
Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Function of the PD-1/PD-L1 Signaling Pathway
2.1. T-Cell Response and Maintenance of Self-Tolerance
2.2. Effects of PD-1 and PD-L1 on Signal Transduction Pathways
2.3. PD1 Activation
2.4. PD1 Expression
2.5. PD-L1 Expression
2.6. Regulation of PD1/PD-L1 Expression in Cancer
3. Landscape of Anti-PD-1/PD-L1 Therapy
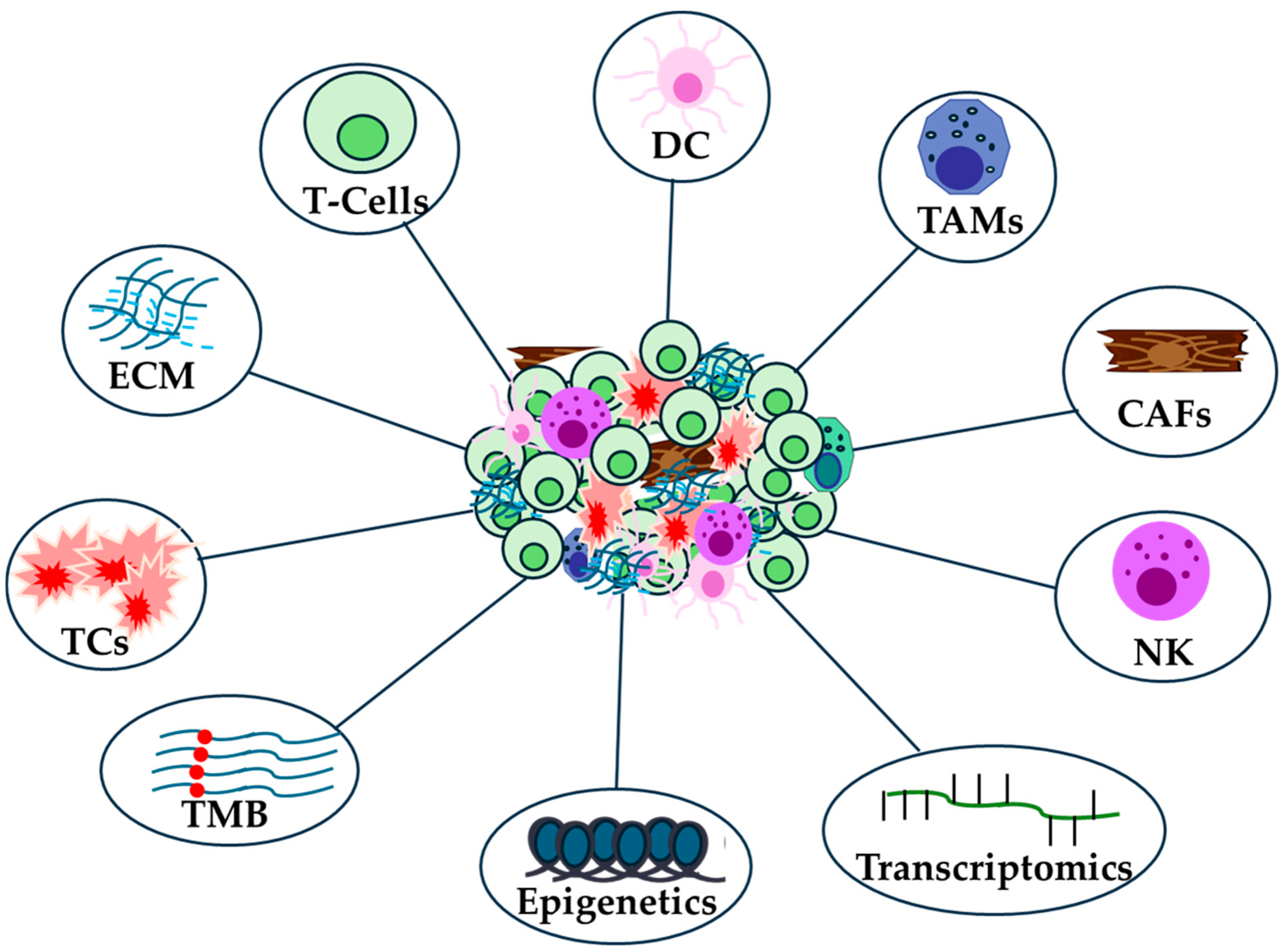
3.1. Immune Landscape
3.2. Genomic Landscape
3.3. Epigenetic Landscape
3.4. Transcriptomic Landscape
4. Antibodies Targeting PD-1/PD-L1
5. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name | Abbreviation | Full Name |
| PD-L1 | Programmed death-ligand 1 | EGFR | Epidermal growth factor receptor |
| PD1 | Programmed cell death protein | DDR | DNA damage repair |
| ICIs | Immune checkpoint inhibitors | ESCC patients | Esophageal squamous cell carcinoma |
| TME | Tumor microenvironment | CHEK2 | Checkpoint kinase 2 |
| ECM | Extracellular matrix | ATM | Ataxia–telangiectasia mutation |
| TAMs | Tumor-associated macrophages | KRAS | Kirsten rat sarcoma virus |
| CAFs | Cancer-associated fibroblasts | TP53 | Tumor Protein P53 |
| TMB | Tumor mutational burden | SMARCA4 | Transcription activator BRG1 |
| Tregs | Regulatory T cells | BC | Breast cancer |
| MAPK | Ras/mitogen-activated protein kinase | FS | Frameshift mutations |
| PI3K | Phosphoinositide 3-kinase | TKIs | Tyrosine kinase inhibitors |
| mTOR | Mammalian target of rapamycin | HLA-I LOH | Human leukocyte antigen-I loss of heterozygosity |
| DC | Dendritic cells | PTEN | Phosphatase and tensin homolog |
| TCs | Ttumor cells | READ | Rectal adenocarcinoma |
| dMMR | Mismatch repair-deficient | METTL3 | Methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit |
| MDSCs | mMyeloid-derived suppressor cells | EVs | Extracellular vesicles |
| MASH-HCC | Steatohepatitis -related hepatocellular carcinoma | CRISPR–Cas9 | Clustered regularly interspaced short palindromic repeats-associated protein 9 |
| SQLE | Squalene epoxidase | TRIM28-SETDB1 | Tripartite Motif Containing 28-SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| mCRC | Metastatic Colorectal Cancer | CD3E | CD3 Epsilon Subunit |
| HCC | Hepatocellular cancer | PDCD1 | Programmed cell death protein 1 |
| PaC | Pancreatic cancer | SV | Synthetic viability |
| ICB | Immune checkpoint blockade | iCCA | Intrahepatic cholangiocarcinoma |
| NK | Natural killer | IL1RN | Interleukin-1 receptor antagonist |
| CPS | Combined positive score | TMEM92 | Transmembrane protein 92 |
| LUAD | Lung adenocarciomaadenocarcinoma | rRCC | Renal cell carcinoma |
| LATPS | Tumor microenvironment prognostic signature | ASPSCR1-TFE3 | Alveolar Soft Part Sarcoma Chromosome Region, Candidate 1–Transcription factor E3 |
| IRGPI | Immune -related gene prognostic index | CTLA4 | Cytotoxic T-lymphocyte associated protein 4 |
| TIME | Tumor immunological microenvironment | FDA | Food and drugs administration |
| NMRGS | NAD+ metabolism-related gene signature | EMA | European medicines agency |
| NSCLC | Non-small -cell lung cancer | AGAs | Actionable oncogenic alterations |
| HNSCC | Head and neck squamous cell carcinoma | VEGF | Vascular endothelial growth factor |
| PDAC | Pancreatic ductal adenocarcinoma | R/M | Recurrent or metastatic |
| IML | Immune -mesenchymal -like | BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| OS | Overall Survival | HER2 | Receptor tyrosine-protein kinase erbB-2 |
| DFS | Disease-free survival | PFS | Progression-free survival |
| NGS | Next-generation sequencing | TNBC | Triple-Negative Breast Cancer |
| TCGA | The Cancer Genome Atlas | SCLC | Small-cell lung cancer |
| IFΝγ | Interferon gamma | ICI | Immune cell infiltration |
| CAMs | Cell adhesion molecules | PBMCs | Blood mononuclear cells |
References
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 Pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Seke Etet, P.F.; Simo Tagne, R.; Vecchio, L.; Lukong, K.E.; Krampera, M. Tumor Microenvironment Uses a Reversible Reprogramming of Mesenchymal Stromal Cells to Mediate Pro-Tumorigenic Effects. Front. Cell Dev. Biol. 2020, 8, 545126. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like Tumor-Associated Macrophages is a Potential Therapeutic Approach to Overcome Antitumor Drug Resistance. NPJ Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, X.; Chen, Z.; Liu, C.; Wu, W.; Zhang, N.; Liu, Z.; Yang, L.; Jiang, Q.; Cheng, Q.; et al. Define Cancer-Associated Fibroblasts (CAFs) in the Tumor Microenvironment: New Opportunities in Cancer Immunotherapy and Advances in Clinical Trials. Mol. Cancer 2023, 22, 159. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Ock, C.Y.; Hwang, J.E.; Keam, B.; Kim, S.B.; Shim, J.J.; Jang, H.J.; Park, S.; Sohn, B.H.; Cha, M.; Ajani, J.A.; et al. Genomic Landscape Associated with Potential Response to Anti-CTLA-4 Treatment in Cancers. Nat. Commun. 2017, 8, 1050. [Google Scholar] [CrossRef] [PubMed]
- Muquith, M.; Espinoza, M.; Elliott, A.; Xiu, J.; Seeber, A.; El-Deiry, W.; Antonarakis, E.S.; Graff, S.L.; Hall, M.J.; Borghaei, H.; et al. Tissue-Specific Thresholds of Mutation Burden Associated with Anti-PD-1/L1 Therapy Benefit and Prognosis in Microsatellite-Stable Cancers. Nat. Cancer 2024, 5, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Cohen, P.R.; Boichard, A.; Frampton, G.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Genomic Landscape of Advanced Basal Cell Carcinoma: Implications for Precision Treatment with Targeted and Immune Therapies. Oncoimmunology 2017, 7, e1404217. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Wang, D.; Wang, A.; Chen, P.; Lin, Y.; Bian, J.; Yang, X.; Zheng, M.; Zhang, H.; Zheng, Y.; et al. A Mutation-Based Gene Set Predicts Survival Benefit after Immunotherapy across Multiple Cancers and Reveals the Immune Response Landscape. Genome Med. 2022, 14, 20. [Google Scholar] [CrossRef]
- Florou, V.; Floudas, C.S.; Maoz, A.; Naqash, A.R.; Norton, C.; Tan, A.C.; Sokol, E.S.; Frampton, G.; Soares, H.P.; Puri, S.; et al. Real-World Pan-Cancer Landscape of Frameshift Mutations and Their Role in Predicting Responses to Immune Checkpoint Inhibitors in Cancers with Low Tumor Mutational Burden. J. Immunother. Cancer 2023, 11, e007440. [Google Scholar] [CrossRef]
- Wang, J.Y.; Xiu, J.; Baca, Y.; Arai, H.; Battaglin, F.; Kawanishi, N.; Soni, S.; Zhang, W.; Millstein, J.; Shields, A.F.; et al. Distinct Genomic Landscapes of Gastroesophageal Adenocarcinoma Depending on PD-L1 Expression Identify Mutations in RAS-MAPK Pathway and TP53 as Potential Predictors of Immunotherapy Efficacy. Ann. Oncol. 2021, 32, 906–916. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 Expression and Tumor Mutational Burden Are Independent Biomarkers in Most Cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Y.; Miao, Q.; Chen, Y. The Therapeutic Potential of PD-1/PD-L1 Pathway on Immune-Related Diseases: Based on the Innate and Adaptive Immune Components. Biomed. Pharmacother. 2023, 167, 115569. [Google Scholar] [CrossRef]
- Hwang, J.R.; Byeon, Y.; Kim, D.; Park, S.G. Recent Insights of T Cell Receptor-Mediated Signaling Pathways for T Cell Activation and Development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Chamoto, K.; Yaguchi, T.; Tajima, M.; Honjo, T. Insights from a 30-Year Journey: Function, Regulation and Therapeutic Modulation of PD1. Nat. Rev. Immunol. 2023, 23, 682–695. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T Cell Exhaustion”. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A Snapshot of the PD-1/PD-L1 Pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping Roles of PD-1 and FoxP3 in Maintaining Immune Tolerance in a Novel Autoimmune Pancreatitis Mouse Model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495. [Google Scholar] [CrossRef]
- Xu, X.; Hou, B.; Fulzele, A.; Masubuchi, T.; Zhao, Y.; Wu, Z.; Hu, Y.; Jiang, Y.; Ma, Y.; Wang, H.; et al. PD-1 and BTLA Regulate T Cell Signaling Differentially and Only Partially through SHP1 and SHP2. J. Cell Biol. 2020, 219, e201905085. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3ζ Signalosome and Downstream Signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol. Cell. Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef]
- Van Der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef]
- Butte, M.J.; Peña-Cruz, V.; Kim, M.J.; Freeman, G.J.; Sharpe, A.H. Interaction of Human PD-L1 and B7-1. Mol. Immunol. 2008, 45, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Y.; Yang, S.; Zeng, B.; Zhang, Z.; Jiao, G.; Zhang, Y.; Cai, L.; Yang, R. Regulation of Arginase I Activity and Expression by Both PD-1 and CTLA-4 on the Myeloid-Derived Suppressor Cells. Cancer Immunol. Immunother. 2009, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 Is a Regulator of Virus-Specific CD8+T Cell Survival in HIV Infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef]
- Bally, A.P.R.; Austin, J.W.; Boss, J.M. Genetic and Epigenetic Regulation of PD-1 Expression. J. Immunol. 2016, 196, 2431–2437. [Google Scholar] [CrossRef]
- Youngblood, B.; Noto, A.; Porichis, F.; Akondy, R.S.; Ndhlovu, Z.M.; Austin, J.W.; Bordi, R.; Procopio, F.A.; Miura, T.; Allen, T.M.; et al. Cutting Edge: Prolonged Exposure to HIV Reinforces a Poised Epigenetic Program for PD-1 Expression in Virus-Specific CD8 T Cells. J. Immunol. 2013, 191, 540–544. [Google Scholar] [CrossRef]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic Virus Infection Enforces Demethylation of the Locus That Encodes PD-1 in Antigen-Specific CD8(+) T Cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef]
- Zhang, N.; Li, M.; Xu, X.; Zhang, Y.; Liu, Y.; Zhao, M.; Li, P.; Chen, J.; Fukuda, T.; Gu, J.; et al. Loss of Core Fucosylation Enhances the Anticancer Activity of Cytotoxic T Lymphocytes by Increasing PD-1 Degradation. Eur. J. Immunol. 2020, 50, 1820–1833. [Google Scholar] [CrossRef]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 Mediates PD-1 Ubiquitination and Regulates Anti-Tumour Immunity of T Cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef]
- Ribas, A.; Hu-Lieskovan, S. What Does PD-L1 Positive or Negative Mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef]
- George, J.; Saito, M.; Tsuta, K.; Iwakawa, R.; Shiraishi, K.; Scheel, A.H.; Uchida, S.; Watanabe, S.I.; Nishikawa, R.; Noguchi, M.; et al. Genomic Amplification of CD274 (PD-L1) in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; De Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A Major Chromatin Regulator Determines Resistance of Tumor Cells to T Cell-Mediated Killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of Essential Genes for Cancer Immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; La Fleur, M.W.; Juneja, V.R.; et al. In Vivo CRISPR Screening Identifies Ptpn2 as a Cancer Immunotherapy Target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 Expression through 3’-UTR Disruption in Multiple Cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms Regulating PD-L1 Expression in Cancers and Associated Opportunities for Novel Small-Molecule Therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The Role of Cancer-Derived MicroRNAs in Cancer Immune Escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Omar, H.A.; El-Serafi, A.T.; Hersi, F.; Arafa, E.S.A.; Zaher, D.M.; Madkour, M.; Arab, H.H.; Tolba, M.F. Immunomodulatory MicroRNAs in Cancer: Targeting Immune Checkpoints and the Tumor Microenvironment. FEBS J. 2019, 286, 3540–3557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pan, S.; Chen, X.; Wang, Z.-w.; Zhu, X. The Role of LncRNAs and CircRNAs in the PD-1/PD-L1 Pathway in Cancer Immunotherapy. Mol. Cancer 2021, 20, 116. [Google Scholar] [CrossRef]
- Liu, W.; Liu, R.; Yuan, R.; Wang, X. MicroRNA-4458 Regulates PD-L1 Expression to Enhance Anti-Tumor Immunity in NSCLC via Targeting STAT3. Mol. Biotechnol. 2021, 63, 1268–1279. [Google Scholar] [CrossRef]
- Chen, K.-b.; Yang, W.; Xuan, Y.; Lin, A.-j. MiR-526b-3p Inhibits Lung Cancer Cisplatin-Resistance and Metastasis by Inhibiting STAT3-Promoted PD-L1. Cell Death Dis. 2021, 12, 748. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by P53 via MiR-34. J. Natl. Cancer Inst. 2015, 108, djv303. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis Is Regulated via MicroRNA-200/ZEB1 Axis Control of Tumour Cell PD-L1 Expression and Intratumoral Immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Yousefi, A.; Sotoodehnejadnematalahi, F.; Nafissi, N.; Zeinali, S.; Azizi, M. MicroRNA-561-3p Indirectly Regulates the PD-L1 Expression by Targeting ZEB1, HIF1A, and MYC Genes in Breast Cancer. Sci. Rep. 2024, 14, 5845. [Google Scholar] [CrossRef]
- Dang, S.; Malik, A.; Chen, J.; Qu, J.; Yin, K.; Cui, L.; Gu, M. LncRNA SNHG15 Contributes to Immuno-Escape of Gastric Cancer Through Targeting MiR141/PD-L1. OncoTargets Ther. 2020, 13, 8547–8556. [Google Scholar] [CrossRef]
- Cheng, S.; Li, F.; Qin, H.; Ping, Y.; Zhao, Q.; Gao, Q.; Song, M.; Qu, J.; Shan, J.; Zhang, K.; et al. Long Noncoding RNA LncNDEPD1 Regulates PD-1 Expression via MiR-3619-5p in CD8+T Cells. J. Immunol. 2022, 208, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.G.; Liang, B.; Liu, J.G.; Jin, S.H.; He, S.S.; Frey, B.; Gu, N.; Fietkau, R.; Hecht, M.; Ma, H.; et al. Identification of 15 LncRNAs Signature for Predicting Survival Benefit of Advanced Melanoma Patients Treated with Anti-PD-1 Monotherapy. Cells 2021, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Toker, J.; Iorgulescu, J.B.; Ling, A.L.; Villa, G.R.; Gadet, J.A.M.A.; Parida, L.; Getz, G.; Wu, C.J.; Reardon, D.A.; Chiocca, E.A.; et al. Clinical Importance of the LncRNA NEAT1 in Cancer Patients Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2023, 29, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Tu, R.; Liu, F.; Zhang, H.; Dai, Z.; Wang, Z.; Luo, P.; He, S.; Xiao, G.; Feng, J.; et al. PD-L1-Related IncRNAs Are Associated with Malignant Characteristics and Immune Microenvironment in Glioma. Aging 2023, 15, 10785–10810. [Google Scholar] [CrossRef]
- Wang, Q.; Li, G.; Ma, X.; Liu, L.; Liu, J.; Yin, Y.; Li, H.; Chen, Y.; Zhang, X.; Zhang, L.; et al. LncRNA TINCR Impairs the Efficacy of Immunotherapy against Breast Cancer by Recruiting DNMT1 and Downregulating MiR-199a-5p via the STAT1-TINCR-USP20-PD-L1 Axis. Cell Death Dis. 2023, 14, 76. [Google Scholar] [CrossRef]
- Salama, E.A.; Adbeltawab, R.E.; El Tayebi, H.M. XIST and TSIX: Novel Cancer Immune Biomarkers in PD-L1-Overexpressing Breast Cancer Patients. Front. Oncol. 2020, 9, 1459. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Chen, M.; Chen, H.; Zhong, Q.; Liang, L.; Li, B. LncRNA TCL6 Correlates with Immune Cell Infiltration and Indicates Worse Survival in Breast Cancer. Breast Cancer 2020, 27, 573–585. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Apollonio, B.; Ioannou, N.; Papazoglou, D.; Ramsay, A.G. Understanding the Immune-Stroma Microenvironment in B Cell Malignancies for Effective Immunotherapy. Front. Oncol. 2021, 11, 626818. [Google Scholar] [CrossRef]
- Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Lynch, S.M.; Rahman, A.; Gallagher, W.M.; Boecxstaens, V.; Marine, J.C.; Cattoretti, G.; van den Oord, J.J.; et al. Mapping the Immune Landscape in Metastatic Melanoma Reveals Localized Cell-Cell Interactions That Predict Immunotherapy Response. Cancer Res. 2022, 82, 3275–3290. [Google Scholar] [CrossRef]
- Guo, J.; Tang, B.; Fu, J.; Zhu, X.; Xie, W.; Wang, N.; Ding, Z.; Song, Z.; Yang, Y.; Xu, G.; et al. High-Plex Spatial Transcriptomic Profiling Reveals Distinct Immune Components and the HLA Class I/DNMT3A/CD8 Modulatory Axis in Mismatch Repair-Deficient Endometrial Cancer. Cell. Oncol. 2024, 47, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Puig-Saus, C.; Sennino, B.; Peng, S.; Wang, C.L.; Pan, Z.; Yuen, B.; Purandare, B.; An, D.; Quach, B.B.; Nguyen, D.; et al. Neoantigen-Targeted CD8+T Cell Responses with PD-1 Blockade Therapy. Nature 2023, 615, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dong, Y.; Xie, S.; Song, Y.; Yu, C.; He, Y.; Wang, Z.; Hu, Q.; Ni, Y.; Ding, L. Immune Checkpoint CD161/LLT1-Associated Immunological Landscape and Diagnostic Value in Oral Squamous Cell Carcinoma. J. Pathol. Clin. Res. 2024, 10, e353. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, X.; Liu, X.; Cai, X.; Shen, T.; Pan, D.; Liang, R.; Ding, R.; Hu, R.; Dong, J.; et al. Single-Cell Sequencing Reveals the Immune Microenvironment Landscape Related to Anti-PD-1 Resistance in Metastatic Colorectal Cancer with High Microsatellite Instability. BMC Med. 2023, 21, 161. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, X.; Wong, C.C.; Zhang, Y.; Pan, Y.; Zhou, Y.; Cheung, A.H.K.; Liu, Y.; Ji, F.; Kang, X.; et al. Targeting Squalene Epoxidase Restores Anti-PD-1 Efficacy in Metabolic Dysfunction-Associated Steatohepatitis-Induced Hepatocellular Carcinoma. Gut 2024, 73, 2023–2036. [Google Scholar] [CrossRef]
- Tan, J.; Fan, W.; Liu, T.; Zhu, B.; Liu, Y.; Wang, S.; Wu, J.; Liu, J.; Zou, F.; Wei, J.; et al. TREM2+ Macrophages Suppress CD8+T-Cell Infiltration after Transarterial Chemoembolisation in Hepatocellular Carcinoma. J. Hepatol. 2023, 79, 126–140. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, Y.; Huang, Y.; Wang, Q.; Kaifi, J.T.; Kimchi, E.T.; Chabu, C.Y.; Liu, Z.; Joshi, T.; Li, G. Single-Cell RNA Sequencing to Characterize the Response of Pancreatic Cancer to Anti-PD-1 Immunotherapy. Transl. Oncol. 2022, 15, 101262. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression—Implications for Anticancer Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Kugel, C.H.; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Kos, K.; Spagnuolo, L.; Isaeva, O.I.; Garner, H.; Wellenstein, M.D.; Bakker, N.; Duits, D.E.M.; Kersten, K.; Klarenbeek, S.; et al. Neoadjuvant Immune Checkpoint Blockade Triggers Persistent and Systemic Treg Activation Which Blunts Therapeutic Efficacy against Metastatic Spread of Breast Tumors. Oncoimmunology 2023, 12, 2201147. [Google Scholar] [CrossRef]
- Yin, Y.; Sakakibara, R.; Honda, T.; Kirimura, S.; Daroonpan, P.; Kobayashi, M.; Ando, K.; Ujiie, H.; Kato, T.; Kaga, K.; et al. High Density and Proximity of CD8+T Cells to Tumor Cells Are Correlated with Better Response to Nivolumab Treatment in Metastatic Pleural Mesothelioma. Thorac. Cancer 2023, 14, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Tanaka, N.; Takamatsu, K.; Hakozaki, K.; Fukumoto, K.; Masuda, T.; Mikami, S.; Shinojima, T.; Kakimi, K.; Tsunoda, T.; et al. Multiplexed Single-Cell Pathology Reveals the Association of CD8 T-Cell Heterogeneity with Prognostic Outcomes in Renal Cell Carcinoma. Cancer Immunol. Immunother. 2021, 70, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Peng, M. Immune Landscape of Distinct Subtypes in Urothelial Carcinoma Based on Immune Gene Profile. Front. Immunol. 2022, 13, 970885. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, X.; Wu, X. Tumor Immune Microenvironment Characterization Identifies Prognosis and Immunotherapy-Related Gene Signatures in Melanoma. Front. Immunol. 2021, 12, 663495. [Google Scholar] [CrossRef] [PubMed]
- Sui, Z.; Wu, X.; Du, L.; Wang, H.; Yuan, L.; Zhang, J.V.; Yu, Z. Characterization of the Immune Cell Infiltration Landscape in Esophageal Squamous Cell Carcinoma. Front. Oncol. 2022, 12, 879326. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, J.; Ren, K.; Tian, X.; Gao, H.; Tian, X.; Zhang, X.; Kan, Q. The Heterogeneity of Immune Cell Infiltration Landscape and Its Immunotherapeutic Implications in Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 861525. [Google Scholar] [CrossRef]
- Huang, J.; Yuan, L.; Huang, W.; Liao, L.; Zhu, X.; Wang, X.; Li, J.; Liang, W.; Wu, Y.; Liu, X.; et al. LATPS, a Novel Prognostic Signature Based on Tumor Microenvironment of Lung Adenocarcinoma to Better Predict Survival and Immunotherapy Response. Front. Immunol. 2022, 13, 1064874. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Feng, Y.; Liu, X.; Wang, Y.; Liu, Y.; Li, H.; Zhang, Y. Decoding the Immune Landscape: A Comprehensive Analysis of Immune-Associated Biomarkers in Cervical Carcinoma and Their Implications for Immunotherapy Strategies. Front. Genet. 2024, 15, 1340569. [Google Scholar] [CrossRef]
- Wang, Z.; Song, J.; Azami, N.L.B.; Sun, M. Identification of a Novel Immune Landscape Signature for Predicting Prognosis and Response of Colon Cancer to Immunotherapy. Front. Immunol. 2022, 13, 802665. [Google Scholar] [CrossRef]
- Jiang, C.; Zhou, Y.; Yan, L.; Zheng, J.; Wang, X.; Li, J.; Jiang, X. A Prognostic NAD+ Metabolism-Related Gene Signature for Predicting Response to Immune Checkpoint Inhibitor in Glioma. Front. Oncol. 2023, 13, 1051641. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, Y.; Wang, F.; Tan, G. A Novel Immune Signature Predicts Immunotherapy Responsiveness and Reveals the Landscape of the Tumor Immune Microenvironment in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2022, 13, 1051051. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.R.; Zhang, J.; Duose, D.Y.; Gonzalez-Kozlova, E.; Redman, M.W.; Chen, H.; Manyam, G.C.; Kumar, G.; Zhang, J.; Song, X.; et al. Multi-Omics Analysis Reveals Immune Features Associated with Immunotherapy Benefit in Patients with Squamous Cell Lung Cancer from Phase III Lung-MAP S1400I Trial. Clin. Cancer Res. 2024, 30, 1655–1668. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Che, X.; Qiu, X.; Li, Z.; Yang, B.; Wang, S.; Hou, K.; Fan, Y.; Qu, X.; Liu, Y. M2 Macrophage Infiltration into Tumor Islets Leads to Poor Prognosis in Non-Small-Cell Lung Cancer. Cancer Manag. Res. 2019, 11, 6125–6138. [Google Scholar] [CrossRef] [PubMed]
- Gavrielatou, N.; Fortis, E.; Spathis, A.; Anastasiou, M.; Economopoulou, P.; Foukas, G.R.P.; Lelegiannis, I.M.; Rusakiewicz, S.; Vathiotis, I.; Aung, T.N.; et al. B-Cell Infiltration Is Associated with Survival Outcomes Following Programmed Cell Death Protein 1 Inhibition in Head and Neck Squamous Cell Carcinoma. Ann. Oncol. 2024, 35, 340–350. [Google Scholar] [CrossRef]
- Chang, T.-G.; Spathis, A.; Schäffer, A.A.; Gavrielatou, N.; Kuo, F.; Jia, D.; Mukherjee, S.; Sievers, C.; Economopoulou, P.; Anastasiou, M.; et al. Tumor and Blood B Cell Abundance Outperforms Established Immune Checkpoint Blockade Response Prediction Signatures in Head and Neck Cancer. Ann. Oncol. 2024, in press. [Google Scholar] [CrossRef]
- Leung, E.L.H.; Li, R.Z.; Fan, X.X.; Wang, L.Y.; Wang, Y.; Jiang, Z.; Huang, J.; Pan, H.D.; Fan, Y.; Xu, H.; et al. Longitudinal High-Dimensional Analysis Identifies Immune Features Associating with Response to Anti-PD-1 Immunotherapy. Nat. Commun. 2023, 14, 5115. [Google Scholar] [CrossRef]
- De Lima, V.A.B.; Borch, A.; Hansen, M.; Draghi, A.; Spanggaard, I.; Rohrberg, K.; Reker Hadrup, S.; Lassen, U.; Svane, I.M. Common Phenotypic Dynamics of Tumor-Infiltrating Lymphocytes across Different Histologies upon Checkpoint Inhibition: Impact on Clinical Outcome. Cytotherapy 2020, 22, 204–213. [Google Scholar] [CrossRef]
- Aoyama, S.; Nakagawa, R.; Nemoto, S.; Perez-Villarroel, P.; Mulé, J.J.; Mailloux, A.W. Checkpoint Blockade Accelerates a Novel Switch from an NKT-Driven TNFα Response toward a T Cell Driven IFN-γ Response within the Tumor Microenvironment. J. Immunother. Cancer 2021, 9, e002269. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; De Sampaio, P.C.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef]
- Li, L.; Shen, L.; Wu, H.; Li, M.; Chen, L.; Zhou, Q.; Ma, J.; Huai, C.; Zhou, W.; Wei, M.; et al. An Integrated Analysis Identifies Six Molecular Subtypes of Pancreatic Ductal Adenocarcinoma Revealing Cellular and Molecular Landscape. Carcinogenesis 2023, 44, 726–740. [Google Scholar] [CrossRef]
- Pich-Bavastro, C.; Yerly, L.; DiDomizio, J.; Tissot-Renaud, S.; Gilliet, M.; Kuonen, F. Activin A-Mediated Polarization of Cancer-Associated Fibroblasts and Macrophages Confers Resistance to Checkpoint Immunotherapy in Skin Cancer. Clin. Cancer Res. 2023, 29, 3498–3513. [Google Scholar] [CrossRef] [PubMed]
- Tunger, A.; Sommer, U.; Wehner, R.; Kubasch, A.S.; Grimm, M.O.; Bachmann, M.P.; Platzbecker, U.; Bornhäuser, M.; Baretton, G.; Schmitz, M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. J. Clin. Med. 2019, 8, 1534. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N.; Molinero, L.; Hamidi, H.; Sehouli, J.; Miller, A.; Moore, K.N.; Taskiran, C.; Bookman, M.; Lindemann, K.; Anderson, C.; et al. Influence of Genomic Landscape on Cancer Immunotherapy for Newly Diagnosed Ovarian Cancer: Biomarker Analyses from the IMagyn050 Randomized Clinical Trial. Clin. Cancer Res. 2023, 29, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Roshan-Zamir, M.; Khademolhosseini, A.; Rajalingam, K.; Ghaderi, A.; Rajalingam, R. The Genomic Landscape of the Immune System in Lung Cancer: Present Insights and Continuing Investigations. Front. Genet. 2024, 15, 1414487. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.; Xu, Y.; Cai, S.; Li, T.; Wang, G.; Li, C.; Zhao, L.; Hu, Y. Co-Occurring Genomic Alterations and Immunotherapy Efficacy in NSCLC. NPJ Precis. Oncol. 2022, 6, 4. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Lavery, J.A.; Brown, S.; de Bruijn, I.; Jee, J.; Tran, T.N.; Rizvi, H.; Arbour, K.C.; Whiting, K.; Shen, R.; et al. The GENIE BPC NSCLC Cohort: A Real-World Repository Integrating Standardized Clinical and Genomic Data for 1846 Patients with Non-Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 3418–3428. [Google Scholar] [CrossRef]
- Zhang, H.; Wen, H.; Zhu, Q.; Zhang, Y.; Xu, F.; Ma, T.; Guo, Y.; Lu, C.; Zhao, X.; Ji, Y.; et al. Genomic Profiling and Associated B Cell Lineages Delineate the Efficacy of Neoadjuvant Anti-PD-1-Based Therapy in Oesophageal Squamous Cell Carcinoma. EBioMedicine 2024, 100, 104971. [Google Scholar] [CrossRef]
- Xu, P.; Gao, Y.; Jiang, S.; Cui, Y.; Xie, Y.; Kang, Z.; Chen, Y.X.; Sun, D.; Fang, J.Y. CHEK2 Deficiency Increase the Response to PD-1 Inhibitors by Affecting the Tumor Immune Microenvironment. Cancer Lett. 2024, 588, 216595. [Google Scholar] [CrossRef]
- Vokes, N.I.; Cobo, A.G.; Fernandez-Chas, M.; Molkentine, D.; Treviño, S.; Druker, V.; Qian, Y.; Patel, S.; Schmidt, S.; Hong, L.; et al. ATM Mutations Associate with Distinct Co-Mutational Patterns and Therapeutic Vulnerabilities in NSCLC. Clin. Cancer Res. 2023, 29, 4958–4972. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The Genomic Landscape of SMARCA4 Alterations and Associations with Outcomes in Patients with Lung Cancer. Clin. Cancer Res. 2020, 26, 5701–5708. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.V.; Goodman, A.M.; Sivakumar, S.; Frampton, G.; Kurzrock, R. Intra-Patient Stability of Tumor Mutational Burden from Tissue Biopsies at Different Time Points in Advanced Cancers. Genome Med. 2021, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Georgoulias, G.; Zaravinos, A. Genomic Landscape of the Immunogenicity Regulation in Skin Melanomas with Diverse Tumor Mutation Burden. Front. Immunol. 2022, 13, 1006665. [Google Scholar] [CrossRef] [PubMed]
- Ravi, A.; Hellmann, M.D.; Arniella, M.B.; Holton, M.; Freeman, S.S.; Naranbhai, V.; Stewart, C.; Leshchiner, I.; Kim, J.; Akiyama, Y.; et al. Genomic and Transcriptomic Analysis of Checkpoint Blockade Response in Advanced Non-Small Cell Lung Cancer. Nat. Genet. 2023, 55, 807–819. [Google Scholar] [CrossRef]
- Kelly, A.D.; Murugesan, K.; Kuang, Z.; Montesion, M.; Ross, J.S.; Albacker, L.A.; Huang, R.S.P.; Lin, D.I.; Demirci, U.; Creeden, J. Pan-Cancer Landscape of CD274 (PD-L1) Rearrangements in 283,050 Patient Samples, Its Correlation with PD-L1 Protein Expression, and Immunotherapy Response. J. Immunother. Cancer 2021, 9, e003550. [Google Scholar] [CrossRef]
- Kim, R.; Hong, J.Y.; Lee, J.; Kwon, G.Y.; Jeong, B.C.; Park, S.H. Genomic Sequencing for Bladder Urothelial Carcinoma and Its Clinical Implications for Immunotherapy. Cancer Res. Treat. 2022, 54, 894–906. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Wu, D.; Liu, Y.; Li, X.; Liu, Y.; Yang, Q.; Liu, Y.; Wu, J.; Tian, C.; Zeng, Y.; Zhao, Z.; et al. Identification of Clonal Neoantigens Derived From Driver Mutations in an EGFR-Mutated Lung Cancer Patient Benefitting From Anti-PD-1. Front. Immunol. 2020, 11, 1366. [Google Scholar] [CrossRef]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292. [Google Scholar] [CrossRef]
- Exposito, F.; Redrado, M.; Houry, M.; Hastings, K.; Molero-Abraham, M.; Lozano, T.; Solorzano, J.L.; Sanz-Ortega, J.; Adradas, V.; Amat, R.; et al. PTEN Loss Confers Resistance to Anti-PD-1 Therapy in Non-Small Cell Lung Cancer by Increasing Tumor Infiltration of Regulatory T Cells. Cancer Res. 2023, 83, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Tien, F.M.; Lu, H.H.; Lin, S.Y.; Tsai, H.C. Epigenetic Remodeling of the Immune Landscape in Cancer: Therapeutic Hurdles and Opportunities. J. Biomed. Sci. 2023, 30, 3. [Google Scholar] [CrossRef] [PubMed]
- Keshari, S.; Barrodia, P.; Singh, A.K. Epigenetic Perspective of Immunotherapy for Cancers. Cells 2023, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic Regulation in the Tumor Microenvironment: Molecular Mechanisms and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Gan, W.; Zong, J.; Hou, Y.; Zhou, M.; Yan, Z.; Li, T.; Lv, S.; Zeng, Z.; Wang, W.; et al. Developing an M5C Regulator-Mediated RNA Methylation Modification Signature to Predict Prognosis and Immunotherapy Efficacy in Rectal Cancer. Front. Immunol. 2023, 14, 1054700. [Google Scholar] [CrossRef]
- Peng, B.; Lin, Y.; Yi, G.; Lin, M.; Xiao, Y.; Qiu, Y.; Yao, W.; Zhou, X.; Liu, Z. Comprehensive Landscape of M6A Regulator-Related Gene Patterns and Tumor Microenvironment Infiltration Characterization in Gastric Cancer. Sci. Rep. 2024, 14, 16404. [Google Scholar] [CrossRef]
- Zeng, C.; Huang, W.; Li, Y.; Weng, H. Roles of METTL3 in Cancer: Mechanisms and Therapeutic Targeting. J. Hematol. Oncol. 2020, 13, 117. [Google Scholar] [CrossRef]
- Ning, J.; Hou, X.; Hao, J.; Zhang, W.; Shi, Y.; Huang, Y.; Ruan, X.; Zheng, X.; Gao, M. METTL3 Inhibition Induced by M2 Macrophage-Derived Extracellular Vesicles Drives Anti-PD-1 Therapy Resistance via M6A-CD70-Mediated Immune Suppression in Thyroid Cancer. Cell Death Differ. 2023, 30, 2265–2279. [Google Scholar] [CrossRef]
- Lin, J.; Guo, D.; Liu, H.; Zhou, W.; Wang, C.; Müller, I.; Kossenkov, A.V.; Drapkin, R.; Bitler, B.G.; Helin, K.; et al. The SETDB1-TRIM28 Complex Suppresses Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 1413–1424. [Google Scholar] [CrossRef]
- Carpen, L.; Falvo, P.; Orecchioni, S.; Mitola, G.; Hillje, R.; Mazzara, S.; Mancuso, P.; Pileri, S.; Raveane, A.; Bertolini, F. A Single-Cell Transcriptomic Landscape of Innate and Adaptive Intratumoral Immunity in Triple Negative Breast Cancer during Chemo- and Immunotherapies. Cell Death Discov. 2022, 8, 106. [Google Scholar] [CrossRef]
- Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers 2020, 12, 1943. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Hsu, C.L.; Chen, Y.H.; Ou, D.L.; Hsu, C.; Tan, C.T. Genomic and Transcriptomic Landscape of an Oral Squamous Cell Carcinoma Mouse Model for Immunotherapy. Cancer Immunol. Res. 2023, 11, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, S.G.; Perry, E.B.; Chen, P.M.; Greenstein, A.; Kaech, S.M.; Townsend, J.P. The Landscape of Novel and Complementary Targets for Immunotherapy: An Analysis of Gene Expression in the Tumor Microenvironment. Oncotarget 2019, 10, 4532–4545. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.H.; Chang, C.J.; Huang, T.W.; Shih, C.M.; Liu, A.J.; Chen, P.H.; Cheng, K.T.; Chen, K.C. Gene Landscape and Correlation between B-Cell Infiltration and Programmed Death Ligand 1 Expression in Lung Adenocarcinoma Patients from The Cancer Genome Atlas Data Set. PLoS ONE 2018, 13, e0208459. [Google Scholar] [CrossRef]
- Liu, M.; Dong, Q.; Chen, B.; Liu, K.; Zhao, Z.; Wang, Y.; Zhuang, S.; Han, H.; Shi, X.; Jin, Z.; et al. Synthetic Viability Induces Resistance to Immune Checkpoint Inhibitors in Cancer Cells. Br. J. Cancer 2023, 129, 1339–1349. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, Y.; Pan, J.; Sang, C.; Lin, Y.; Dong, L.; Shen, X.; Wu, Y.; Song, G.; Ji, S.; et al. An Inflammatory Checkpoint Generated by IL1RN Splicing Offers Therapeutic Opportunity for KRAS-Mutant Intrahepatic Cholangiocarcinoma. Cancer Discov. 2023, 13, 2248–2269. [Google Scholar] [CrossRef]
- Zhang, S.; Wan, X.; Lv, M.; Li, C.; Chu, Q.; Wang, G. TMEM92 Acts as an Immune-Resistance and Prognostic Marker in Pancreatic Cancer from the Perspective of Predictive, Preventive, and Personalized Medicine. EPMA J. 2022, 13, 519–534. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, Y.; Hu, X.; Yin, X.; Chen, Y.; Chen, J.; Liu, H.; Liu, H.; Liang, J.; Zhang, X.; et al. Patients with ASPSCR1-TFE3 Fusion Achieve Better Response to ICI Based Combination Therapy among TFE3-Rearranged Renal Cell Carcinoma. Mol. Cancer 2024, 23, 132. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, H.; Li, H.; Dou, W.; Wang, J.; Zhang, J.; Liu, T.; Wu, Y.; Liu, Y.; Wang, X. Characterization of Stem Cell Landscape and Identification of Stemness-Relevant Prognostic Gene Signature to Aid Immunotherapy in Colorectal Cancer. Stem Cell Res. Ther. 2022, 13, 244. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-Line Nivolumab plus Chemotherapy versus Chemotherapy Alone for Advanced Gastric, Gastro-Oesophageal Junction, and Oesophageal Adenocarcinoma (CheckMate 649): A Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Powles, T.; Stus, V.; Gafanov, R.; Nosov, D.; Waddell, T.; Alekseev, B.; Pouliot, F.; Melichar, B.; Soulières, D.; et al. Pembrolizumab Plus Axitinib Versus Sunitinib as First-Line Treatment of Advanced Renal Cell Carcinoma: 43-Month Follow-up of the Phase 3 KEYNOTE-426 Study. Eur. Urol. 2023, 84, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Brave, M.H.; Maguire, W.F.; Weinstock, C.; Zhang, H.; Gao, X.; Li, F.; Yu, J.; Fu, W.; Zhao, H.; Pierce, W.F.; et al. FDA Approval Summary: Enfortumab Vedotin plus Pembrolizumab for Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2024, 30, 4815–4821. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Kelley, R.K.; Lau, G.; Kudo, M.; Sukeepaisarnjaroen, W.; Yarchoan, M.; De Toni, E.N.; Furuse, J.; Kang, Y.K.; et al. Four-Year Overall Survival Update from the Phase III HIMALAYA Study of Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. Ann. Oncol. 2024, 35, 448–457. [Google Scholar] [CrossRef]
- Oh, D.Y.; He, A.R.; Bouattour, M.; Okusaka, T.; Qin, S.; Chen, L.T.; Kitano, M.; Lee, C.-k.; Kim, J.W.; Chen, M.H.; et al. Durvalumab or Placebo plus Gemcitabine and Cisplatin in Participants with Advanced Biliary Tract Cancer (TOPAZ-1): Updated Overall Survival from a Randomised Phase 3 Study. Lancet Gastroenterol. Hepatol. 2024, 9, 694–704. [Google Scholar] [CrossRef]
- Powles, T.; Park, S.H.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Ullén, A.; Loriot, Y.; Sridhar, S.S.; Sternberg, C.N.; Bellmunt, J.; et al. Avelumab First-Line Maintenance for Advanced Urothelial Carcinoma: Results From the JAVELIN Bladder 100 Trial After ≥ 2 Years of Follow-Up. J. Clin. Oncol. 2023, 41, 3486–3492. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer Consensus Statement on Immunotherapy for the Treatment of Squamous Cell Carcinoma of the Head and Neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus Trastuzumab and Chemotherapy for HER2-Positive Gastric or Gastro-Oesophageal Junction Adenocarcinoma: Interim Analyses from the Phase 3 KEYNOTE-811 Randomised Placebo-Controlled Trial. Lancet 2023, 402, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Solimando, A.G.; Fasano, R.; Argentiero, A.; Malerba, E.; Buonavoglia, A.; Lupo, L.G.; De Re, V.; Silvestris, N.; Racanelli, V. The Evolving Role of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma Treatment. Vaccines 2021, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Susca, N.; Argentiero, A.; Brunetti, O.; Leone, P.; De Re, V.; Fasano, R.; Krebs, M.; Petracci, E.; Azzali, I.; et al. Second-Line Treatments for Advanced Hepatocellular Carcinoma: A Systematic Review and Bayesian Network Meta-Analysis. Clin. Exp. Med. 2022, 22, 65–74. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.-H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N.; et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 385, 683–694. [Google Scholar] [CrossRef]
- Van der Heijden, M.S.; Sonpavde, G.; Powles, T.; Necchi, A.; Burotto, M.; Schenker, M.; Sade, J.P.; Bamias, A.; Beuzeboc, P.; Bedke, J.; et al. Nivolumab plus Gemcitabine-Cisplatin in Advanced Urothelial Carcinoma. N. Engl. J. Med. 2023, 389, 1778–1789. [Google Scholar] [CrossRef]
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef]
- Cescon, D.W.; Schmid, P.; Rugo, H.S.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Health-Related Quality of Life with Pembrolizumab plus Chemotherapy vs Placebo plus Chemotherapy for Advanced Triple-Negative Breast Cancer: KEYNOTE-355. J. Natl. Cancer. Inst. 2024, 116, 717–727. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Sznol, M. Resistance Mechanisms to Checkpoint Inhibitors. Curr. Opin. Immunol. 2021, 69, 47–55. [Google Scholar] [CrossRef]

{kind=link}
| Clinical Trial (Phase III) | Drug Name (PD-1) | Cancer Type | Primary End Points | Response Rate (%) | Sample Size | FDA Approval (Yes/No) | Reference |
|---|---|---|---|---|---|---|---|
| CheckMate-067 | Nivolumab + Ipilimumab vs. Nivolumab vs. Ipilimumab | Melanoma | PFS, OS | 58 | 945 | Yes | [145] |
| Keynote-189 | Pembrolizumab + Chemotherapy vs. Chemotherapy + Placebo | NSCLC (Non-squamous) | OS, PFS | 48.3 | 616 | Yes | [147] |
| Keynote-048 | Pembrolizumab + Chemotherapy vs. Pembrolizumab vs. Chemotherapy + Cetuximab | Head and Neck Squamous Cell Carcinoma (HNSCC) | PFS, OS | 16.9 | 882 | Yes | [148] |
| CheckMate-141 | Nivolumab vs. Chemotherapy | HNSCC | OS | 13.3 | 361 | Yes | [149] |
| Keynote-177 | Pembrolizumab vs. Chemotherapy + Bevacizumab or cetuximab | Colorectal Cancer (MSI-H/dMMR) | PFS, OS | 43.8 | 307 | Yes | [150] |
| CheckMate-649 | Nivolumab + Chemotherapy vs. Chemotherapy | Gastric/Esophageal Cancer | OS, PFS | 47.4 | 1581 | Yes | [151] |
| CheckMate-214 | Nivolumab + Ipilimumab vs. Sunitinib | Renal Cell Carcinoma | OS, PFS, ORR | 42 | 1096 | Yes | [152] |
| EV-302 | Pembrolizumab + Enfortumab vedotin vs. Chemotherapy | Urothelial Cancer 1st Line | PFS, OS | 44 | 608 | Yes | [153] |
| Clinical Trial (Phase III) | Drug Name (PD-L1) | Cancer Type | Primary End Points | Response Rate (%) | Sample Size | FDA Approval (Yes/No) | Reference |
|---|---|---|---|---|---|---|---|
| IMpower150 | Atezolizumab + Bevacizumab + Chemotherapy vs. Bevacizumab + Chemotherapy | NSCLC 1st Line | PFS, OS | 63 | 1202 | Yes | [155] |
| PACIFIC | Durvalumab vs. Placebo after Concurrent Chemoradiotherapy | NSCLC (Stage III) | OS, PFS | 28.4 | 713 | Yes | [156] |
| HIMALAYA | Durvalumab + Tremelimumab (STRIDE) vs. Durvalumab vs. Sorafenib | Hepatocellular Carcinoma (HCC) | OS for STRIDE vs. Sorafenib | 20.1 | 1171 | Yes | [157] |
| TOPAZ-1 | Durvalumab + Chemotherapy vs. Chemotherapy + Placebo | Biliary Tract Cancer | OS | 26.7 | 685 | Yes | [158] |
| JAVELIN 100 | Avelumab vs. Best Supportive Care as Maintenance Therapy after 4–6 cycles Chemotherapy | Urothelial Cancer | OS | 53.2 | 700 | Yes | [159] |
| IMbrave050 | Atezolizumab + Bevacizumab vs. Active Surveillance | Hepatocellular Carcinoma (HCC) | 33 | 662 | Yes | [160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strati, A.; Adamopoulos, C.; Kotsantis, I.; Psyrri, A.; Lianidou, E.; Papavassiliou, A.G. Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers. Int. J. Mol. Sci. 2025, 26, 1235. https://doi.org/10.3390/ijms26031235
Strati A, Adamopoulos C, Kotsantis I, Psyrri A, Lianidou E, Papavassiliou AG. Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers. International Journal of Molecular Sciences. 2025; 26(3):1235. https://doi.org/10.3390/ijms26031235
Chicago/Turabian StyleStrati, Areti, Christos Adamopoulos, Ioannis Kotsantis, Amanda Psyrri, Evi Lianidou, and Athanasios G. Papavassiliou. 2025. "Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers" International Journal of Molecular Sciences 26, no. 3: 1235. https://doi.org/10.3390/ijms26031235
APA StyleStrati, A., Adamopoulos, C., Kotsantis, I., Psyrri, A., Lianidou, E., & Papavassiliou, A. G. (2025). Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers. International Journal of Molecular Sciences, 26(3), 1235. https://doi.org/10.3390/ijms26031235