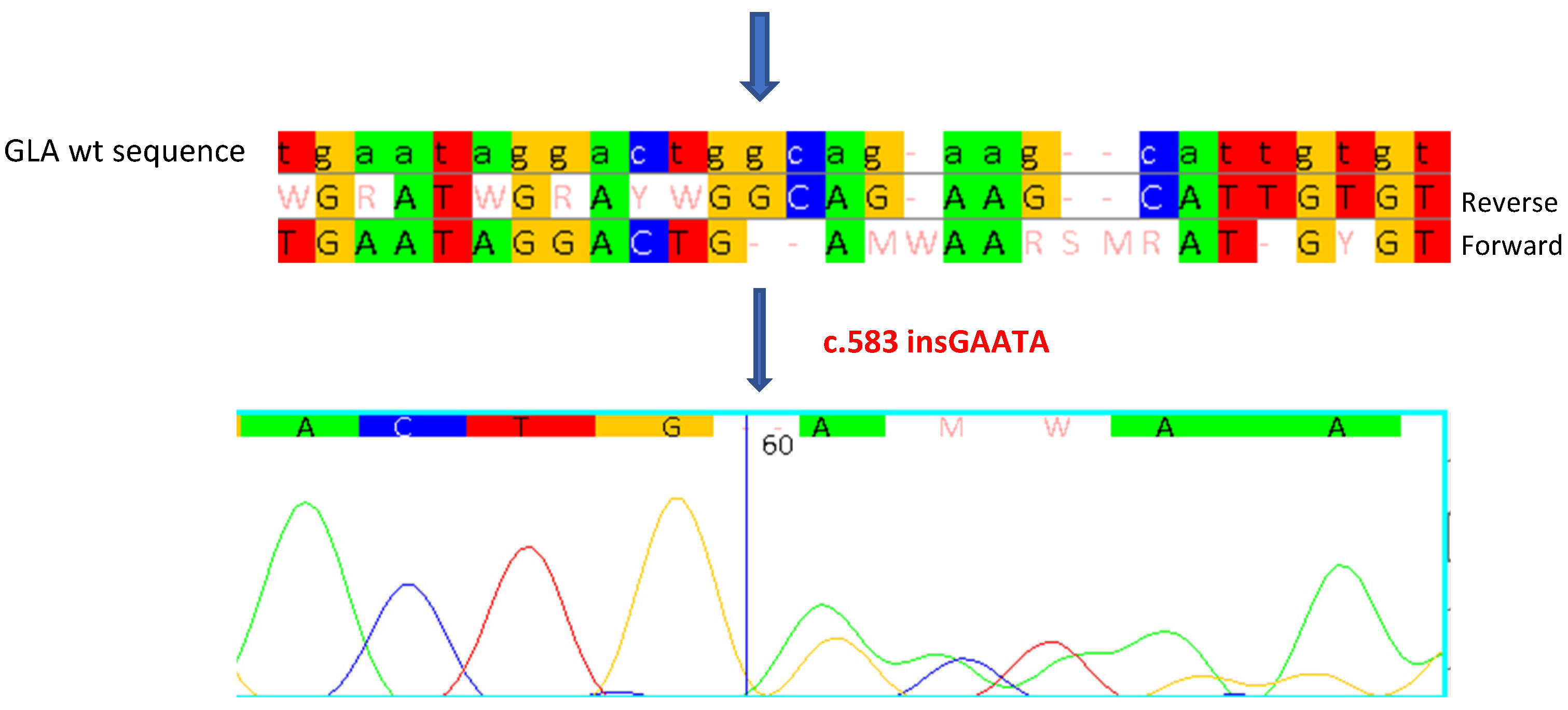
2.3. CASE 3: Mutation c.583insGAATA
2.4. CASE 4: Mutation p.Y207X+p.A143T
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genetic Analysis
4.3. α-Galactosidase A Activity Assay
4.4. Lyso-Gb3 Determination
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. Alpha-galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Bertoldi, G.; Caputo, I.; Driussi, G.; Stefanelli, L.F.; Di Vico, V.; Carraro, G.; Nalesso, F.; Calò, L.A. Biochemical Mechanisms beyond Glycosphingolipid Accumulation in Fabry Disease: Might They Provide Additional Therapeutic Treatments? J. Clin. Med. 2023, 12, 2063. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA gene and LysoGb3: Is it really Anderson-Fabry disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M.J.; Kasper, D.C. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease. Mol. Genet. Metab. 2018, 123, 148–153. [Google Scholar] [CrossRef]
- Kramer, J.; Weidemann, F. Biomarkers for diagnosing and staging of Fabry disease. Curr. Med. Chem. 2018, 25, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Gambarin, F.I.; Disabella, E.; Narula, J.; Diegoli, M.; Grasso, M.; Serio, A.; Favalli, B.M.; Agozzino, M.; Tavazzi, L.; Fraser, A.G.; et al. When should cardiologists suspect Anderson-Fabry disease? Am. J. Cardiol. 2010, 6, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.L.; Vieira, T.A.; Giugliani, R.; Schwartz, I.V. Expression of the disease on female carriers of X-linked lysosomal disorders: A brief review. Orphanet J. Rare Dis. 2010, 5, 14. [Google Scholar] [CrossRef]
- Hoffmann, B.; Mayatepek, E. Fabry disease-often seen, rarely diagnosed. Dtsch. Arztebl. Int. 2009, 106, 440–447. [Google Scholar] [PubMed]
- Duro, G.; Anania, M.; Zizzo, C.; Francofonte, D.; Giacalone, I.; D’Errico, A.; Marsana, E.M.; Colomba, P. Disgnosis of Fabry Disease Using Alpha-Galactosidase A Activity or LysoGb3 in Blood Fails to Identify Up to Two Thirds of Female Patients. Int. J. Mol. Sci. 2024, 25, 5158. [Google Scholar] [CrossRef]
- Nakao, S.; Kodama, C.; Takenaka, T.; Tanaka, A.; Yasumoto, Y.; Yoshida, A.; Kanzaki, T.; Enriquez, A.L.; Eng, C.M.; Tanaka, H.; et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003, 64, 801–807. [Google Scholar] [PubMed]
- Morey, C.; Avner, P. Genetics and epigenetics of the X chromosome. Ann. N. Y Acad. Sci. 2010, 1214, E18–E33. [Google Scholar]
- Lidove, O.; Kaminsky, P.; Hachulla, E.; Leguy-Seguin, V.; Lavigne, C.; Marie, I.; Maillot, F.; Serratrice, C.; Masseau, A.; Chérin, P.; et al. Fabry disease ‘The New Great Imposter’: Results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin. Genet. 2012, 81, 571–577. [Google Scholar] [PubMed]
- Metha, A. New developments in the management of Anderson-Fabry disease. QJM 2002, 95, 647–653. [Google Scholar]
- The Human Gene Mutation Database. 2021. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GLA (accessed on 3 September 2021).
- Rocchetti, M.T.; Spadaccino, F.; Catalano, V.; Zaza, G.; Stallone, G.; Fiocco, D.; Netti, G.S.; Ranieri, E. Metabolic Fingerprinting of Fabry Disease: Diagnostic and Prognostic Aspects. Metabolites 2022, 2, 703. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Oliveira, J.P.; Bichet, D.G.; Yoo, H.W.; Hopkin, R.J.; Lemay, R.; Politei, J.; Wanner, C.; Wilcox, W.R.; Warnock, D.G. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispeciality Fabry disease genotype-phenotype workgroup. J. Med. Genet. 2020, 57, 542–551. [Google Scholar] [CrossRef]
- Marchesoni, C.L.; Roa, N.; Pardal, A.M.; Neumann, P.; Cáceres, G.; Martínez, P.; Kisinovsky, I.; Bianchi, S.; Tarabuso, A.L.; Reisin, R.C. Misdiagnosis in Fabry disease. J. Pediatr. 2010, 156, 828–831. [Google Scholar]
- Rigold, M.; Concolino, D.; Morrone, A.; Pieruzzi, F.; Ravaglia, R.; Furlan, F.; Santus, F.; Strisciuglio, P.; Torti, G.; Parini, G. Intrafamilia phenotypic variability in four families with Anderson-Fabry disease. Clin. Genet. 2014, 86, 258–263. [Google Scholar]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef]
- De Brabander, I.; Yperzeele, L.; Ceuterick-De Groote, C.; Brouns, R.; Baker, R.; Belachew, S.; Delbecq, J.; De Keulenaer, G.; Dethy, S.; Eyskens, F.; et al. Phenotypical characterization of α-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young. Clin. Neurol. Neurosurg. 2013, 115, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M.; van der Beergh Weerman, M.A.; Florquin, S.; Kok, W.E.M.; Lekanne Deprez, R.H.; Timmermans, J.; Linthorst, G.E. Diagnostic dilemmas in Fabry disease: A case series study on GLA mutations on unknow clinical significance. Clin. Genet. 2015, 88, 161–166. [Google Scholar] [CrossRef]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef]
- Spinelli, L.; Pisani, A.; Sabbatini, M.; Petretta, M.; Andreucci, M.V.; Procaccini, D.; Lo Surdo, N.; Federico, S.; Cianciaruso, B. Enzyme replacement therapy with algasidase beta improves cardiac involvement in Fabry’s disease. Clin. Genet. 2004, 66, 158–165. [Google Scholar] [PubMed]
- Yoshimitsu, M.; Higuchi, K.; Miyata, M.; Devine, S.; Mattman, A.; Sirrs, S.; Medin, J.A.; Tei, C.; Takenaka, T. Identification of novel mutations in the α-galactosidase A gene in patients with Fabry disease: Pitfalls of mutation analyses in patients with low α-galactosidase A activity. J. Cardiol. 2011, 57, 345–353. [Google Scholar]
- Altarescu, G.M.; Goldfarb, L.G.; Park, K.Y.; Kanescki, C.; Jeffries, N.; Litvak, S.; Nagle, J.W.; Schiffmann, R. Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin. Genet. 2001, 60, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.; Baron, K.; Widmer, U.; Deegan, P.; Neumann, H.P.H.; Sunder-Plassmann, G.; Johansson, J.O.; Whybra, C.; Ries, M.; Pastores, G.M.; et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum. Mutat. 2005, 25, 412. [Google Scholar] [CrossRef]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar]
- Whybra, C.; Kampmann, C.; Willers, I.; Davies, J.; Winchester, B.; Kriegsmann, J.; Bruhl, K.; Gal, A.; Bunge, S.; Beck, M. Anderson-Fabry disease: Clinical manifestations of disease in female heterozygotes. J. Inherit. Metab. Dis. 2001, 24, 715–724. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Fabry registry. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Mutation | Alpha-Galactosidase A Activity (nmol/mL/h) Normal Range: >3.0 nmol/mL/h | Lyso-Gb3 in Blood (nmol/L) Normal Range: <2.3 nmol/L | Clinical Signs |
|---|---|---|---|---|---|---|
| Proband | F | 51 | p.G261C | 1.4 | 2.31 | Conduction defects, tortuosity of retinal blood vessels, hypertension, TIA event, cornea verticillata, albuminuria, and proteinuria |
| Brother | M | 54 | p.G261C | 0.7 | 29.9 | Acroparesthesia in childhood, abdominal pain with gastrointestinal manifestations, intolerance to heat and/or cold, LVH, tortuosity of retinal blood vessels, hearing loss, and hypertension |
| Sister | F | 53 | p.G261C | 1.8 | 4.88 | LVH, hypertension, hearing loss, and cornea verticillata |
| Patient | Sex | Age | Mutation | Alpha-Galactosidase A Activity (nmol/mL/h) Normal Range: >3.0 nmol/mL/h | Lyso-Gb3 in Blood (nmol/L) Normal Range: <2.3 nmol/L | Clinical Signs |
|---|---|---|---|---|---|---|
| Proband | M | 41 | p.W262C | 0.1 | 93.27 | Infantile acroparesthesia, hypo-anhidrosis, angiokeratoma, recurrent fever |
| Mother | F | 67 | p.W262C | 5.8 | 12.34 | Hypo-anhidrosis, LVH |
| Patient | Sex | Age | Mutation | Alpha-Galactosidase A Activity (nmol/mL/h) Normal Range: >3.0 nmol/mL/h | Lyso-Gb3 in Blood (nmol/L) Normal Range: <2.3 nmol/L | Clinical Signs |
|---|---|---|---|---|---|---|
| Proband | F | 49 | c.583insGAATA | 4.6 | 12.47 | Renal replacement treatment, albuminuria, proteinuria, azotemia, conduction defects, LVH, recurrent headaches, acroparesthesias, angiokeratomas |
| Patient | Sex | Age | Mutation | Alpha-Galactosidase A Activity (nmol/mL/h) Normal Range: >3.0 nmol/mL/h | Lyso-Gb3 in Blood (nmol/L) Normal Range: <2.3 nmol/L | Clinical Signs |
|---|---|---|---|---|---|---|
| Proband | F | 39 | p.A143T + p.Y207X | 3.2 | 9.92 | Angiokeratoma, heat or cold intolerance, cornea verticillata |
| Mother | F | 74 | p.A143T + p.Y207X | 18.6 | 7.89 | Clinical study in progress |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anania, M.; Pieruzzi, F.; Giacalone, I.; Trezzi, B.; Marsana, E.M.; Roggero, L.; Francofonte, D.; Stefanoni, M.; Vinci, M.; Zizzo, C.; et al. Identification of Four New Mutations in the GLA Gene Associated with Anderson–Fabry Disease. Int. J. Mol. Sci. 2025, 26, 473. https://doi.org/10.3390/ijms26020473
Anania M, Pieruzzi F, Giacalone I, Trezzi B, Marsana EM, Roggero L, Francofonte D, Stefanoni M, Vinci M, Zizzo C, et al. Identification of Four New Mutations in the GLA Gene Associated with Anderson–Fabry Disease. International Journal of Molecular Sciences. 2025; 26(2):473. https://doi.org/10.3390/ijms26020473
Chicago/Turabian StyleAnania, Monia, Federico Pieruzzi, Irene Giacalone, Barbara Trezzi, Emanuela Maria Marsana, Letizia Roggero, Daniele Francofonte, Michele Stefanoni, Martina Vinci, Carmela Zizzo, and et al. 2025. "Identification of Four New Mutations in the GLA Gene Associated with Anderson–Fabry Disease" International Journal of Molecular Sciences 26, no. 2: 473. https://doi.org/10.3390/ijms26020473
APA StyleAnania, M., Pieruzzi, F., Giacalone, I., Trezzi, B., Marsana, E. M., Roggero, L., Francofonte, D., Stefanoni, M., Vinci, M., Zizzo, C., Zora, M., Di Chiara, T., Duro, G., Duro, G., & Colomba, P. (2025). Identification of Four New Mutations in the GLA Gene Associated with Anderson–Fabry Disease. International Journal of Molecular Sciences, 26(2), 473. https://doi.org/10.3390/ijms26020473