
Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH2Mab-250 in Human Breast Cancer Xenografts

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
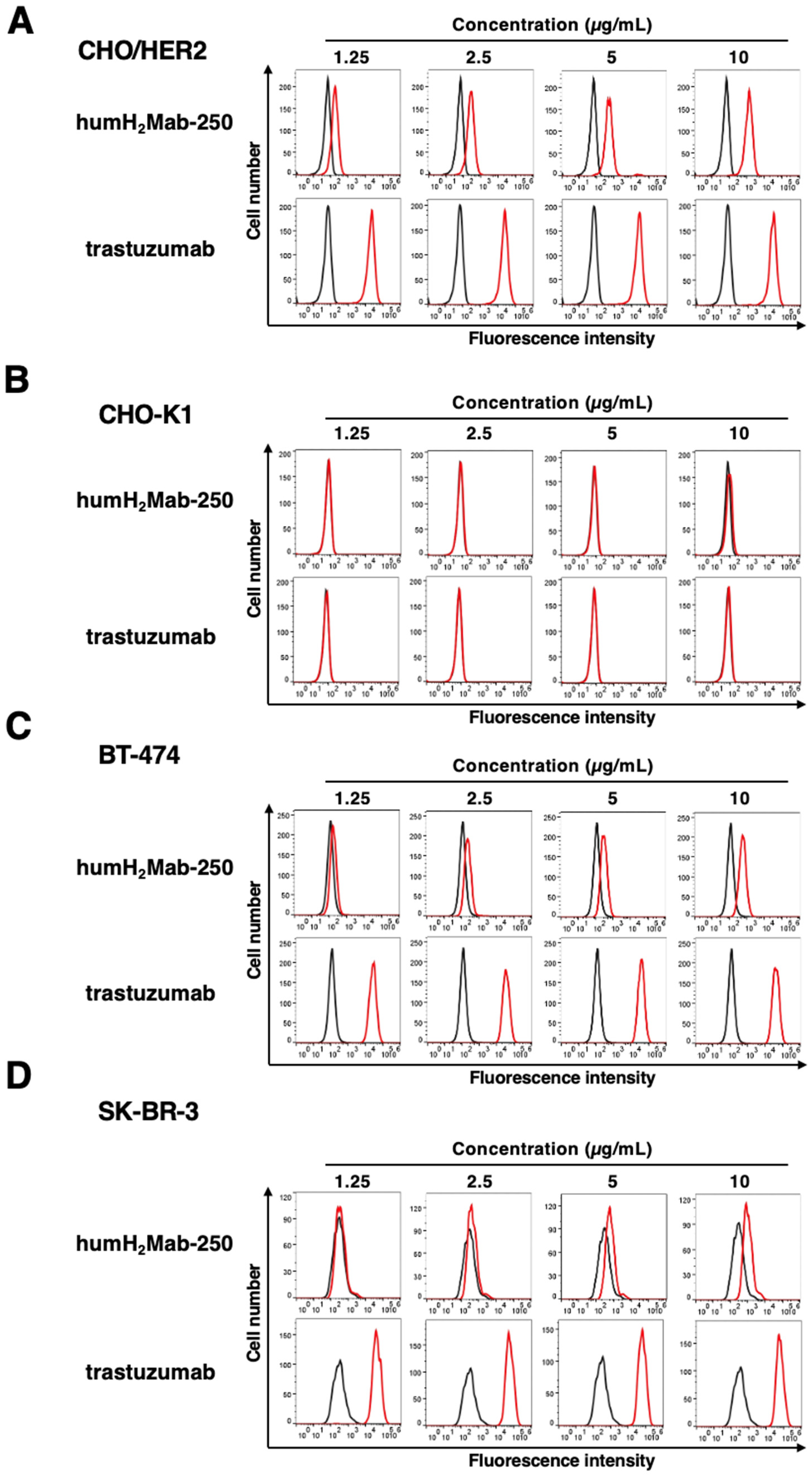
2.1. Humanized Anti-HER2 mAb (humH2Mab-250)
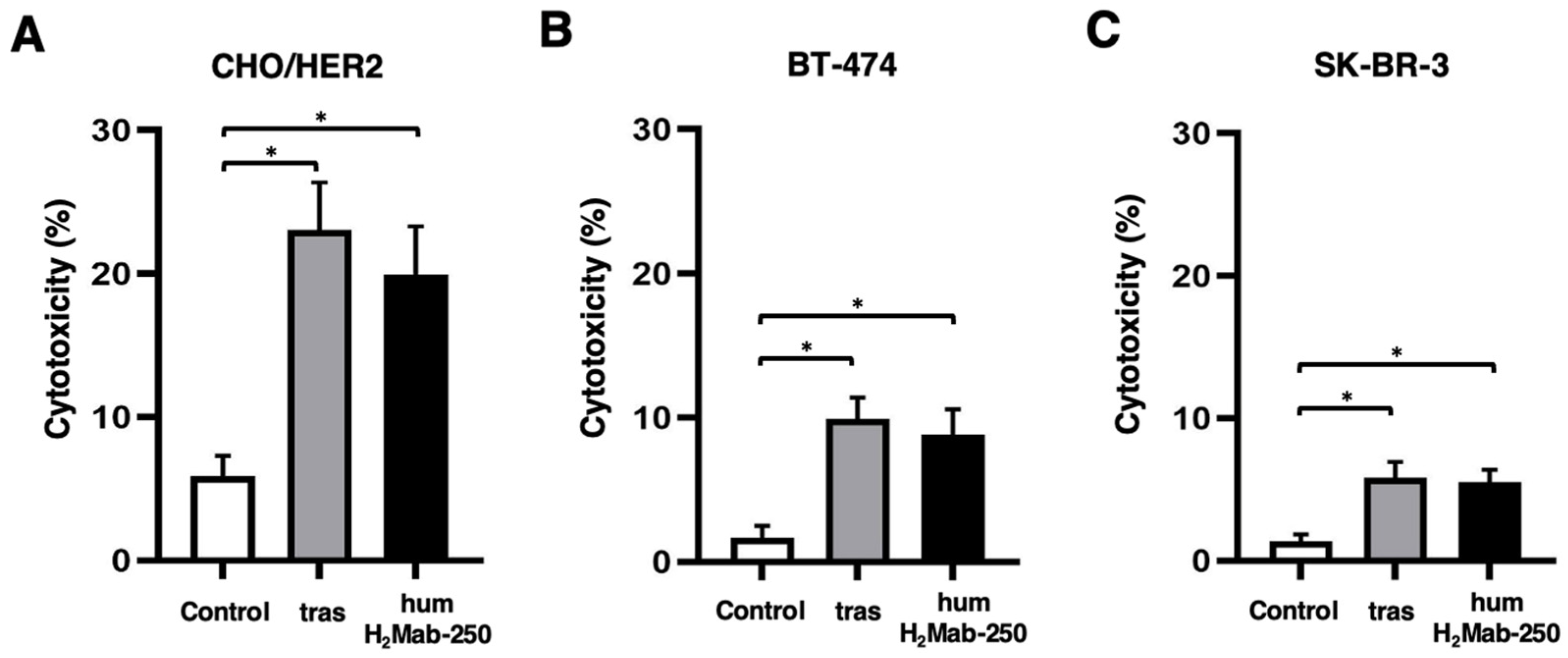
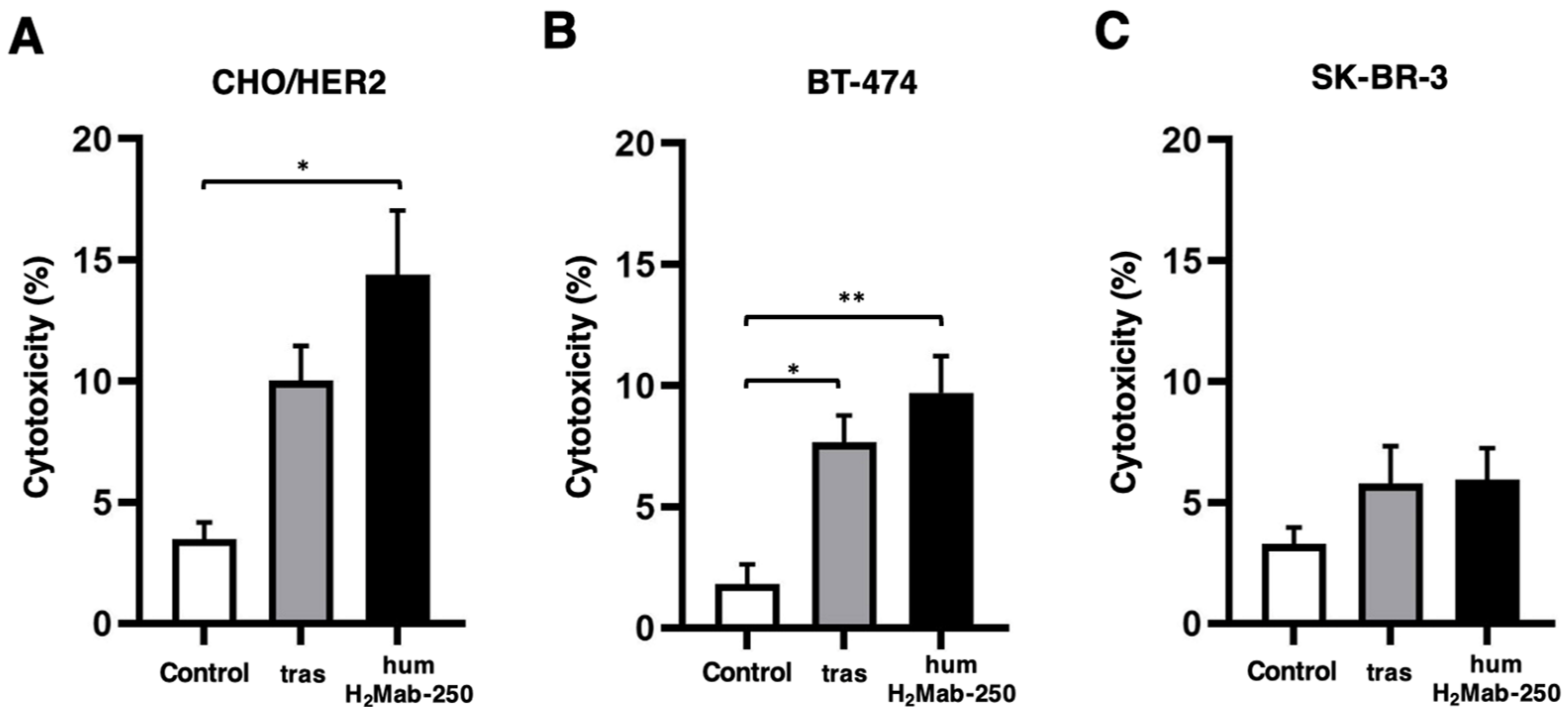
2.2. ADCC and CDC by humH2Mab-250 Against HER2-Positive Cells
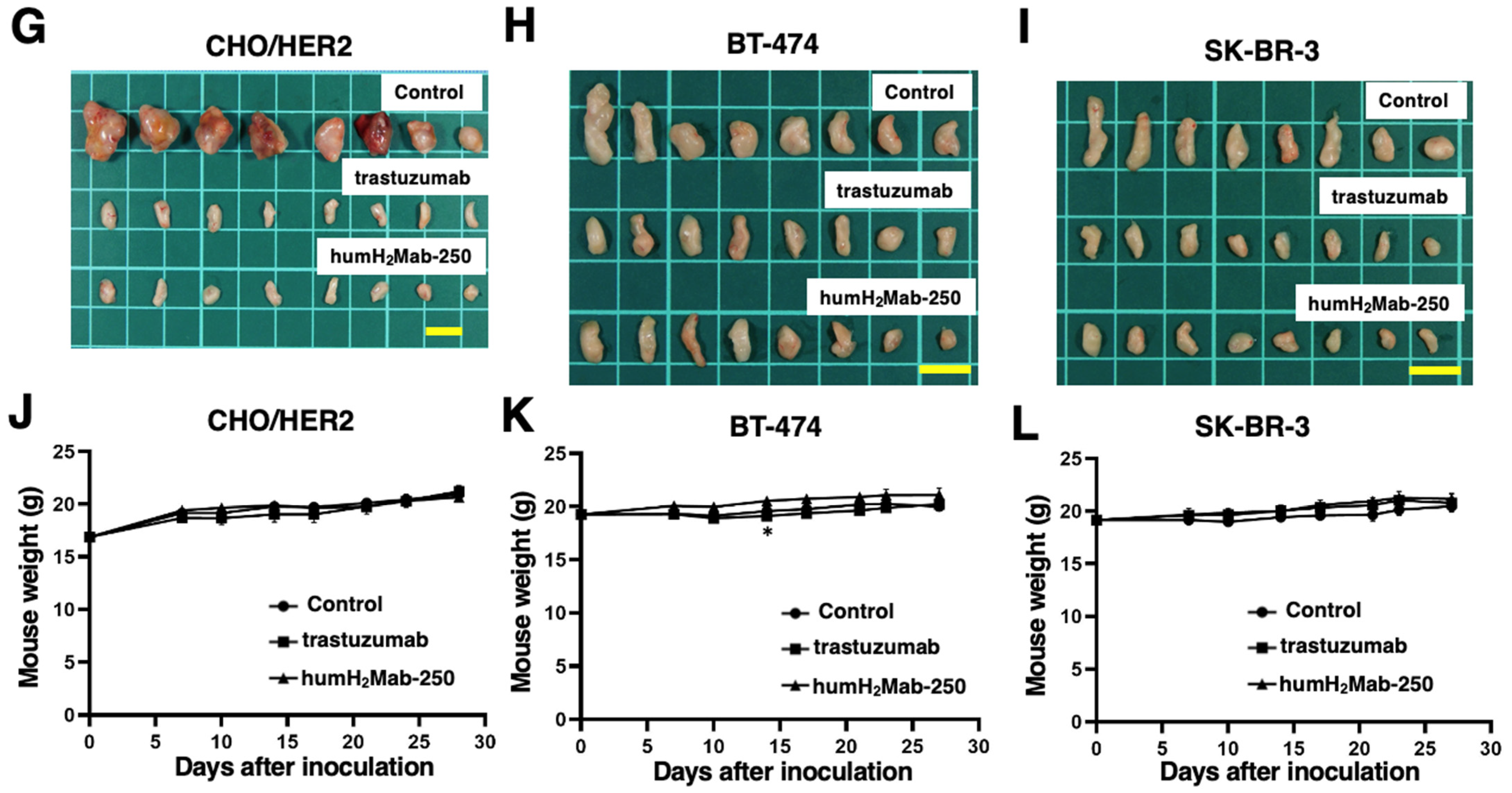
2.3. Antitumor Effects of humH2Mab-250 Against CHO/HER2, BT-474, and SK-BR-3 Xenografts
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Recombinant mAb Production
4.3. Animal Experiments
4.4. Flow Cytometry
4.5. ADCC
4.6. CDC
4.7. Antitumor Activity of humH2Mab-250 in Xenografts of CHO/HER2, BT-474, and SK-BR-3
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef]
- Raja, A.; Kasana, A.; Verma, V. Next-Generation Therapeutic Antibodies for Cancer Treatment: Advancements, Applications, and Challenges. Mol. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Konig, M.F.; Pardoll, D.M.; Bettegowda, C.; Papadopoulos, N.; Wright, K.M.; Gabelli, S.B.; Ho, M.; van Elsas, A.; Zhou, S. Cancer therapy with antibodies. Nat. Rev. Cancer 2024, 24, 399–426. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Pegram, M.; Hsu, S.; Lewis, G.; Pietras, R.; Beryt, M.; Sliwkowski, M.; Coombs, D.; Baly, D.; Kabbinavar, F.; Slamon, D. Inhibitory effects of combinations of HER-2/neu antibody and chemotherapeutic agents used for treatment of human breast cancers. Oncogene 1999, 18, 2241–2251. [Google Scholar] [CrossRef]
- Pietras, R.J.; Pegram, M.D.; Finn, R.S.; Maneval, D.A.; Slamon, D.J. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 1998, 17, 2235–2249. [Google Scholar] [CrossRef]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998, 58, 2825–2831. [Google Scholar] [PubMed]
- Tsao, L.C.; Force, J.; Hartman, Z.C. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res. 2021, 81, 4641–4651. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. HER2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619–4625. [Google Scholar] [CrossRef]
- Pous, A.; Notario, L.; Hierro, C.; Layos, L.; Bugés, C. HER2-Positive Gastric Cancer: The Role of Immunotherapy and Novel Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 11403. [Google Scholar] [CrossRef] [PubMed]
- Balestra, A.; Larsimont, D.; Noël, J.C. HER2 Amplification in p53-Mutated Endometrial Carcinomas. Cancers 2023, 15, 1435. [Google Scholar] [CrossRef]
- Diver, E.J.; Foster, R.; Rueda, B.R.; Growdon, W.B. The Therapeutic Challenge of Targeting HER2 in Endometrial Cancer. Oncologist 2015, 20, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Felip, E. HER2 driven non-small cell lung cancer (NSCLC): Potential therapeutic approaches. Transl. Lung Cancer Res. 2013, 2, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Sullivan, I.; Abdayem, P.; Planchard, D. Targeting HER2 in non-small-cell lung cancer (NSCLC): A glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open 2021, 6, 100260. [Google Scholar] [CrossRef]
- Nasioudis, D.; Gysler, S.; Latif, N.; Cory, L.; Giuntoli, R.L., 2nd; Kim, S.H.; Simpkins, F.; Martin, L.; Ko, E.M. Molecular landscape of ERBB2/HER2 gene amplification among patients with gynecologic malignancies; clinical implications and future directions. Gynecol. Oncol. 2024, 180, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mark, C.; Lee, J.S.; Cui, X.; Yuan, Y. Antibody-Drug Conjugates in Breast Cancer: Current Status and Future Directions. Int. J. Mol. Sci. 2023, 24, 13726. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Bang, Y.J.; Iwasa, S.; Sugimoto, N.; Ryu, M.H.; Sakai, D.; Chung, H.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. [Google Scholar] [CrossRef]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody-drug conjugates come of age in oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Beurskens, F.J.; Lindorfer, M.A.; Farooqui, M.; Beum, P.V.; Engelberts, P.; Mackus, W.J.; Parren, P.W.; Wiestner, A.; Taylor, R.P. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J. Immunol. 2012, 188, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S. Ofatumumab: A novel monoclonal anti-CD20 antibody. Pharmgenom. Pers. Med. 2010, 3, 51–59. [Google Scholar] [CrossRef]
- Manches, O.; Lui, G.; Chaperot, L.; Gressin, R.; Molens, J.P.; Jacob, M.C.; Sotto, J.J.; Leroux, D.; Bensa, J.C.; Plumas, J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood 2003, 101, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.J.; Wang, Z.; Liao, J.; Liu, M.; Zhong, X.R.; Zheng, H.; Wang, Y.P. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol. Lett. 2017, 14, 2961–2969. [Google Scholar] [CrossRef]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; van der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; van den Brink, E.N.; van den Brakel, J.H.N.; Rademaker, H.J.; van Kessel, B.; van den Noort, J.; et al. DuoHexaBody-CD37(®), a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 30. [Google Scholar] [CrossRef]
- Arimori, T.; Mihara, E.; Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Takagi, J.; Kato, Y. Locally misfolded HER2 expressed on cancer cells is a promising target for development of cancer-specific antibodies. Structure 2024, 32, 536–549.e535. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Suzuki, H.; Kato, Y. Establishment of a Novel Cancer-Specific Anti-HER2 Monoclonal Antibody H(2)Mab-250/H(2)CasMab-2 for Breast Cancers. Monoclon. Antib. Immunodiagn. Immunother. 2024, 43, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Anti-HER2 Cancer-Specific mAb, H(2)Mab-250-hG(1), Possesses Higher Complement-Dependent Cytotoxicity than Trastuzumab. Int. J. Mol. Sci. 2024, 25, 8386. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.K.; Suzuki, H.; Ohishi, T.; Nakamura, T.; Tanaka, T.; Kato, Y. A Cancer-Specific Monoclonal Antibody against HER2 Exerts Antitumor Activities in Human Breast Cancer Xenograft Models. Int. J. Mol. Sci. 2024, 25, 1941. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Yamamoto, Y.; Sato, K.; Okemoto-Nakamura, Y.; Shimizu, Y.; Ogawa, M.; Onodera, T.; Takahashi, Y.; Wakita, T.; Kaneko, M.K.; et al. Overcoming antibody-resistant SARS-CoV-2 variants with bispecific antibodies constructed using non-neutralizing antibodies. iScience 2024, 27, 109363. [Google Scholar] [CrossRef] [PubMed]
- Yamane-Ohnuki, N.; Kinoshita, S.; Inoue-Urakubo, M.; Kusunoki, M.; Iida, S.; Nakano, R.; Wakitani, M.; Niwa, R.; Sakurada, M.; Uchida, K.; et al. Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Ortiz Buijsse, A.; Vink, T.; Leusen, J.H.; Bleeker, W.K.; Parren, P.W. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 2012, 189, 3430–3438. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Hiemstra, I.H.; Santegoets, K.C.M.; Janmaat, M.L.; De Goeij, B.; Ten Hagen, W.; van Dooremalen, S.; Boross, P.; van den Brakel, J.; Bosgra, S.; Andringa, G.; et al. Preclinical anti-tumour activity of HexaBody-CD38, a next-generation CD38 antibody with superior complement-dependent cytotoxic activity. EBioMedicine 2023, 93, 104663. [Google Scholar] [CrossRef]
- Diwanji, D.; Trenker, R.; Thaker, T.M.; Wang, F.; Agard, D.A.; Verba, K.A.; Jura, N. Structures of the HER2-HER3-NRG1beta complex reveal a dynamic dimer interface. Nature 2021, 600, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark. Res. 2020, 8, 18. [Google Scholar] [CrossRef]
- An, L.; Lin, Y.; Deng, B.; Yin, Z.; Zhao, D.; Ling, Z.; Wu, T.; Zhao, Y.; Chang, A.H.; Tong, C.; et al. Humanized CD19 CAR-T cells in relapsed/refractory B-ALL patients who relapsed after or failed murine CD19 CAR-T therapy. BMC Cancer 2022, 22, 393. [Google Scholar] [CrossRef]
- Michelozzi, I.M.; Gomez-Castaneda, E.; Pohle, R.V.C.; Cardoso Rodriguez, F.; Sufi, J.; Puigdevall Costa, P.; Subramaniyam, M.; Kirtsios, E.; Eddaoudi, A.; Wu, S.W.; et al. Activation priming and cytokine polyfunctionality modulate the enhanced functionality of low-affinity CD19 CAR T cells. Blood Adv. 2023, 7, 1725–1738. [Google Scholar] [CrossRef]
- Caraballo Galva, L.D.; Jiang, X.; Hussein, M.S.; Zhang, H.; Mao, R.; Brody, P.; Peng, Y.; He, A.R.; Kehinde-Ige, M.; Sadek, R.; et al. Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against HCC. Hepatology 2022, 76, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.S.; Dobrenkov, K.; Pankov, D.; Xu, X.L.; Cheung, N.K. Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology 2017, 6, e1320625. [Google Scholar] [CrossRef]
- Zhang, Y.; Patel, R.P.; Kim, K.H.; Cho, H.; Jo, J.C.; Jeong, S.H.; Oh, S.Y.; Choi, Y.S.; Kim, S.H.; Lee, J.H.; et al. Safety and efficacy of a novel anti-CD19 chimeric antigen receptor T cell product targeting a membrane-proximal domain of CD19 with fast on- and off-rates against non-Hodgkin lymphoma: A first-in-human study. Mol. Cancer 2023, 22, 200. [Google Scholar] [CrossRef] [PubMed]
- Hosking, M.; Shirinbak, S.; Omilusik, K.; Chandra, S.; Gentile, A.; Kennedy, S.; Loter, L.; Ibitokou, S.; Ecker, C.; Brookhouser, N.; et al. 268 Development of FT825/ONO-8250: An off-the-shelf CAR-T cell with preferential HER2 targeting and engineered to enable multi-antigen targeting, improve trafficking, and overcome immunosuppression. J. ImmunoTherapy Cancer 2023, 11, A307. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; Lindorfer, M.A.; van de Winkel, J.G.; Parren, P.W.; Taylor, R.P. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): Considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 2009, 183, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. A Cancer-Specific Monoclonal Antibody against Podocalyxin Exerted Antitumor Activities in Pancreatic Cancer Xenografts. Int. J. Mol. Sci. 2023, 25, 161. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneko, M.K.; Suzuki, H.; Ohishi, T.; Nakamura, T.; Yanaka, M.; Tanaka, T.; Kato, Y. Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH2Mab-250 in Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2025, 26, 1079. https://doi.org/10.3390/ijms26031079
Kaneko MK, Suzuki H, Ohishi T, Nakamura T, Yanaka M, Tanaka T, Kato Y. Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH2Mab-250 in Human Breast Cancer Xenografts. International Journal of Molecular Sciences. 2025; 26(3):1079. https://doi.org/10.3390/ijms26031079
Chicago/Turabian StyleKaneko, Mika K., Hiroyuki Suzuki, Tomokazu Ohishi, Takuro Nakamura, Miyuki Yanaka, Tomohiro Tanaka, and Yukinari Kato. 2025. "Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH2Mab-250 in Human Breast Cancer Xenografts" International Journal of Molecular Sciences 26, no. 3: 1079. https://doi.org/10.3390/ijms26031079
APA StyleKaneko, M. K., Suzuki, H., Ohishi, T., Nakamura, T., Yanaka, M., Tanaka, T., & Kato, Y. (2025). Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH2Mab-250 in Human Breast Cancer Xenografts. International Journal of Molecular Sciences, 26(3), 1079. https://doi.org/10.3390/ijms26031079