

Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
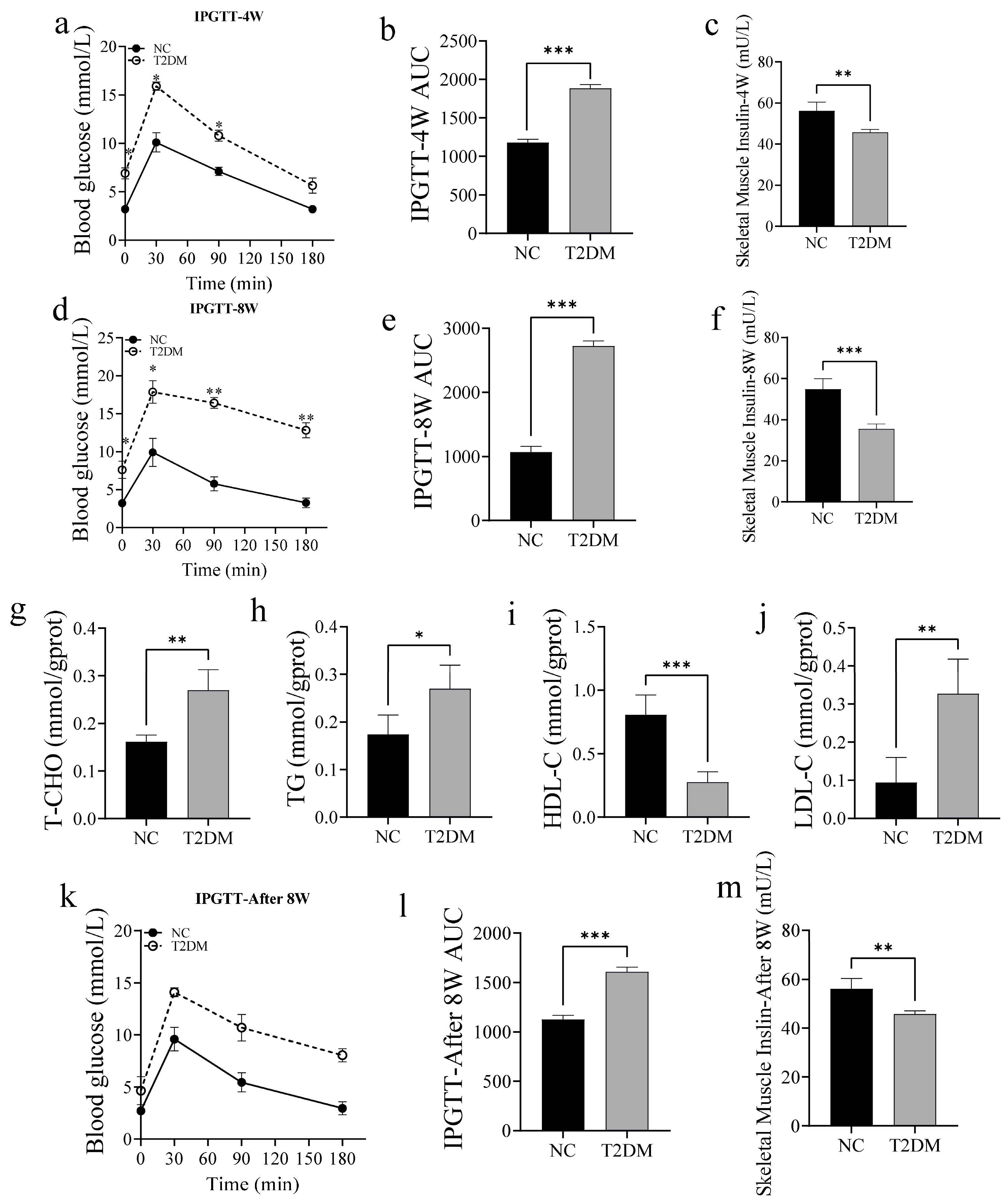
2.1. High-Glucose Treatment Induces T2DM in Zebrafish
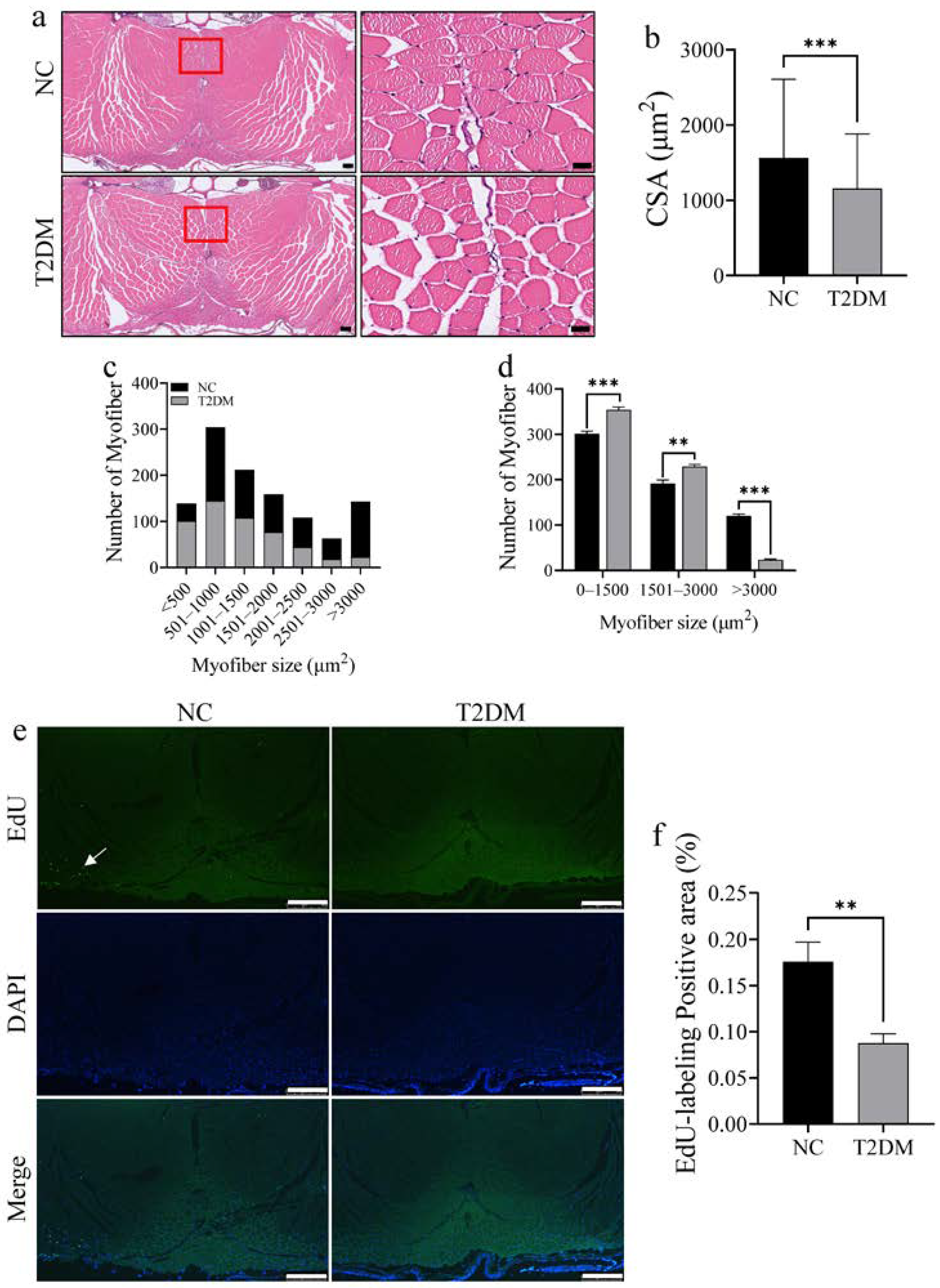
2.2. Loss of Skeletal Muscle Mass and Decreased Proliferative Ability in T2DM Zebrafish
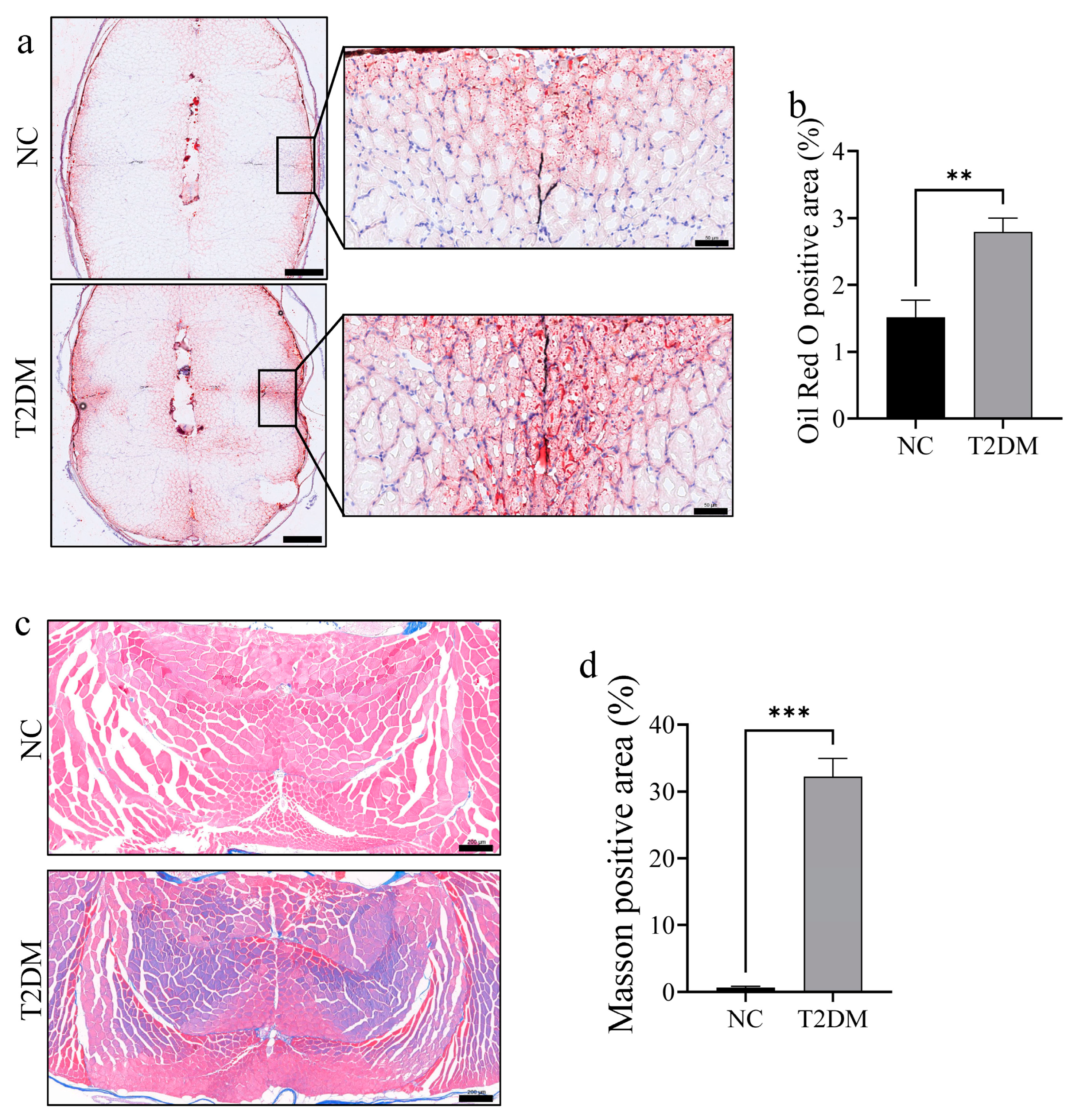
2.3. T2DM Zebrafish Exhibit Increased Lipid Accumulation and Fibrosis in Skeletal Muscle
2.4. Skeletal Muscle Function Is Suppressed in T2DM Zebrafish
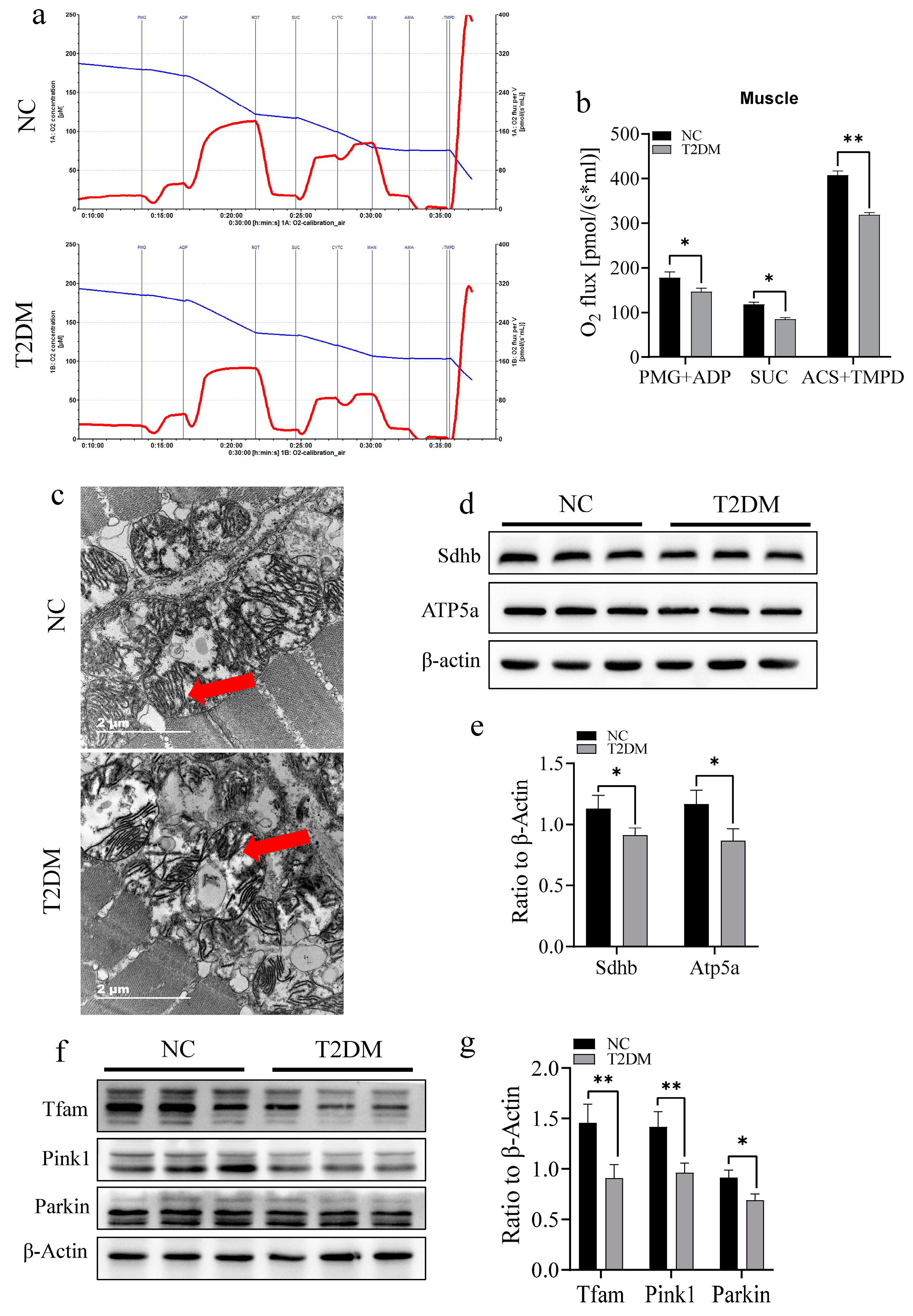
2.5. T2DM Impairs Mitochondrial Function and Induced an Inflammatory Response in the Skeletal Muscle of Zebrafish
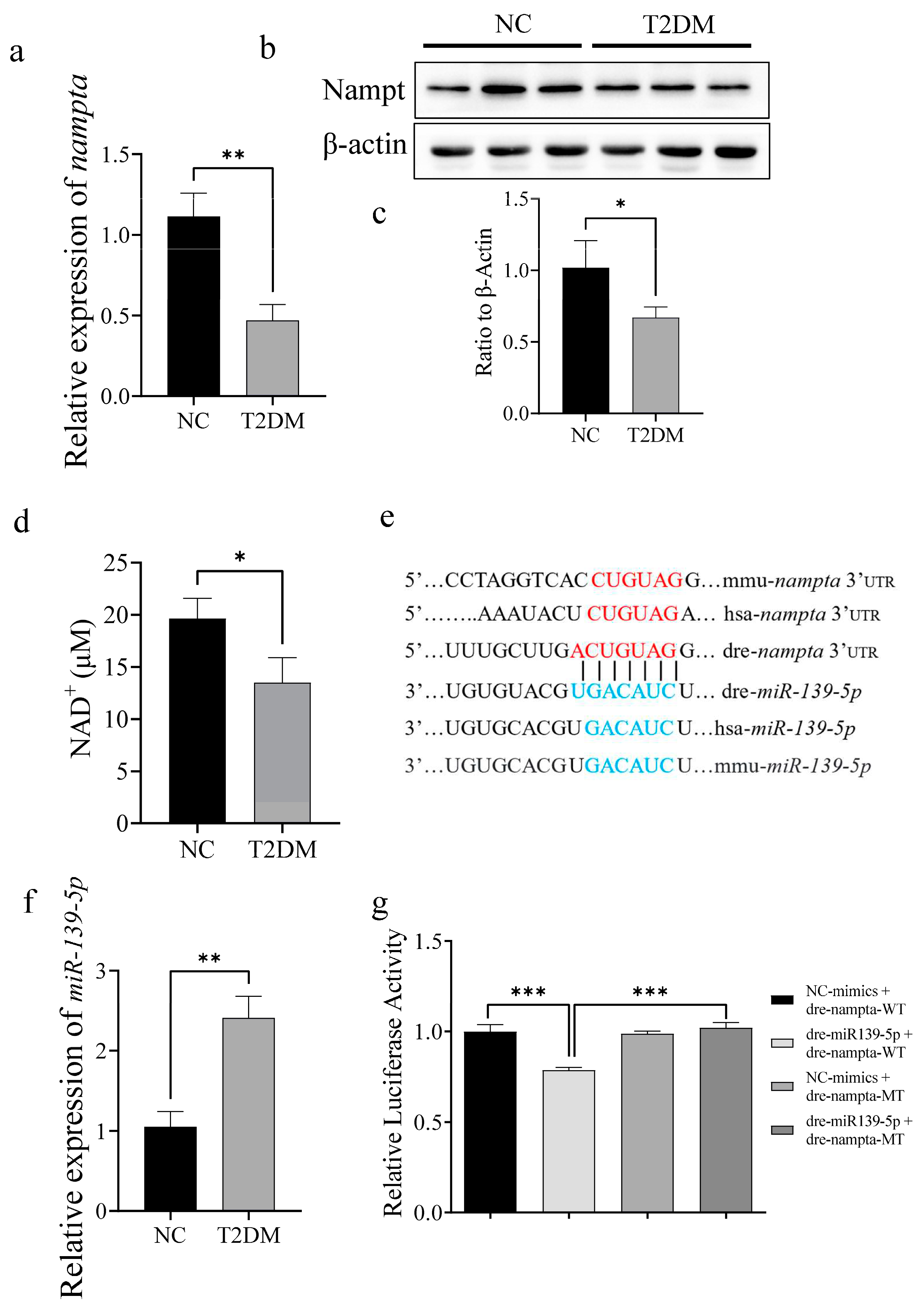
2.6. T2DM Induces Mitochondrial Dysfunction via the miR-139a-5p/NAMPT Pathway
3. Discussion
4. Materials and Methods
4.1. Experimental Subjects and Grouping
4.2. Zebrafish Exercise Capacity Test
4.3. Intraperitoneal Glucose Tolerance Test and Whole-Body Biochemical Analysis
4.4. Sample Collection
4.5. Mitochondrial Respiration Detection
4.6. Western Blotting
4.7. Detection of Tissue NAD+ Content
4.8. Transmission Electron Microscopy
4.9. Dual-Luciferase Reporter Gene Assay
4.10. EdU Cell Proliferation Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Available online: https://idf.org/about-diabetes/diabetes-facts-figures/ (accessed on 5 January 2024).
- Blonde, L. Epidemiology, costs, consequences, and pathophysiology of type 2 diabetes: An american epidemic. Ochsner J. 2001, 3, 126–131. [Google Scholar] [PubMed]
- Hulett, N.A.; Scalzo, R.L.; Reusch, J.E.B. Glucose Uptake by Skeletal Muscle within the Contexts of Type 2 Diabetes and Exercise: An Integrated Approach. Nutrients 2022, 14, 647. [Google Scholar] [CrossRef]
- Park, S.W.; Goodpaster, B.H.; Strotmeyer, E.S.; de Rekeneire, N.; Harris, T.B.; Schwartz, A.V.; Tylavsky, F.A.; Newman, A.B. Decreased muscle strength and quality in older adults with type 2 diabetes: The health, aging, and body composition study. Diabetes 2006, 55, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Leenders, M.; Verdijk, L.B.; van der Hoeven, L.; Adam, J.J.; van Kranenburg, J.; Nilwik, R.; van Loon, L.J. Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J. Am. Med. Dir. Assoc. 2013, 14, 585–592. [Google Scholar] [CrossRef]
- Hirata, Y.; Nomura, K.; Senga, Y.; Okada, Y.; Kobayashi, K.; Okamoto, S.; Minokoshi, Y.; Imamura, M.; Takeda, S.; Hosooka, T.; et al. Hyperglycemia induces skeletal muscle atrophy via a WWP1/KLF15 axis. JCI Insight 2019, 4, e124952. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Ryan, A.J.; Mudaliar, S.R.; Henry, R.R. Altered Myokine Secretion Is an Intrinsic Property of Skeletal Muscle in Type 2 Diabetes. PLoS ONE 2016, 11, e0158209. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.M.F.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schon, M.R.; Bluher, M.; Punkt, K. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef]
- Metter, E.J.; Talbot, L.A.; Schrager, M.; Conwit, R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B359–B365. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsoe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.D.S.; Falcao, K.V.G.; Amaral, I.P.G.; Leite, A.C.R.; Bezerra, R.S. Mitochondria as targets for toxicity and metabolism research using zebrafish. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129634. [Google Scholar] [CrossRef]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.P.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Alshahrani, A.; AlDubayee, M.; Zahra, M.; Alsebayel, F.M.; Alammari, N.; Alsudairy, F.; Almajed, M.; Aljada, A. Differential Expression of Human N-Alpha-Acetyltransferase 40 (hNAA40), Nicotinamide Phosphoribosyltransferase (NAMPT) and Sirtuin-1 (SIRT-1) Pathway in Obesity and T2DM: Modulation by Metformin and Macronutrient Intake. Diabetes Metab. Syndr. Obes. 2019, 12, 2765–2774. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Frederick, D.W.; Loro, E.; Liu, L.; Davila, A., Jr.; Chellappa, K.; Silverman, I.M.; Quinn, W.J., 3rd; Gosai, S.J.; Tichy, E.D.; Davis, J.G.; et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016, 24, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Agerholm, M.; Dall, M.; Jensen, B.A.H.; Prats, C.; Madsen, S.; Basse, A.L.; Graae, A.S.; Risis, S.; Goldenbaum, J.; Quistorff, B.; et al. Perturbations of NAD+ salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E377–E395. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kuang, G.; Wu, Y.; Ou, C. Emerging roles of exosomal miRNAs in diabetes mellitus. Clin. Transl. Med. 2021, 11, e468. [Google Scholar] [CrossRef]
- Brandao-Lima, P.N.; de Carvalho, G.B.; Payolla, T.B.; Sarti, F.M.; Fisberg, R.M.; Malcomson, F.C.; Mathers, J.C.; Rogero, M.M. Circulating microRNAs Showed Specific Responses according to Metabolic Syndrome Components and Sex of Adults from a Population-Based Study. Metabolites 2022, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Gut, P. Metabolic insights from zebrafish genetics, physiology, and chemical biology. Cell Mol. Life Sci. 2015, 72, 2249–2260. [Google Scholar] [CrossRef]
- Schlegel, A.; Stainier, D.Y. Lessons from “lower” organisms: What worms, flies, and zebrafish can teach us about human energy metabolism. PLoS Genet. 2007, 3, e199. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.C.; Warram, J.H.; Krolewski, A.S.; Bergman, R.N.; Soeldner, J.S.; Kahn, C.R. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: Results of a 25-year follow-up study. Lancet 1992, 340, 925–929. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.J.; Erskine, R.M.; Morse, C.I.; Winwood, K.; Onambele-Pearson, G. The impact of obesity on skeletal muscle strength and structure through adolescence to old age. Biogerontology 2016, 17, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.M.F.; Gingrich, M.A.; Hawke, T.J. Considering Type 1 Diabetes as a Form of Accelerated Muscle Aging. Exerc. Sport. Sci. Rev. 2019, 47, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.M.F.; Perry, C.G.R.; Hawke, T.J. Diabetic Myopathy: Current molecular understanding of this novel neuromuscular disorder. Curr. Opin. Neurol. 2017, 30, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.M.F.; Bellissimo, C.A.; Hughes, M.C.; Ramos, S.V.; Laham, R.; Perry, C.G.R.; Hawke, T.J. Sexual dimorphism in human skeletal muscle mitochondrial bioenergetics in response to type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E44–E51. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Heras, G.; Lauschke, V.M.; Mi, J.; Tian, G.; Gastaldello, S. High glucose-induced oxidative stress accelerates myogenesis by altering SUMO reactions. Exp. Cell Res. 2020, 395, 112234. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, R.; Nair, K.S. Skeletal muscle mitochondrial dysfunction & diabetes. Indian. J. Med. Res. 2007, 125, 399–410. [Google Scholar]
- Xiao Liang, K. Interplay of mitochondria and diabetes: Unveiling novel therapeutic strategies. Mitochondrion 2024, 75, 101850. [Google Scholar] [CrossRef]
- Vamecq, J.; Dessein, A.F.; Fontaine, M.; Briand, G.; Porchet, N.; Latruffe, N.; Andreolotti, P.; Cherkaoui-Malki, M. Mitochondrial dysfunction and lipid homeostasis. Curr. Drug Metab. 2012, 13, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Parhofer, K.G. Interaction between Glucose and Lipid Metabolism: More than Diabetic Dyslipidemia. Diabetes Metab. J. 2015, 39, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Doumas, M.; Imprialos, K.P.; Stavropoulos, K.; Georgianou, E.; Katsimardou, A.; Karagiannis, A. Diabetes and lipid metabolism. Hormones 2018, 17, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, C.; Wang, T.; Sun, L.; Qin, L.; Ju, J. miR-139-5p promotes neovascularization in diabetic retinopathy by regulating the phosphatase and tensin homolog. Arch. Pharm. Res. 2021, 44, 205–218. [Google Scholar] [CrossRef]
- Luo, Y.F.; Wan, X.X.; Zhao, L.L.; Guo, Z.; Shen, R.T.; Zeng, P.Y.; Wang, L.H.; Yuan, J.J.; Yang, W.J.; Yue, C.; et al. MicroRNA-139-5p upregulation is associated with diabetic endothelial cell dysfunction by targeting c-jun. Aging 2020, 13, 1186–1211. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Gambardella, J.; Sardu, C.; Lombardi, A.; Santulli, G. Functional Role of miR-155 in the Pathogenesis of Diabetes Mellitus and Its Complications. Noncoding RNA 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Petzold, S.; Korner, A.; Imai, S.; Kiess, W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. 2009, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, P.; Huang, X.; Shuai, W.; Liu, L.; Zhang, S.; Zhao, R.; Hu, X.; Wang, G. From Rate-Limiting Enzyme to Therapeutic Target: The Promise of NAMPT in Neurodegenerative Diseases. Front. Pharmacol. 2022, 13, 920113. [Google Scholar] [CrossRef]
- Houry, D.; Raasakka, A.; Ferrario, E.; Niere, M.; Bifulco, E.; Kursula, P.; Ziegler, M. Identification of structural determinants of nicotinamide phosphoribosyl transferase (NAMPT) activity and substrate selectivity. J. Struct. Biol. 2023, 215, 108004. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Nishimura, Y.; Zang, L.; Hirano, M.; Shimada, Y.; Wang, Z.; Umemoto, N.; Kuroyanagi, J.; Nishimura, N.; Tanaka, T. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol. 2010, 10, 21. [Google Scholar] [CrossRef]
- Asaoka, Y.; Terai, S.; Sakaida, I.; Nishina, H. The expanding role of fish models in understanding non-alcoholic fatty liver disease. Dis. Model. Mech. 2013, 6, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kinkel, M.D.; Prince, V.E. On the diabetic menu: Zebrafish as a model for pancreas development and function. Bioessays 2009, 31, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.; Connaughton, V.; Arneson, L.S. Induction of hyperglycaemia in zebrafish (Danio rerio) leads to morphological changes in the retina. Acta Diabetol. 2007, 44, 157–163. [Google Scholar] [CrossRef]
- Connaughton, V.P.; Baker, C.; Fonde, L.; Gerardi, E.; Slack, C. Alternate Immersion in an External Glucose Solution Differentially Affects Blood Sugar Values in Older Versus Younger Zebrafish Adults. Zebrafish 2016, 13, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.H.; Chen, B.; Zhang, J.P. Intracellular Insulin and Impaired Autophagy in a Zebrafish model and a Cell Model of Type 2 diabetes. Int. J. Biol. Sci. 2017, 13, 985–995. [Google Scholar] [CrossRef]
- Ko, J.H.; Rodriguez, I.; Joo, S.W.; Kim, H.G.; Lee, Y.G.; Kang, T.H.; Baek, N.I. Synergistic Effect of Two Major Components of Malva verticillata in the Recovery of Alloxan-Damaged Pancreatic Islet Cells in Zebrafish. J. Med. Food 2019, 22, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.L.; Liu, K.C.; Sheng, W.L.; Xia, Q.; Wang, R.C.; Chen, X.Q.; Zhang, Y. Effects of streptozotocin on pancreatic islet beta-cell apoptosis and glucose metabolism in zebrafish larvae. Fish. Physiol. Biochem. 2020, 46, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Lai, K.; Zhong, Q.; Demin, K.A.; Kalueff, A.V.; Song, C. High-glucose/high-cholesterol diet in zebrafish evokes diabetic and affective pathogenesis: The role of peripheral and central inflammation, microglia and apoptosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109752. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Shimada, Y.; Nishimura, N. Development of a Novel Zebrafish Model for Type 2 Diabetes Mellitus. Sci. Rep. 2017, 7, 1461. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Lawrence, C. The husbandry of zebrafish (Danio rerio): A review. Aquaculture 2007, 269, 1–20. [Google Scholar] [CrossRef]
- Sanni, O.; Fasemore, T.; Nkomozepi, P. Non-Genetic-Induced Zebrafish Model for Type 2 Diabetes with Emphasis on Tools in Model Validation. Int. J. Mol. Sci. 2023, 25, 240. [Google Scholar] [CrossRef]
- Chen, Z.L.; Guo, C.; Zou, Y.Y.; Feng, C.; Yang, D.X.; Sun, C.C.; Wen, W.; Jian, Z.J.; Zhao, Z.; Xiao, Q.; et al. Aerobic exercise enhances mitochondrial homeostasis to counteract D-galactose-induced sarcopenia in zebrafish. Exp. Gerontol. 2023, 180, 112265. [Google Scholar] [CrossRef]
- Patyal, P.; Nguyen, B.; Zhang, X.; Azhar, G.; Ameer, F.S.; Verma, A.; Crane, J.; Kc, G.; Che, Y.; Wei, J.Y. Rho/SRF Inhibitor Modulates Mitochondrial Functions. Int. J. Mol. Sci. 2022, 23, 11536. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Zhou, Z.; Deng, Q.; Zou, Y.; Wang, B.; Huang, S.; Tian, J.; Zheng, L.; Peng, X.; Tang, C. Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway. Int. J. Mol. Sci. 2025, 26, 752. https://doi.org/10.3390/ijms26020752
Chen Z, Zhou Z, Deng Q, Zou Y, Wang B, Huang S, Tian J, Zheng L, Peng X, Tang C. Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway. International Journal of Molecular Sciences. 2025; 26(2):752. https://doi.org/10.3390/ijms26020752
Chicago/Turabian StyleChen, Zhanglin, Zuoqiong Zhou, Qinhua Deng, Yunyi Zou, Bihan Wang, Shuaiwang Huang, Jiaqi Tian, Lan Zheng, Xiyang Peng, and Changfa Tang. 2025. "Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway" International Journal of Molecular Sciences 26, no. 2: 752. https://doi.org/10.3390/ijms26020752
APA StyleChen, Z., Zhou, Z., Deng, Q., Zou, Y., Wang, B., Huang, S., Tian, J., Zheng, L., Peng, X., & Tang, C. (2025). Type 2 Diabetes Induces Mitochondrial Dysfunction in Zebrafish Skeletal Muscle Leading to Diabetic Myopathy via the miR-139-5p/NAMPT Pathway. International Journal of Molecular Sciences, 26(2), 752. https://doi.org/10.3390/ijms26020752