
Pyrrolysine Aminoacyl-tRNA Synthetase as a Tool for Expanding the Genetic Code

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pyrrolysine Aminoacyl-tRNA Synthetase
3. PylRS Enzyme Groups
4. Modification of the Structure of Native PylRS Variants to Extend the Range of Incorporated ncAAs
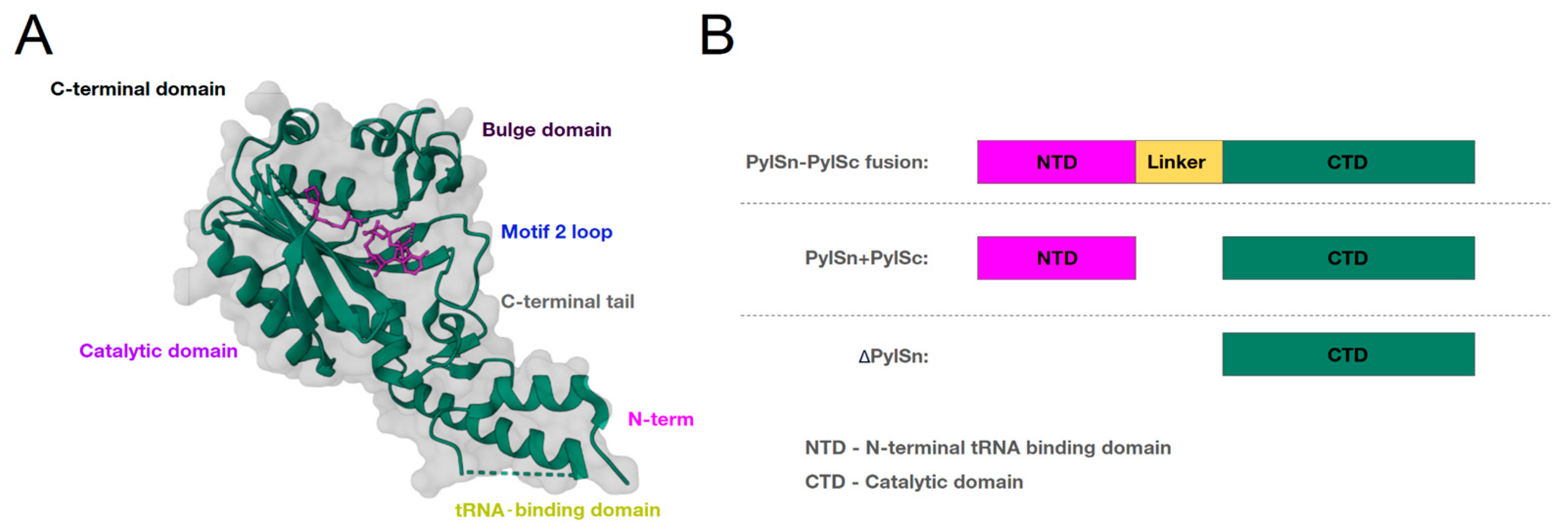
4.1. Changes to the C-Domain
4.2. Changes to the N-Domain
4.3. Comparisons of the Efficiency of Aminoacylation by Modified Synthetases
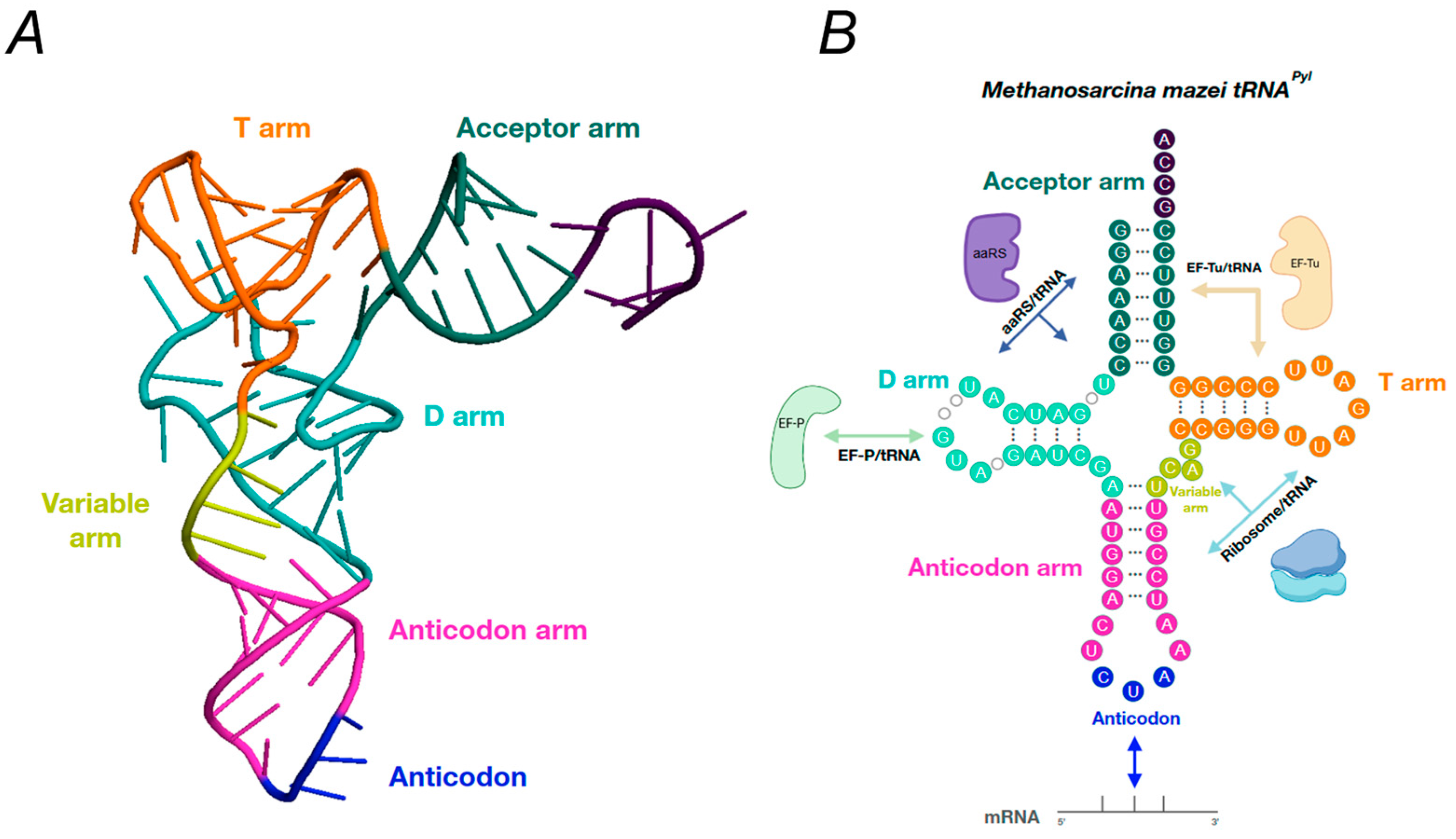
5. Pyl tRNAs
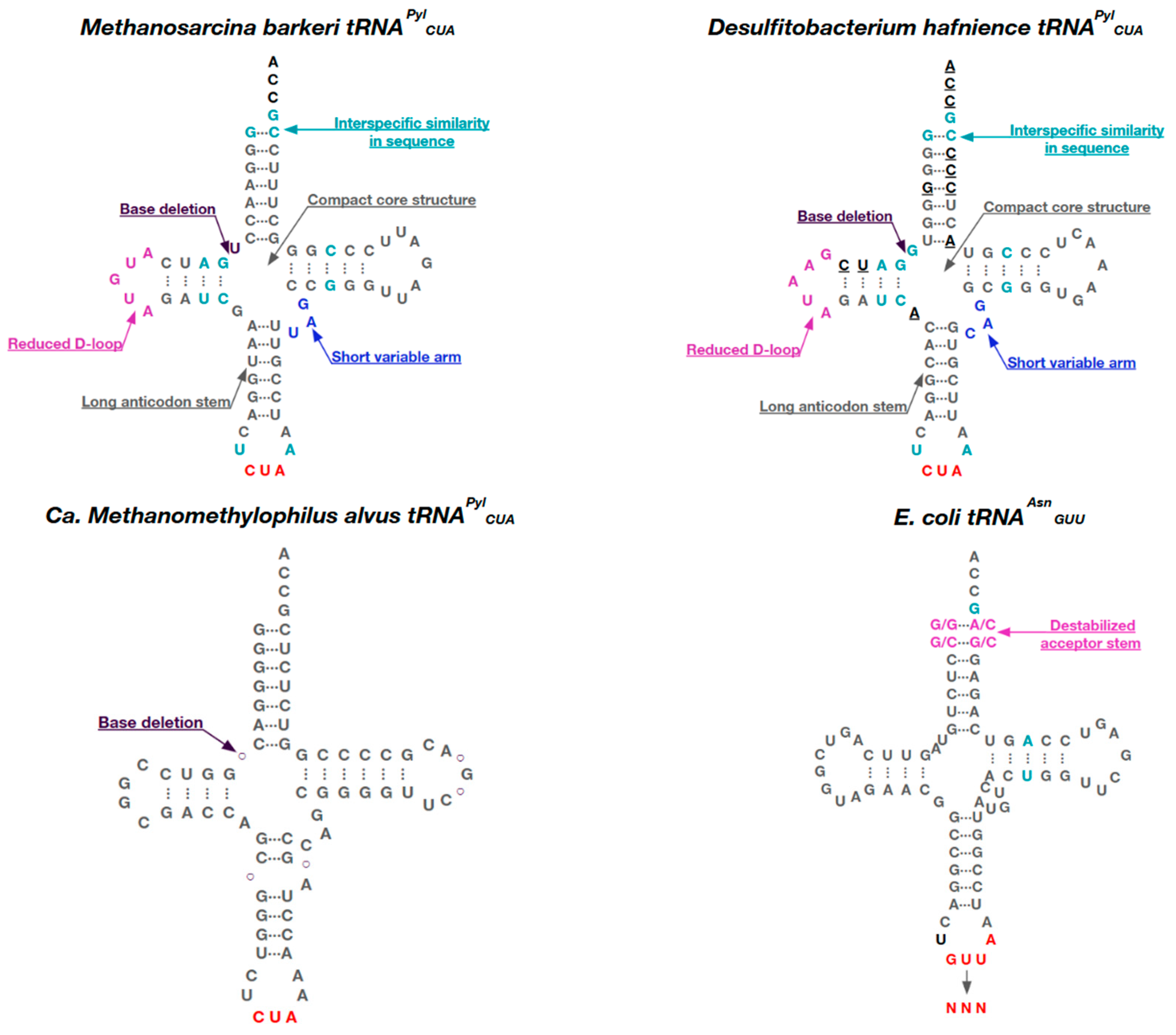
6. tRNAPyl Engineering
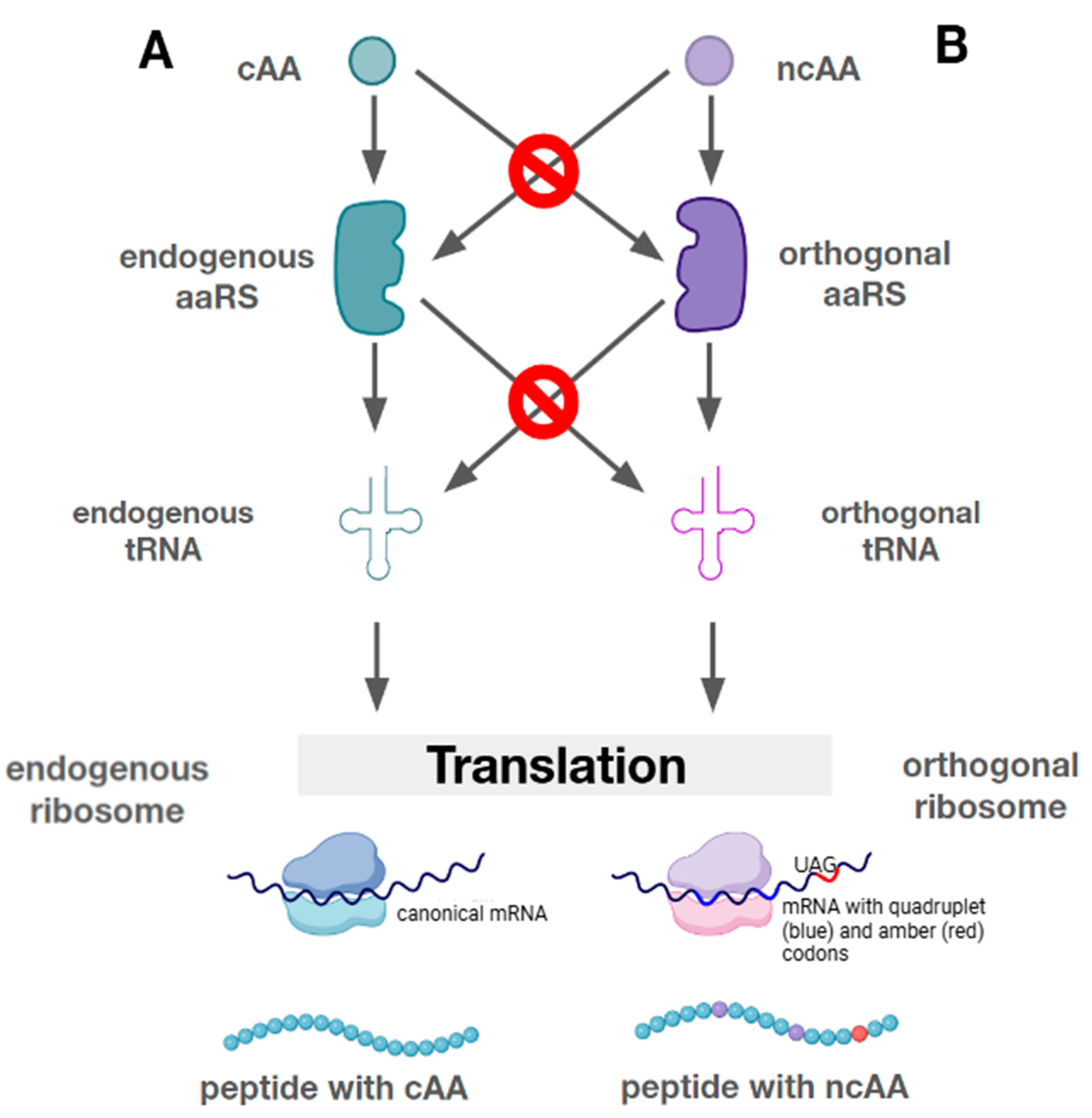
7. Mutually Orthogonal Systems
8. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aaRS | Aminoacyl-tRNA synthetase |
| ATP | Adenosine triphosphate |
| cAA | Canonical amino acid |
| CTD | C-terminal domain |
| Dh-PylSc | Desulfitobacterium hafniense PylSc |
| Eco LeuRS | Leucyl-tRNA synthetase of E. coli |
| Eco TyrRS | Tyrosyl-tRNA synthetase of E. coli |
| EF-Tu | Elongation factor thermo unstable, a prokaryotic elongation factor that is responsible for catalyzing the binding of aminoacyl-tRNA to the ribosome |
| ESI-MS | Electrospray ionization mass spectrometry |
| FSY | Fluorosulfate L-tyrosine |
| GCE | Genetic code expansion |
| GTP | Guanosine-5′-triphosphate |
| HMET1 | Halophilic euryarchaeal methanogen Candidatus Methanohalarchaeum thermophilum |
| ISO4-G1 | Methanogenic archaeon |
| LC | Liquid chromatography |
| LUCA | Last universal common ancestor |
| MAGE | Multiplexed automated genome engineering |
| Ma-PylRS | Methanomethylophilus alvus PylRS |
| Mb-PylRS | Methanosarcina barkeri PylRS |
| MCPs | Macrocyclic peptide drugs |
| Mm-PylRS | Methanosarcina mazei PylRS |
| MtmB | Methanosarcina barkeri monomethylamine methyltransferase |
| mutRNA | Mutated RNA |
| NAD | Nicotinamide adenine dinucleotide |
| ncAAs | Non-canonical amino acids |
| NTD | N-terminal domain |
| PACE | Phage assisted continuous evolution |
| PANCE | Phage assisted non-continuous evolution |
| PD-1 | Programmed cell death 1 |
| PheRS | Phenylalanine-tRNA synthetase |
| Pyl | Pyrrolysine |
| PylB | The pyrrolysine cluster gene encoding the enzyme S-adenosyl-L-methionine |
| PylC | The pyrrolysine cluster gene encoding a member of the carbamoyl phosphate synthetase family |
| PylD | The pyrrolysine cluster gene encoding NAD-dependent dehydrogenase |
| PylRS | Pyrrolysyl-tRNA synthetase |
| PylRS-AA | Variant of pyrrolysine synthesis with replacement of amino acids at positions 348 and 346 with alanine |
| PylS | Pyrrolysine cluster gene encoding tRNA pyrrolysine synthetase |
| PylSc | Catalytic domain of PylRS |
| PylSn | A group of PylRSs containing N- and T-terminal domains |
| PylSn+PylSc | A group of PylRSs that is formed from two separate domains expressed as different proteins from different genes |
| PylSn–PylSc | A group of PylRSs whose domains are fused by a linker |
| PylT | Pyrrolysine cluster gene encoding pyrrolysine tRNA |
| RNA | Ribonucleic acid |
| RS | tRNA synthetase |
| tRNA | Transfer RNA |
| tRNAPyl | Transfer RNA that carries the amino acid residue pyrrolysine |
| ΔPylSn | A group of pyrrolysine synthetases containing only a C-terminal domain |
| Ψ | Pseudouridine |
References
- Blight, S.K.; Larue, R.C.; Mahapatra, A.; Longstaff, D.G.; Chang, E.; Zhao, G.; Kang, P.T.; Green-Church, K.B.; Chan, M.K.; Krzycki, J.A. Direct Charging of tRNA(CUA) with Pyrrolysine in Vitro and in Vivo. Nature 2004, 431, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xin, X.; Zhang, Y.; Li, S.; Zhao, X.; Li, S.; Xu, Z. Advances in Biosynthesis of Non-Canonical Amino Acids (ncAAs) and the Methods of ncAAs Incorporation into Proteins. Molecules 2023, 28, 6745. [Google Scholar] [CrossRef] [PubMed]
- Enninful, G.N.; Kuppusamy, R.; Tiburu, E.K.; Kumar, N.; Willcox, M.D.P. Non-Canonical Amino Acid Bioincorporation into Antimicrobial Peptides and Its Challenges. J. Pept. Sci. 2024, 30, e3560. [Google Scholar] [CrossRef]
- Gillane, R.; Daygon, D.; Khalil, Z.G.; Marcellin, E. Biosynthesis of Novel Non-Proteinogenic Amino Acids β-Hydroxyenduracididine and β-Methylphenylalanine in Escherichia coli. Front. Bioeng. Biotechnol. 2024, 12, 1468974. [Google Scholar] [CrossRef]
- Natter Perdiguero, A.; Deliz Liang, A. Practical Approaches to Genetic Code Expansion with Aminoacyl-tRNA Synthetase/tRNA Pairs. Chimia 2024, 78, 22–31. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, T. Therapeutic Applications of Genetic Code Expansion. Synth. Syst. Biotechnol. 2018, 3, 150. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Q.; Klauser, P.C.; Li, M.; Zheng, F.; Wang, N.; Li, X.; Zhang, Q.; Fu, X.; Wang, Q.; et al. Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 2020, 182, 85–97.e16. [Google Scholar] [CrossRef]
- Zampaloni, C.; Mattei, P.; Bleicher, K.; Winther, L.; Thäte, C.; Bucher, C.; Adam, J.-M.; Alanine, A.; Amrein, K.E.; Baidin, V.; et al. A Novel Antibiotic Class Targeting the Lipopolysaccharide Transporter. Nature 2024, 625, 566. [Google Scholar] [CrossRef]
- Arias, M.; Haney, E.F.; Hilchie, A.L.; Corcoran, J.A.; Hyndman, M.E.; Hancock, R.E.W.; Vogel, H.J. Selective Anticancer Activity of Synthetic Peptides Derived from the Host Defence Peptide Tritrpticin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183228. [Google Scholar] [CrossRef]
- Pagar, A.D.; Patil, M.D.; Flood, D.T.; Yoo, T.H.; Dawson, P.E.; Yun, H. Recent Advances in Biocatalysis with Chemical Modification and Expanded Amino Acid Alphabet. Chem. Rev. 2021, 121, 6173–6245. [Google Scholar] [CrossRef]
- Jin, X.; Park, O.-J.; Hong, S.H. Incorporation of Non-Standard Amino Acids into Proteins: Challenges, Recent Achievements, and Emerging Applications. Appl. Microbiol. Biotechnol. 2019, 103, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Nödling, A.R.; Spear, L.A.; Williams, T.L.; Luk, L.Y.P.; Tsai, Y.-H. Using Genetically Incorporated Unnatural Amino Acids to Control Protein Functions in Mammalian Cells. Essays Biochem. 2019, 63, 237–266. [Google Scholar] [CrossRef]
- de la Torre, D.; Chin, J.W. Reprogramming the Genetic Code. Nat. Rev. Genet. 2021, 22, 169–184. [Google Scholar] [CrossRef]
- Andrews, J.; Gan, Q.; Fan, C. “Not-so-Popular” Orthogonal Pairs in Genetic Code Expansion. Protein Sci. 2023, 32, e4559. [Google Scholar] [CrossRef]
- Melnikov, S.V.; Söll, D. Aminoacyl-tRNA Synthetases and tRNAs for an Expanded Genetic Code: What Makes Them Orthogonal? Int. J. Mol. Sci. 2019, 20, 1929. [Google Scholar] [CrossRef]
- Italia, J.S.; Latour, C.; Wrobel, C.J.J.; Chatterjee, A. Resurrecting the Bacterial Tyrosyl-tRNA Synthetase/tRNA Pair for Expanding the Genetic Code of Both E. Coli and Eukaryotes. Cell Chem. Biol. 2018, 25, 1304–1312.e5. [Google Scholar] [CrossRef]
- Liu, W.; Brock, A.; Chen, S.; Chen, S.; Schultz, P.G. Genetic Incorporation of Unnatural Amino Acids into Proteins in Mammalian Cells. Nat. Methods 2007, 4, 239–244. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, H.; Chen, Y.; Zhang, B.; Yang, Y.; Zang, J.; Wu, J.; Lin, S. Chimeric Design of Pyrrolysyl-tRNA Synthetase/tRNA Pairs and Canonical Synthetase/tRNA Pairs for Genetic Code Expansion. Nat. Commun. 2020, 11, 3154. [Google Scholar] [CrossRef]
- Dunkelmann, D.L.; Willis, J.C.W.; Beattie, A.T.; Chin, J.W. Engineered Triply Orthogonal Pyrrolysyl-tRNA Synthetase/tRNA Pairs Enable the Genetic Encoding of Three Distinct Non-Canonical Amino Acids. Nat. Chem. 2020, 12, 535–544. [Google Scholar] [CrossRef]
- Johnson, D.B.F.; Xu, J.; Shen, Z.; Takimoto, J.K.; Schultz, M.D.; Schmitz, R.J.; Xiang, Z.; Ecker, J.R.; Briggs, S.P.; Wang, L. RF1 Knockout Allows Ribosomal Incorporation of Unnatural Amino Acids at Multiple Sites. Nat. Chem. Biol. 2011, 7, 779–786. [Google Scholar] [CrossRef]
- Povolotskaya, I.S.; Kondrashov, F.A.; Ledda, A.; Vlasov, P.K. Stop Codons in Bacteria Are Not Selectively Equivalent. Biol. Direct 2012, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically Recoded Organisms Expand Biological Functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Phelps, S.S.; Gaudin, C.; Yoshizawa, S.; Benitez, C.; Fourmy, D.; Joseph, S. Translocation of a tRNA with an Extended Anticodon through the Ribosome. J. Mol. Biol. 2006, 360, 610–622. [Google Scholar] [CrossRef]
- Xi, Z.; Davis, L.; Baxter, K.; Tynan, A.; Goutou, A.; Greiss, S. Using a Quadruplet Codon to Expand the Genetic Code of an Animal. Nucleic Acids Res. 2021, 50, 4801. [Google Scholar] [CrossRef]
- Mukai, T.; Lajoie, M.J.; Englert, M.; Söll, D. Rewriting the Genetic Code. Annu. Rev. Microbiol. 2017, 71, 557–577. [Google Scholar] [CrossRef]
- Krahn, N.; Tharp, J.M.; Crnković, A.; Söll, D. Engineering Aminoacyl-tRNA Synthetases for Use in Synthetic Biology. Enzymes 2020, 48, 351–395. [Google Scholar] [CrossRef]
- Nozawa, K.; O’Donoghue, P.; Gundllapalli, S.; Araiso, Y.; Ishitani, R.; Umehara, T.; Söll, D.; Nureki, O. Pyrrolysyl-tRNA Synthetase-tRNA(Pyl) Structure Reveals the Molecular Basis of Orthogonality. Nature 2009, 457, 1163–1167. [Google Scholar] [CrossRef]
- Borrel, G.; Gaci, N.; Peyret, P.; O’Toole, P.W.; Gribaldo, S.; Brugère, J.-F. Unique Characteristics of the Pyrrolysine System in the 7th Order of Methanogens: Implications for the Evolution of a Genetic Code Expansion Cassette. Archaea 2014, 2014, 374146. [Google Scholar] [CrossRef]
- Suzuki, T.; Miller, C.; Guo, L.-T.; Ho, J.M.L.; Bryson, D.I.; Wang, Y.-S.; Liu, D.R.; Söll, D. Crystal Structures Reveal an Elusive Functional Domain of Pyrrolysyl-tRNA Synthetase. Nat. Chem. Biol. 2017, 13, 1261. [Google Scholar] [CrossRef]
- Kavran, J.M.; Gundllapalli, S.; O’Donoghue, P.; Englert, M.; Söll, D.; Steitz, T.A. Structure of Pyrrolysyl-tRNA Synthetase, an Archaeal Enzyme for Genetic Code Innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Crystallographic Studies on Multiple Conformational States of Active-Site Loops in Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2008, 378, 634–652. [Google Scholar] [CrossRef]
- Ambrogelly, A.; Gundllapalli, S.; Herring, S.; Polycarpo, C.; Frauer, C.; Söll, D. Pyrrolysine Is Not Hardwired for Cotranslational Insertion at UAG Codons. Proc. Natl. Acad. Sci. USA 2007, 104, 3141–3146. [Google Scholar] [CrossRef]
- Guo, L.-T.; Amikura, K.; Jiang, H.-K.; Mukai, T.; Fu, X.; Wang, Y.-S.; O’Donoghue, P.; Söll, D.; Tharp, J.M. Ancestral Archaea Expanded the Genetic Code with Pyrrolysine. J. Biol. Chem. 2022, 298, 102521. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, H.; Shen, Y.; Fu, X. Update of the Pyrrolysyl-tRNA Synthetase/tRNAPyl Pair and Derivatives for Genetic Code Expansion. J. Bacteriol. 2023, 205, e0038522. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Fang, X.; Wallace, A.L.; Wu, B.; Liu, W.R. A Rationally Designed Pyrrolysyl-tRNA Synthetase Mutant with a Broad Substrate Spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Fang, X.; Chen, H.-Y.; Wu, B.; Wang, Z.U.; Hilty, C.; Liu, W.R. Genetic Incorporation of Twelve Meta-Substituted Phenylalanine Derivatives Using a Single Pyrrolysyl-tRNA Synthetase Mutant. ACS Chem. Biol. 2013, 8, 405–415. [Google Scholar] [CrossRef]
- Jiang, H.-K.; Lee, M.-N.; Tsou, J.-C.; Chang, K.-W.; Tseng, H.-W.; Chen, K.-P.; Li, Y.-K.; Wang, Y.-S. Linker and N-Terminal Domain Engineering of Pyrrolysyl-tRNA Synthetase for Substrate Range Shifting and Activity Enhancement. Front. Bioeng. Biotechnol. 2020, 8, 235. [Google Scholar] [CrossRef]
- Jiang, R.; Krzycki, J.A. PylSn and the Homologous N-Terminal Domain of Pyrrolysyl-tRNA Synthetase Bind the tRNA That Is Essential for the Genetic Encoding of Pyrrolysine. J. Biol. Chem. 2012, 287, 32738–32746. [Google Scholar] [CrossRef]
- Giegé, R.; Sissler, M.; Florentz, C. Universal Rules and Idiosyncratic Features in tRNA Identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef]
- Beattie, A.T.; Dunkelmann, D.L.; Chin, J.W. Quintuply Orthogonal Pyrrolysyl-tRNA Synthetase/tRNAPyl Pairs. Nat. Chem. 2023, 15, 948–959. [Google Scholar] [CrossRef]
- Dunkelmann, D.L.; Chin, J.W. Engineering Pyrrolysine Systems for Genetic Code Expansion and Reprogramming. Chem. Rev. 2024, 124, 11008. [Google Scholar] [CrossRef] [PubMed]
- Tharp, J.M.; Vargas-Rodriguez, O.; Schepartz, A.; Söll, D. Genetic Encoding of Three Distinct Noncanonical Amino Acids Using Reprogrammed Initiator and Nonsense Codons. ACS Chem. Biol. 2021, 16, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Wang, Y.-S.; Nakamura, A.; Guo, L.-T.; Söll, D.; Umehara, T. Pyrrolysyl-tRNA Synthetase Variants Reveal Ancestral Aminoacylation Function. FEBS Lett. 2013, 587, 3243. [Google Scholar] [CrossRef]
- Koch, N.G.; Budisa, N. Evolution of Pyrrolysyl-tRNA Synthetase: From Methanogenesis to Genetic Code Expansion. Chem. Rev. 2024, 124, 9580–9608. [Google Scholar] [CrossRef]
- Awawdeh, A.; Tapia, A.; Alshawi, S.A.; Dawodu, O.; Gaier, S.A.; Specht, C.; Beaudoin, J.-D.; Tharp, J.M.; Vargas-Rodriguez, O. Efficient Suppression of Premature Termination Codons with Alanine by Engineered Chimeric Pyrrolysine tRNAs. Nucleic Acids Res. 2024, 52, 14244–14259. [Google Scholar] [CrossRef]
- Mariotti, M.; Lobanov, A.V.; Guigo, R.; Gladyshev, V.N. SECISearch3 and Seblastian: New Tools for Prediction of SECIS Elements and Selenoproteins. Nucleic Acids Res. 2013, 41, e149. [Google Scholar] [CrossRef]
- Srinivasan, G.; James, C.M.; Krzycki, J.A. Pyrrolysine Encoded by UAG in Archaea: Charging of a UAG-Decoding Specialized tRNA. Science 2002, 296, 1459–1462. [Google Scholar] [CrossRef]
- Burke, S.A.; Lo, S.L.; Krzycki, J.A. Clustered Genes Encoding the Methyltransferases of Methanogenesis from Monomethylamine. J. Bacteriol. 1998, 180, 3432–3440. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Seki, E.; Tanabe, H.; Fujii, Y.; Sakamoto, K.; Yokoyama, S. Crystal Structure of Pyrrolysyl-tRNA Synthetase from a Methanogenic Archaeon ISO4-G1 and Its Structure-Based Engineering for Highly-Productive Cell-Free Genetic Code Expansion with Non-Canonical Amino Acids. Int. J. Mol. Sci. 2023, 24, 6256. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. High Content of Proteins Containing 21st and 22nd Amino Acids, Selenocysteine and Pyrrolysine, in a Symbiotic Deltaproteobacterium of Gutless Worm Olavius Algarvensis. Nucleic Acids Res. 2007, 35, 4952–4963. [Google Scholar] [CrossRef]
- Quitterer, F.; List, A.; Eisenreich, W.; Bacher, A.; Groll, M. Crystal Structure of Methylornithine Synthase (PylB): Insights into the Pyrrolysine Biosynthesis. Angew. Chem. Int. Ed. Engl. 2012, 51, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.A.; Zhang, L.; Green-Church, K.B.; Krzycki, J.A. The Complete Biosynthesis of the Genetically Encoded Amino Acid Pyrrolysine from Lysine. Nature 2011, 471, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Krzycki, J.A. The Path of Lysine to Pyrrolysine. Curr. Opin. Chem. Biol. 2013, 17, 619–625. [Google Scholar] [CrossRef]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [Google Scholar] [CrossRef]
- Polycarpo, C.R.; Herring, S.; Bérubé, A.; Wood, J.L.; Söll, D.; Ambrogelly, A. Pyrrolysine Analogues as Substrates for Pyrrolysyl-tRNA Synthetase. FEBS Lett. 2006, 580, 6695–6700. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Umehara, T.; Sakamoto, K.; Yokoyama, S. Expanded Genetic Code Technologies for Incorporating Modified Lysine at Multiple Sites. Chembiochem 2014, 15, 2181–2187. [Google Scholar] [CrossRef]
- Englert, M.; Moses, S.; Hohn, M.; Ling, J.; O’Donoghue, P.; Söll, D. Aminoacylation of tRNA 2′- or 3′-Hydroxyl by Phosphoseryl- and Pyrrolysyl-tRNA Synthetases. FEBS Lett. 2013, 587, 3360–3364. [Google Scholar] [CrossRef]
- Tharp, J.M.; Ehnbom, A.; Liu, W.R. tRNAPyl: Structure, Function, and Applications. RNA Biol. 2018, 15, 441–452. [Google Scholar] [CrossRef]
- Willis, J.C.W.; Chin, J.W. Mutually Orthogonal Pyrrolysyl-tRNA Synthetase/tRNA Pairs. Nat. Chem. 2018, 10, 831–837. [Google Scholar] [CrossRef]
- Meng, K.; Chung, C.Z.; Söll, D.; Krahn, N. Unconventional Genetic Code Systems in Archaea. Front. Microbiol. 2022, 13, 1007832. [Google Scholar] [CrossRef]
- Fladischer, P.; Weingartner, A.; Blamauer, J.; Darnhofer, B.; Birner-Gruenberger, R.; Kardashliev, T.; Ruff, A.J.; Schwaneberg, U.; Wiltschi, B. A Semi-Rationally Engineered Bacterial Pyrrolysyl-tRNA Synthetase Genetically Encodes Phenyl Azide Chemistry. Biotechnol. J. 2019, 14, e1800125. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Iraha, F.; Ohtake, K.; Sakamoto, K. Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes. Molecules 2018, 23, 2460. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; O’Toole, P.W.; Harris, H.M.; Peyret, P.; Brugère, J.-F.; Gribaldo, S. Phylogenomic Data Support a Seventh Order of Methylotrophic Methanogens and Provide Insights into the Evolution of Methanogenesis. Genome Biol. Evol. 2013, 5, 1769. [Google Scholar] [CrossRef]
- Borrel, G.; Parisot, N.; Harris, H.M.B.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative Genomics Highlights the Unique Biology of Methanomassiliicoccales, a Thermoplasmatales-Related Seventh Order of Methanogenic Archaea That Encodes Pyrrolysine. BMC Genom. 2014, 15, 679. [Google Scholar] [CrossRef]
- Mihajlovski, A.; Alric, M.; Brugère, J.-F. A Putative New Order of Methanogenic Archaea Inhabiting the Human Gut, as Revealed by Molecular Analyses of the mcrA Gene. Res. Microbiol. 2008, 159, 516–521. [Google Scholar] [CrossRef]
- Meineke, B.; Heimgärtner, J.; Lafranchi, L.; Elsässer, S.J. Methanomethylophilus Alvus Mx1201 Provides Basis for Mutual Orthogonal Pyrrolysyl tRNA/Aminoacyl-tRNA Synthetase Pairs in Mammalian Cells. ACS Chem. Biol. 2018, 13, 3087. [Google Scholar] [CrossRef]
- Beránek, V.; Willis, J.C.W.; Chin, J.W. An Evolved Methanomethylophilus Alvus Pyrrolysyl-tRNA Synthetase/tRNA Pair Is Highly Active and Orthogonal in Mammalian Cells. Biochemistry 2019, 58, 387–390. [Google Scholar] [CrossRef]
- Cozannet, M.; Borrel, G.; Roussel, E.; Moalic, Y.; Allioux, M.; Sanvoisin, A.; Toffin, L.; Alain, K. New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments. Microorganisms 2020, 9, 30. [Google Scholar] [CrossRef]
- Matassi, G. Horizontal Gene Transfer Drives the Evolution of Rh50 Permeases in Prokaryotes. BMC Evol. Biol. 2017, 17, 2. [Google Scholar] [CrossRef]
- Fournier, G.P.; Huang, J.; Gogarten, J.P. Horizontal Gene Transfer from Extinct and Extant Lineages: Biological Innovation and the Coral of Life. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2229–2239. [Google Scholar] [CrossRef]
- Fu, C.; Chen, Q.; Zheng, F.; Yang, L.; Li, H.; Zhao, Q.; Wang, X.; Wang, L.; Wang, Q. Genetically Encoding a Lipidated Amino Acid for Extension of Protein Half-Life in Vivo. Angew. Chem. Int. Ed. Engl. 2019, 58, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yang, B.; Fu, C.; Zhu, H.; Zheng, F.; Kobayashi, T.; Liu, J.; Li, S.; Ma, C.; Wang, P.G.; et al. Genetically Encoding Fluorosulfate-l-Tyrosine To React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J. Am. Chem. Soc. 2018, 140, 4995–4999. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jacinto, M.P.; Zeng, Y.; Yu, Z.; Qu, J.; Liu, W.R.; Lin, Q. Genetically Encoded 2-Aryl-5-Carboxytetrazoles for Site-Selective Protein Photo-Cross-Linking. J. Am. Chem. Soc. 2017, 139, 6078–6081. [Google Scholar] [CrossRef]
- Li, S.; Wang, N.; Yu, B.; Sun, W.; Wang, L. Genetically Encoded Chemical Crosslinking of Carbohydrate. Nat. Chem. 2023, 15, 33–42. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Russell, W.K.; Wang, Z.; Wan, W.; Dodd, L.E.; Pai, P.-J.; Russell, D.H.; Liu, W.R. The de Novo Engineering of Pyrrolysyl-tRNA Synthetase for Genetic Incorporation of L-Phenylalanine and Its Derivatives. Mol. Biosyst. 2011, 7, 714–717. [Google Scholar] [CrossRef]
- Crnković, A.; Suzuki, T.; Söll, D.; Reynolds, N.M. Pyrrolysyl-tRNA Synthetase, an Aminoacyl-tRNA Synthetase for Genetic Code Expansion. Croat. Chem. Acta 2016, 89, 163. [Google Scholar] [CrossRef]
- Takimoto, J.K.; Dellas, N.; Noel, J.P.; Wang, L. Stereochemical Basis for Engineered Pyrrolysyl-tRNA Synthetase and the Efficient in Vivo Incorporation of Structurally Divergent Non-Native Amino Acids. ACS Chem. Biol. 2011, 6, 733–743. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep Engineering of Pyrrolysyl-tRNA Synthetase to Genetically Encode N(Epsilon)-(o-Azidobenzyloxycarbonyl) Lysine for Site-Specific Protein Modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Kuratani, M.; Seki, E.; Hino, N.; Sakamoto, K.; Yokoyama, S. Structural Basis for Genetic-Code Expansion with Bulky Lysine Derivatives by an Engineered Pyrrolysyl-tRNA Synthetase. Cell Chem. Biol. 2019, 26, 936–949.e13. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Weber, A.; Pott, M.; Welte, W.; Summerer, D. Structural Basis of Furan-Amino Acid Recognition by a Polyspecific Aminoacyl-tRNA-Synthetase and Its Genetic Encoding in Human Cells. Chembiochem 2014, 15, 1755–1760. [Google Scholar] [CrossRef]
- Umehara, T.; Kim, J.; Lee, S.; Guo, L.-T.; Söll, D.; Park, H.-S. N-Acetyl Lysyl-tRNA Synthetases Evolved by a CcdB-Based Selection Possess N-Acetyl Lysine Specificity in Vitro and in Vivo. FEBS Lett. 2012, 586, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Recognition of Non-Alpha-Amino Substrates by Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2009, 385, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Passioura, T.; Suga, H. Reprogramming the Genetic Code in Vitro. Trends Biochem. Sci. 2014, 39, 400–408. [Google Scholar] [CrossRef]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically Encoding N(Epsilon)-Acetyllysine in Recombinant Proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef]
- Cho, C.-C.D.; Blankenship, L.R.; Ma, X.; Xu, S.; Liu, W. Pyrrolysyl-tRNA Synthetase Activity Can Be Improved by a P188 Mutation That Stabilizes the Full-Length Enzyme. J. Mol. Biol. 2022, 434, 167453. [Google Scholar] [CrossRef]
- Mukai, T.; Yamaguchi, A.; Ohtake, K.; Takahashi, M.; Hayashi, A.; Iraha, F.; Kira, S.; Yanagisawa, T.; Yokoyama, S.; Hoshi, H.; et al. Reassignment of a Rare Sense Codon to a Non-Canonical Amino Acid in Escherichia coli. Nucleic Acids Res. 2015, 43, 8111–8122. [Google Scholar] [CrossRef]
- Sharma, V.; Zeng, Y.; Wang, W.W.; Qiao, Y.; Kurra, Y.; Liu, W.R. Evolving the N-Terminal Domain of Pyrrolysyl-tRNA Synthetase for Improved Incorporation of Noncanonical Amino Acids. Chembiochem 2018, 19, 26–30. [Google Scholar] [CrossRef]
- Guo, L.-T.; Wang, Y.-S.; Nakamura, A.; Eiler, D.; Kavran, J.M.; Wong, M.; Kiessling, L.L.; Steitz, T.A.; O’Donoghue, P.; Söll, D. Polyspecific Pyrrolysyl-tRNA Synthetases from Directed Evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16724–16729. [Google Scholar] [CrossRef]
- Fan, C.; Xiong, H.; Reynolds, N.M.; Söll, D. Rationally Evolving tRNAPyl for Efficient Incorporation of Noncanonical Amino Acids. Nucleic Acids Res. 2015, 43, e156. [Google Scholar] [CrossRef]
- Jiang, H.-K.; Weng, J.-H.; Wang, Y.-H.; Tsou, J.-C.; Chen, P.-J.; Ko, A.-L.A.; Söll, D.; Tsai, M.-D.; Wang, Y.-S. Rational Design of the Genetic Code Expansion Toolkit for in Vivo Encoding of D-Amino Acids. Front. Genet. 2023, 14, 1277489. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.-T.; Miller, C.; Söll, D.; Liu, D.R. Continuous Directed Evolution of Aminoacyl-tRNA Synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Amiram, M.; Haimovich, A.D.; Fan, C.; Wang, Y.-S.; Aerni, H.-R.; Ntai, I.; Moonan, D.W.; Ma, N.J.; Rovner, A.J.; Hong, S.H.; et al. Evolution of Translation Machinery in Recoded Bacteria Enables Multi-Site Incorporation of Nonstandard Amino Acids. Nat. Biotechnol. 2015, 33, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cho, S.; Kim, J.-C.; Park, H.-S. tRNA Engineering Strategies for Genetic Code Expansion. Front. Genet. 2024, 15, 1373250. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.M.; Vargas-Rodriguez, O.; Söll, D.; Crnković, A. The Central Role of tRNA in Genetic Code Expansion. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3001–3008. [Google Scholar] [CrossRef]
- Polycarpo, C.; Ambrogelly, A.; Bérubé, A.; Winbush, S.M.; McCloskey, J.A.; Crain, P.F.; Wood, J.L.; Söll, D. An Aminoacyl-tRNA Synthetase That Specifically Activates Pyrrolysine. Proc. Natl. Acad. Sci. USA 2004, 101, 12450–12454. [Google Scholar] [CrossRef]
- Gaston, M.A.; Jiang, R.; Krzycki, J.A. Functional Context, Biosynthesis, and Genetic Encoding of Pyrrolysine. Curr. Opin. Microbiol. 2011, 14, 342–349. [Google Scholar] [CrossRef]
- Sterner, T.; Jansen, M.; Hou, Y.M. Structural and Functional Accommodation of Nucleotide Variations at a Conserved tRNA Tertiary Base Pair. RNA 1995, 1, 841. [Google Scholar]
- Cervettini, D.; Tang, S.; Fried, S.D.; Willis, J.C.W.; Funke, L.F.H.; Colwell, L.J.; Chin, J.W. Rapid Discovery and Evolution of Orthogonal Aminoacyl-tRNA Synthetase-tRNA Pairs. Nat. Biotechnol. 2020, 38, 989–999. [Google Scholar] [CrossRef]
- Giegé, R.; Eriani, G. The tRNA Identity Landscape for Aminoacylation and Beyond. Nucleic Acids Res. 2023, 51, 1528–1570. [Google Scholar] [CrossRef]
- Krahn, N.; Zhang, J.; Melnikov, S.V.; Tharp, J.M.; Villa, A.; Patel, A.; Howard, R.J.; Gabir, H.; Patel, T.R.; Stetefeld, J.; et al. tRNA Shape Is an Identity Element for an Archaeal Pyrrolysyl-tRNA Synthetase from the Human Gut. Nucleic Acids Res. 2024, 52, 513–524. [Google Scholar] [CrossRef]
- DeBenedictis, E.A.; Carver, G.D.; Chung, C.Z.; Söll, D.; Badran, A.H. Multiplex Suppression of Four Quadruplet Codons via tRNA Directed Evolution. Nat. Commun. 2021, 12, 5706. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Melançon, C.E.; Lee, H.S.; Groff, D.; Schultz, P.G. Evolution of Amber Suppressor tRNAs for Efficient Bacterial Production of Proteins Containing Nonnatural Amino Acids. Angew. Chem. Int. Ed. Engl. 2009, 48, 9148–9151. [Google Scholar] [CrossRef] [PubMed]
- Schmied, W.H.; Elsässer, S.J.; Uttamapinant, C.; Chin, J.W. Efficient Multisite Unnatural Amino Acid Incorporation in Mammalian Cells via Optimized Pyrrolysyl tRNA Synthetase/tRNA Expression and Engineered eRF1. J. Am. Chem. Soc. 2014, 136, 15577–15583. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kawai, G.; Yokogawa, T.; Hayashi, N.; Kumazawa, Y.; Ueda, T.; Nishikawa, K.; Hirao, I.; Miura, K.; Watanabe, K. Higher-Order Structure of Bovine Mitochondrial tRNA(SerUGA): Chemical Modification and Computer Modeling. Nucleic Acids Res. 1994, 22, 5378. [Google Scholar] [CrossRef]
- Serfling, R.; Lorenz, C.; Etzel, M.; Schicht, G.; Böttke, T.; Mörl, M.; Coin, I. Designer tRNAs for Efficient Incorporation of Non-Canonical Amino Acids by the Pyrrolysine System in Mammalian Cells. Nucleic Acids Res. 2018, 46, 1–10. [Google Scholar] [CrossRef]
- Jahn, M.; Rogers, M.J.; Söll, D. Anticodon and Acceptor Stem Nucleotides in tRNA(Gln) Are Major Recognition Elements for E. Coli Glutaminyl-tRNA Synthetase. Nature 1991, 352, 258–260. [Google Scholar] [CrossRef]
- Sakamoto, K.; Hayashi, A. Synthetic Tyrosine tRNA Molecules with Noncanonical Secondary Structures. Int. J. Mol. Sci. 2018, 20, 92. [Google Scholar] [CrossRef]
- Miller, C.; Bröcker, M.J.; Prat, L.; Ip, K.; Chirathivat, N.; Feiock, A.; Veszprémi, M.; Söll, D. A Synthetic tRNA for EF-Tu Mediated Selenocysteine Incorporation in Vivo and in Vitro. FEBS Lett. 2015, 589, 2194. [Google Scholar] [CrossRef]
- Thyer, R.; Robotham, S.A.; Brodbelt, J.S.; Ellington, A.D. Evolving tRNA(Sec) for Efficient Canonical Incorporation of Selenocysteine. J. Am. Chem. Soc. 2015, 137, 46–49. [Google Scholar] [CrossRef]
- Mukai, T.; Vargas-Rodriguez, O.; Englert, M.; Tripp, H.J.; Ivanova, N.N.; Rubin, E.M.; Kyrpides, N.C.; Söll, D. Transfer RNAs with Novel Cloverleaf Structures. Nucleic Acids Res. 2017, 45, 2776–2785. [Google Scholar] [CrossRef]
- Mukai, T.; Sevostyanova, A.; Suzuki, T.; Fu, X.; Söll, D. A Facile Method for Producing Selenocysteine-Containing Proteins. Angew. Chem. Int. Ed. Engl. 2018, 57, 7215–7219. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, Z.; Huang, Y.; Liu, W.R. Catalyst-Free and Site-Specific One-Pot Dual-Labeling of a Protein Directed by Two Genetically Incorporated Noncanonical Amino Acids. Chembiochem 2012, 13, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Sun, S.B.; Furman, J.L.; Xiao, H.; Schultz, P.G. A Versatile Platform for Single- and Multiple-Unnatural Amino Acid Mutagenesis in Escherichia coli. Biochemistry 2013, 52, 1828–1837. [Google Scholar] [CrossRef]
- Meineke, B.; Heimgärtner, J.; Caridha, R.; Block, M.F.; Kimler, K.J.; Pires, M.F.; Landreh, M.; Elsässer, S.J. Dual Stop Codon Suppression in Mammalian Cells with Genomically Integrated Genetic Code Expansion Machinery. Cell Rep. Methods 2023, 3, 100626. [Google Scholar] [CrossRef]
- Seki, E.; Yanagisawa, T.; Kuratani, M.; Sakamoto, K.; Yokoyama, S. Fully Productive Cell-Free Genetic Code Expansion by Structure-Based Engineering of Methanomethylophilus Alvus Pyrrolysyl-tRNA Synthetase. ACS Synth. Biol. 2020, 9, 718–732. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, X.; Zhao, Q.; Mukai, T.; Vargas-Rodriguez, O.; Zhang, H.; Zhang, Y.; Wassel, P.; Amikura, K.; Maupin-Furlow, J.; et al. The tRNA Discriminator Base Defines the Mutual Orthogonality of Two Distinct Pyrrolysyl-tRNA Synthetase/tRNAPyl Pairs in the Same Organism. Nucleic Acids Res. 2022, 50, 4601–4615. [Google Scholar] [CrossRef]
- Dunkelmann, D.L.; Oehm, S.B.; Beattie, A.T.; Chin, J.W. A 68-Codon Genetic Code to Incorporate Four Distinct Non-Canonical Amino Acids Enabled by Automated Orthogonal mRNA Design. Nat. Chem. 2021, 13, 1110–1117. [Google Scholar] [CrossRef]
- Italia, J.S.; Addy, P.S.; Erickson, S.B.; Peeler, J.C.; Weerapana, E.; Chatterjee, A. Mutually Orthogonal Nonsense-Suppression Systems and Conjugation Chemistries for Precise Protein Labeling at up to Three Distinct Sites. J. Am. Chem. Soc. 2019, 141, 6204–6212. [Google Scholar] [CrossRef]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding Multiple Unnatural Amino Acids via Evolution of a Quadruplet-Decoding Ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef]
- Anderson, J.C.; Wu, N.; Santoro, S.W.; Lakshman, V.; King, D.S.; Schultz, P.G. An Expanded Genetic Code with a Functional Quadruplet Codon. Proc. Natl. Acad. Sci. USA 2004, 101, 7566–7571. [Google Scholar] [CrossRef]
- Shi, N.; Tong, L.; Lin, H.; Zheng, Z.; Zhang, H.; Dong, L.; Yang, Y.; Shen, Y.; Xia, Q. Optimizing eRF1 to Enable the Genetic Encoding of Three Distinct Noncanonical Amino Acids in Mammalian Cells. Adv. Biol. 2022, 6, e2200092. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dakhnevich, A.; Kazakova, A.; Iliushin, D.; Ivanov, R.A. Pyrrolysine Aminoacyl-tRNA Synthetase as a Tool for Expanding the Genetic Code. Int. J. Mol. Sci. 2025, 26, 539. https://doi.org/10.3390/ijms26020539
Dakhnevich A, Kazakova A, Iliushin D, Ivanov RA. Pyrrolysine Aminoacyl-tRNA Synthetase as a Tool for Expanding the Genetic Code. International Journal of Molecular Sciences. 2025; 26(2):539. https://doi.org/10.3390/ijms26020539
Chicago/Turabian StyleDakhnevich, Anastasia, Alisa Kazakova, Danila Iliushin, and Roman A. Ivanov. 2025. "Pyrrolysine Aminoacyl-tRNA Synthetase as a Tool for Expanding the Genetic Code" International Journal of Molecular Sciences 26, no. 2: 539. https://doi.org/10.3390/ijms26020539
APA StyleDakhnevich, A., Kazakova, A., Iliushin, D., & Ivanov, R. A. (2025). Pyrrolysine Aminoacyl-tRNA Synthetase as a Tool for Expanding the Genetic Code. International Journal of Molecular Sciences, 26(2), 539. https://doi.org/10.3390/ijms26020539