
CNEURO-201, an Anti-amyloidogenic Agent and σ1-Receptor Agonist, Improves Cognition in the 3xTg Mouse Model of Alzheimer’s Disease by Multiple Actions in the Pathology

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
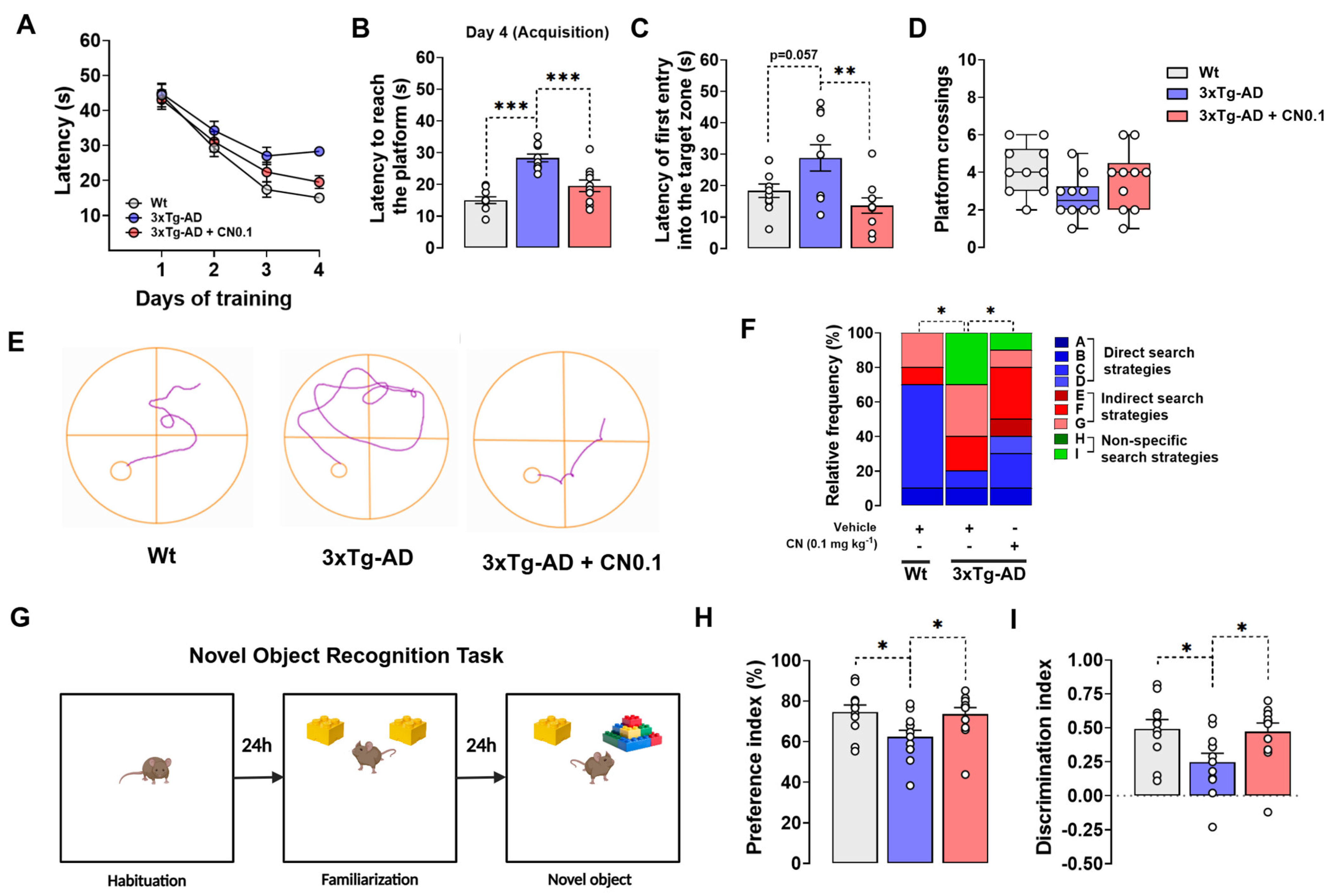
2.1. CNEURO-201 Greatly Improves the Cognitive Functions of 3xTg-AD Mice
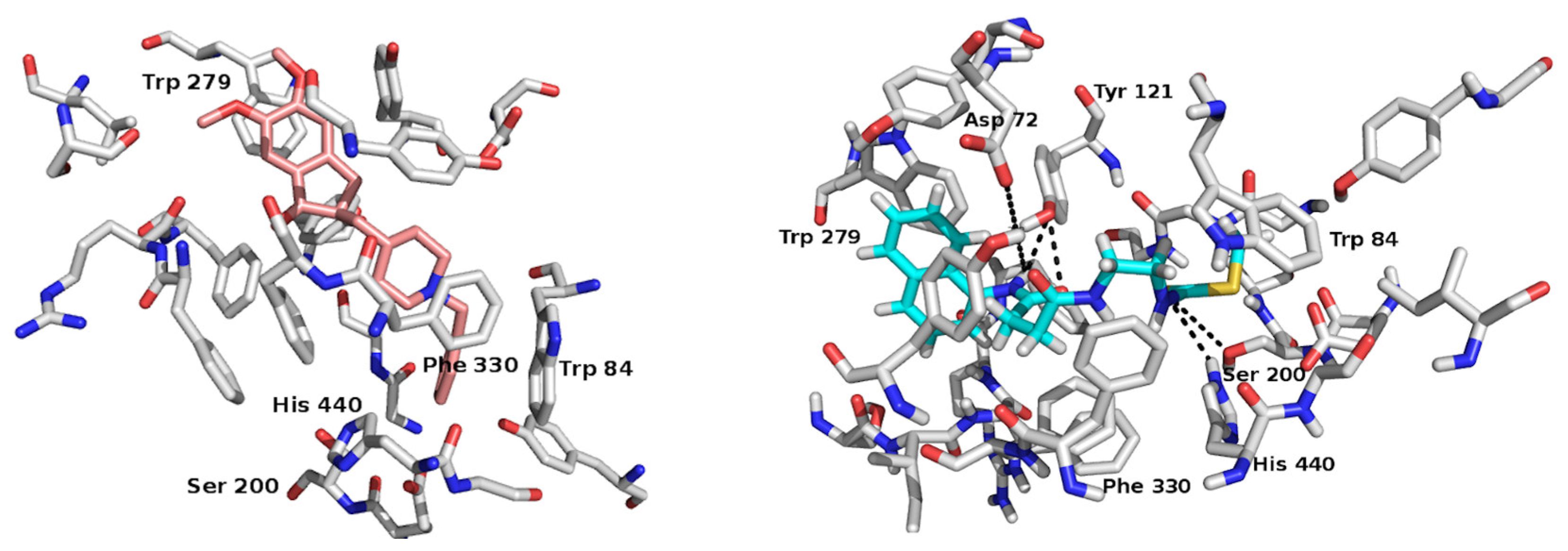
2.2. In Silico Study: A Potential Interaction Between CNEURO-201 and Acetylcholinesterase
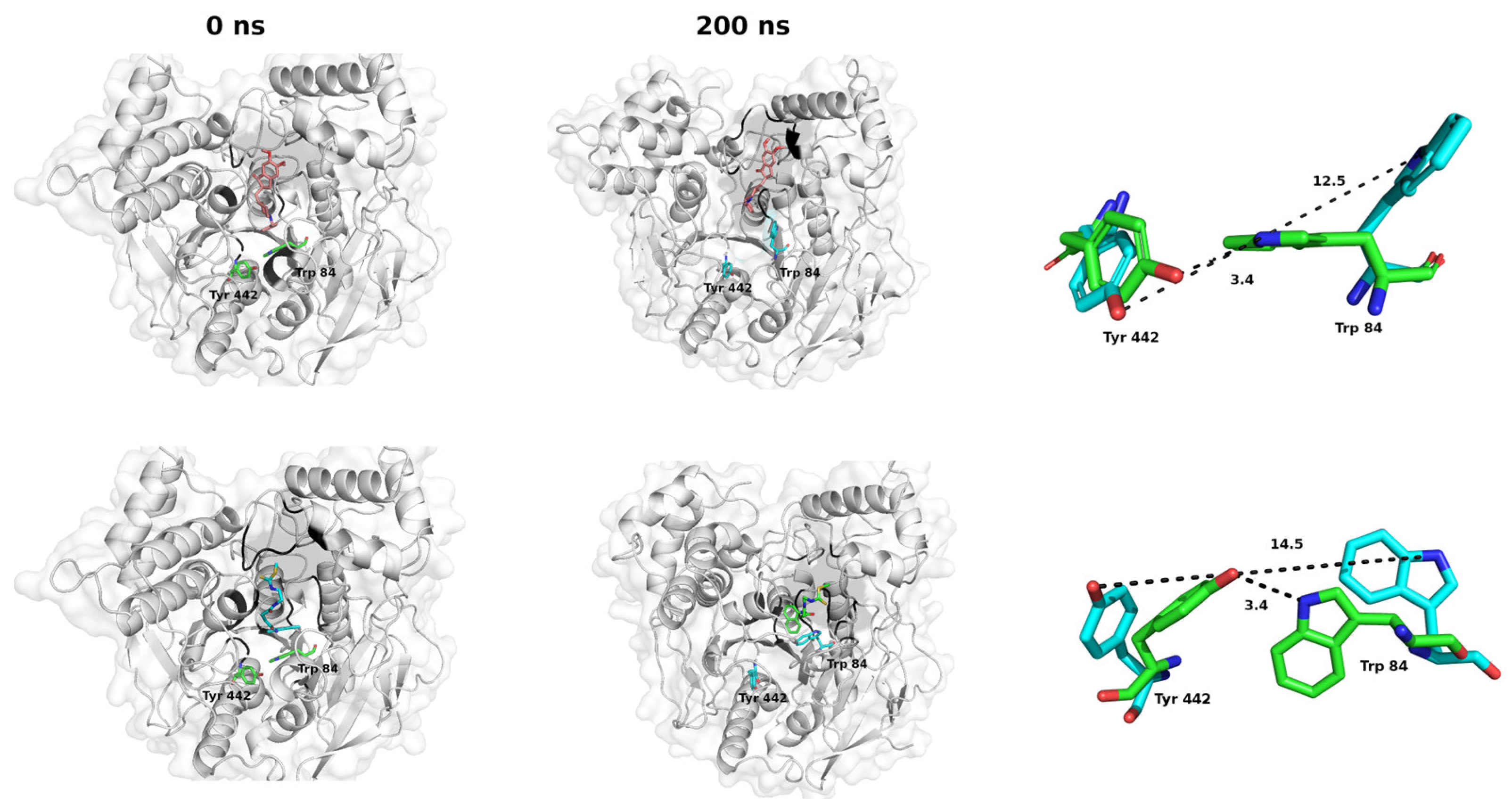
2.3. Molecular Dynamics Studies: Temporal Stability Evaluation of the CNEURO-201/AChE System
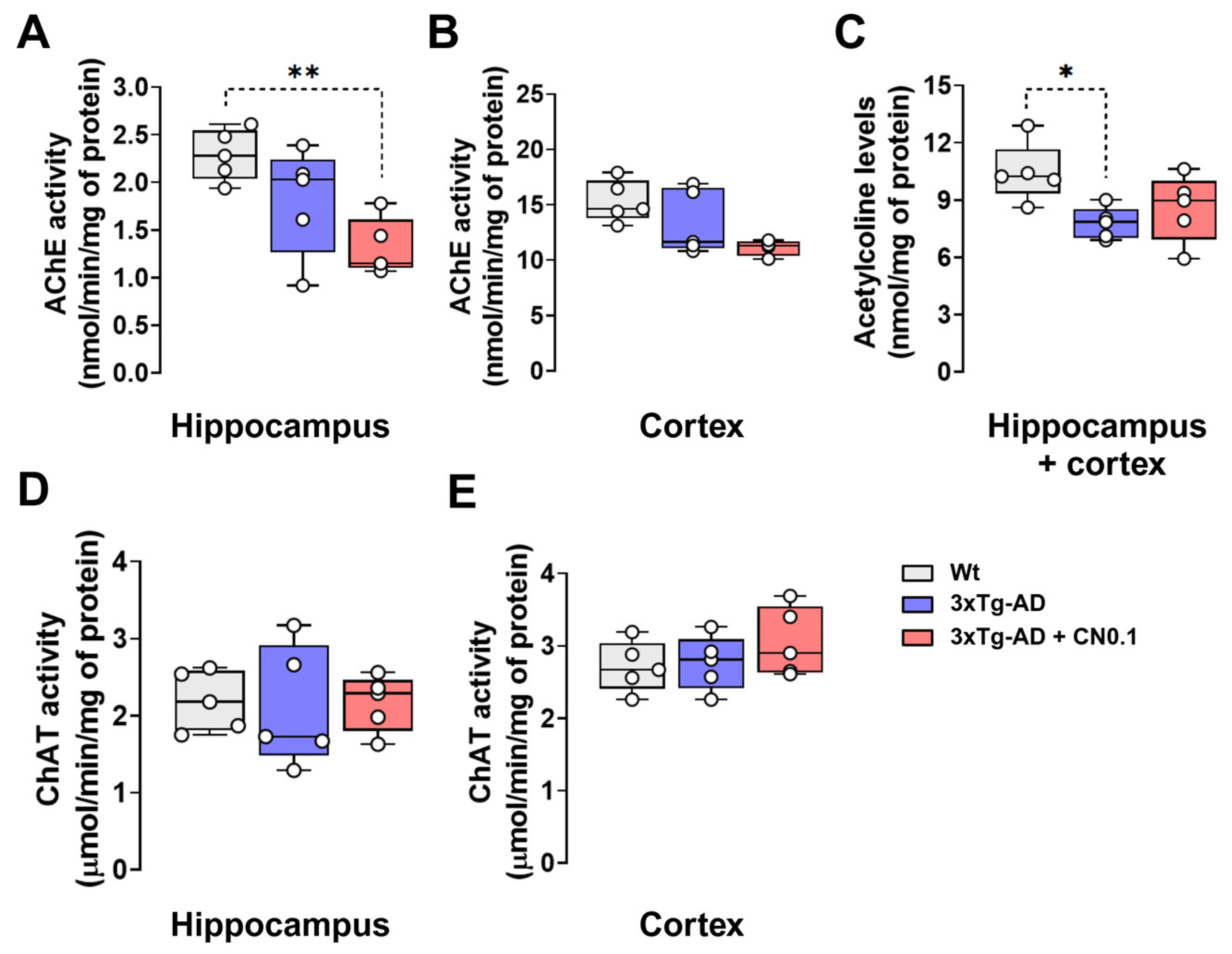
2.4. CNEURO-201 Reduces Acetylcholinesterase Activity and Acetylcholine Decay in the Hippocampus and Cortex of 3xTg-AD Mice
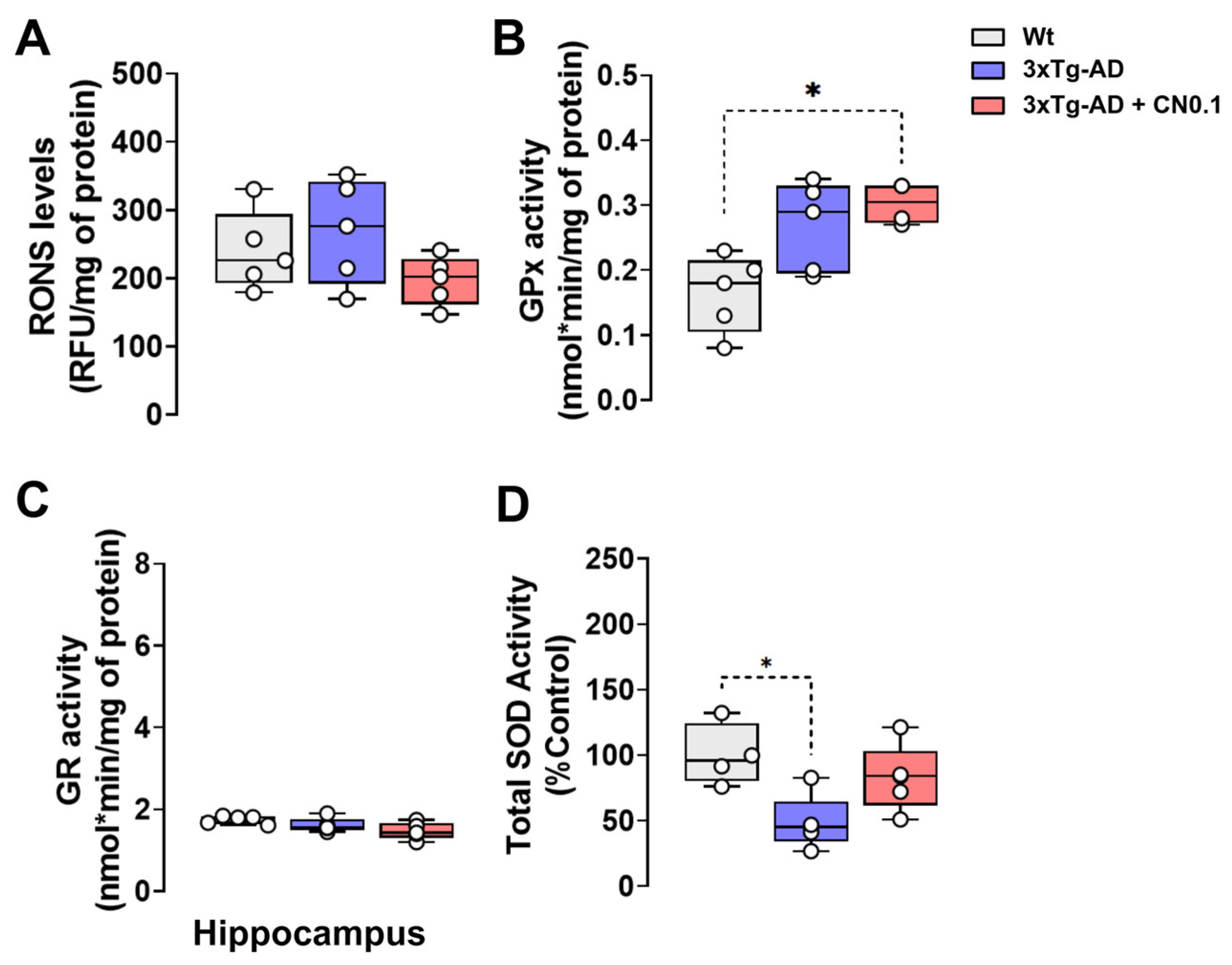
2.5. CNEURO-201 Influences Antioxidant Enzymatic Activity in the Hippocampus of 3xTg-AD Mice
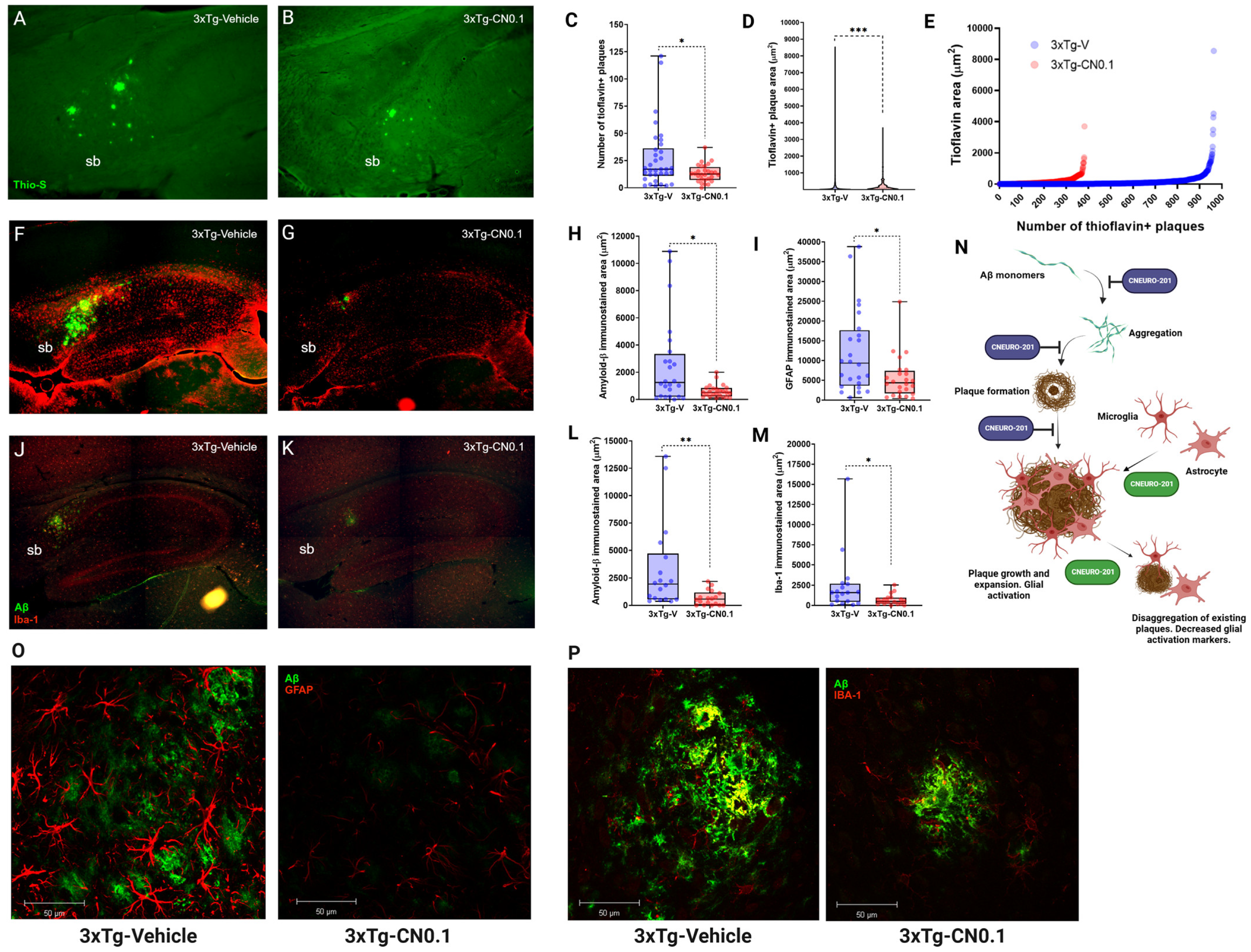
2.6. CNEURO-201 Decreases Expansion and Promotes Dissagregation of Amyloid Plaques Together with Reductions in Glial Activation Markers in the Subiculum of Aged 3xTg-AD Mice
3. Discussion
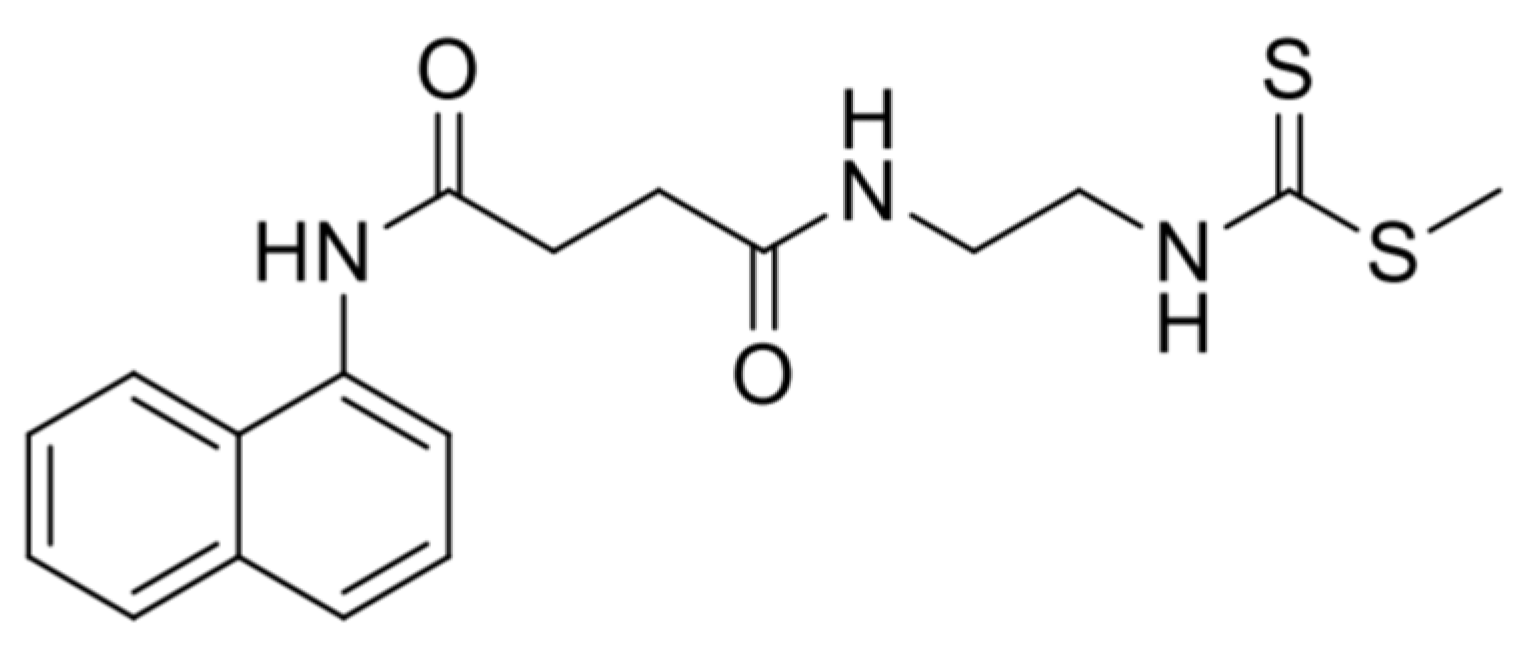
4. Materials and Methods
4.1. Molecular Docking
4.2. Molecular Dynamics Simulations
4.3. Experimental Animals
4.4. Pharmacological Treatments
4.5. Morris Water Maze Task
4.6. Novel Object Recognition Task
4.7. Tissue Isolation and Preparation
4.8. Acetylcholinesterase Activity
4.9. Acetylcholine Quantification
4.10. Choline Acetyltransferase Activity
4.11. Determination of Reactive Oxygen/Nitrogen Species
4.12. Glutathione Peroxydase Activity
4.13. Glutathione Reductase Activity
4.14. Total Superoxide Dismutase Activity
4.15. Thioflavin-S Staining
4.16. Immunofluorescence
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 5 October 2024).
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Song, M. Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target. Molecules 2024, 29, 5744. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Soares, C.; Da Ros, L.U.; Machado, L.S.; Rocha, A.; Lazzarotto, G.; Carello-Collar, G.; De Bastiani, M.A.; Ferrari-Souza, J.P.; Lussier, F.Z.; Souza, D.O.; et al. The glutamatergic system in Alzheimer’s disease: A systematic review with meta-analysis. Mol Psychiatry 2024, 29, 2261–2273. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Pelucchi, S.; Gardoni, F.; Di Luca, M.; Marcello, E. Synaptic dysfunction in early phases of Alzheimer’s Disease. Handb. Clin. Neurol. 2022, 184, 417–438. [Google Scholar] [CrossRef]
- Meftah, S.; Gan, J. Alzheimer’s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Front. Synaptic Neurosci. 2023, 15, 1129036. [Google Scholar] [CrossRef]
- Bermejo-Pareja, F.; Del Ser, T. Controversial Past, Splendid Present, Unpredictable Future: A Brief Review of Alzheimer Disease History. J. Clin. Med. 2024, 13, 536. [Google Scholar] [CrossRef]
- Walker, L.C. Aβ Plaques. Free Neuropathol. 2020, 1, 1–31. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. In 2024, the amyloid-cascade-hypothesis still remains a working hypothesis, no less but certainly no more. Front. Aging Neurosci. 2024, 16, 1459224. [Google Scholar] [CrossRef]
- Michno, W.; Stringer, K.M.; Enzlein, T.; Passarelli, M.K.; Escrig, S.; Vitanova, K.; Wood, J.; Blennow, K.; Zetterberg, H.; Meibom, A.; et al. Following spatial Aβ aggregation dynamics in evolving Alzheimer’s disease pathology by imaging stable isotope labeling kinetics. Sci. Adv. 2021, 7, eabg4855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Yadollahikhales, G.; Rojas, J.C. Anti-Amyloid Immunotherapies for Alzheimer’s Disease: A 2023 Clinical Update. Neurotherapeutics 2023, 20, 914–931. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Perry, E.K.; Gibson, P.H.; Blessed, G.; Perry, R.H.; Tomlinson, B.E. Neurotransmitter enzyme abnormalities in senile dementia. Choline acetyltransferase and glutamic acid decarboxylase activities in necropsy brain tissue. J. Neurol. Sci. 1977, 34, 247–265. [Google Scholar] [CrossRef]
- Rylett, R.J.; Ball, M.J.; Colhoun, E.H. Evidence for high affinity choline transport in synaptosomes prepared from hippocampus and neocortex of patients with Alzheimer’s disease. Brain Res. 1983, 289, 169–175. [Google Scholar] [CrossRef]
- Nilsson, L.; Nordberg, A.; Hardy, J.; Wester, P.; Winblad, B. Physostigmine restores 3H-acetylcholine efflux from Alzheimer brain slices to normal level. J. Neural Transm. 1986, 67, 275–285. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current Understanding of the Physiopathology, Diagnosis and Therapeutic Approach to Alzheimer’s Disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef] [PubMed]
- Gajendra, K.; Pratap, G.K.; Poornima, D.V.; Shantaram, M.; Ranjita, G. Natural acetylcholinesterase inhibitors: A multi-targeted therapeutic potential in Alzheimer’s disease. Eur. J. Med. Chem. 2024, 11, 100154. [Google Scholar] [CrossRef]
- Sablón-Carrazana, M.; Fernández, I.; Bencomo, A.; Lara-Martínez, R.; Rivera-Marrero, S.; Domínguez, G.; Pérez-Perera, R.; Jiménez-García, L.F.; Altamirano-Bustamante, N.F.; Diaz-Delgado, M.; et al. Drug Development in Conformational Diseases: A Novel Family of Chemical Chaperones that Bind and Stabilise Several Polymorphic Amyloid Structures. PLoS ONE 2015, 10, e0135292. [Google Scholar] [CrossRef]
- Fernández-Gómez, I.; Sablón-Carrazana, M.; Bencomo-Martínez, A.; Domínguez, G.; Lara-Martínez, R.; Altamirano-Bustamante, N.F.; Jiménez-García, L.F.; Pasten-Hidalgo, K.; Castillo-Rodríguez, R.A.; Altamirano, P.; et al. Diabetes Drug Discovery: hIAPP1-37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules 2018, 23, 686. [Google Scholar] [CrossRef]
- Rivera-Marrero, S.; Bencomo-Martínez, A.; Orta Salazar, E.; Sablón-Carrazana, M.; García-Pupo, L.; Zoppolo, F.; Arredondo, F.; Dapueto, R.; Daniela Santi, M.; Kreimerman, I.; et al. A new naphthalene derivative with anti-amyloidogenic activity as potential therapeutic agent for Alzheimer’s disease. Bioorg. Med. Chem. 2020, 28, 115700. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Mercerón-Martínez, D.; Alacán Ricardo, L.; Bejerano Pina, A.; Orama Rojo, N.; Expósito Seco, A.; Vega Hurtado, Y.; Estupiñán Días, B.; Fernández, I.; García Pupo, L.; Sablón Carrazana, M.; et al. Amylovis-201 enhances physiological memory formation and rescues memory and hippocampal cell loss in a streptozotocin-induced Alzheimer’s disease animal model. Brain Res. 2024, 1831, 148848. [Google Scholar] [CrossRef]
- García-Pupo, L.; Crouzier, L.; Bencomo-Martínez, A.; Meunier, J.; Morilleau, A.; Delprat, B.; Sablón-Carrazana, M.; Menéndez-Soto del Valle, R.; Maurice, T.; Rodríguez-Tanty, C. Amylovis-201 is a new dual-target ligand, acting as an anti-amyloidogenic compound and a potent agonist of the σ1 chaperone protein. Acta Pharm. Sin. B 2024, 14, 4345–4359. [Google Scholar] [CrossRef]
- Gallagher, M.; Koh, M.T. Episodic memory on the path to Alzheimer’s disease. Curr. Opin. Neurobiol. 2011, 21, 929–934. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Alisaraie, L.; Fels, G. Molecular docking study on the “back door” hypothesis for product clearance in acetylcholinesterase. J. Mol. Model. 2006, 12, 348–354. [Google Scholar] [CrossRef]
- Bartolucci, C.; Perola, E.; Cellai, L.; Brufani, M.; Lamba, D. “Back door” opening implied by the crystal structure of a carbamoylated acetylcholinesterase. Biochemistry 1999, 38, 5714–5719. [Google Scholar] [CrossRef]
- Kronman, C.; Ordentlich, A.; Barak, D.; Velan, B.; Shafferman, A. The “back door” hypothesis for product clearance in acetylcholinesterase challenged by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 27819–27822. [Google Scholar] [CrossRef]
- Xu, Y.; Colletier, J.P.; Weik, M.; Qin, G.; Jiang, H.; Silman, I.; Sussman, J.L. Long route or shortcut? A molecular dynamics study of traffic of thiocholine within the active-site gorge of acetylcholinesterase. Biophys. J. 2010, 99, 4003–4011. [Google Scholar] [CrossRef]
- Cheng, S.; Song, W.; Yuan, X.; Xu, Y. Gorge Motions of Acetylcholinesterase Revealed by Microsecond Molecular Dynamics Simulations. Sci. Rep. 2017, 7, 3219. [Google Scholar] [CrossRef]
- Richter, J.A.; Perry, E.K.; Tomlinson, B.E. Acetylcholine and choline levels in post-mortem human brain tissue: Preliminary observations in Alzheimer’s disease. Life Sci. 1980, 26, 1683–1689. [Google Scholar] [CrossRef]
- Raiteri, M. Functional pharmacology in human brain. Pharmacol. Rev. 2006, 58, 162–193. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim Care Companion CNS Disord. 2013, 15, PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; Navarrete, F.; Ginsberg, S.D.; Manzanares, J.; Sáez-Valero, J.; García-Ayllón, M.S. Increased Expression of Readthrough Acetylcholinesterase Variants in the Brains of Alzheimer’s Disease Patients. J. Alzheimers Dis. 2016, 53, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Pappas, B.A.; Bayley, P.J.; Bui, B.K.; Hansen, L.A.; Thal, L.J. Choline acetyltransferase activity and cognitive domain scores of Alzheimer’s patients. Neurobiol Aging 2000, 21, 11–17. [Google Scholar] [CrossRef]
- Ikonomovic, M.D.; Mufson, E.J.; Wuu, J.; Bennett, D.A.; DeKosky, S.T. Reduction of choline acetyltransferase activity in primary visual cortex in mild to moderate Alzheimer’s disease. Arch. Neurol. 2005, 62, 425–430. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Javonillo, D.I.; Tran, K.M.; Phan, J.; Hingco, E.; Kramár, E.A.; da Cunha, C.; Forner, S.; Kawauchi, S.; Milinkeviciute, G.; Gomez-Arboledas, A.; et al. Systematic Phenotyping and Characterization of the 3xTg-AD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2022, 15, 785276. [Google Scholar] [CrossRef]
- Shin, J.; Park, S.; Lee, H.; Kim, Y. Thioflavin-positive tau aggregates complicating quantification of amyloid plaques in the brain of 5XFAD transgenic mouse model. Sci. Rep. 2021, 11, 1617. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Goldman, D.P.; Simmons-Stern, N.R.; Ponton, E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimers Dement. 2022, 18, 469–477. [Google Scholar] [CrossRef]
- Parums, D.V. A Review of the Current Status of Disease-Modifying Therapies and Prevention of Alzheimer’s Disease. Med. Sci. Monit. 2024, 30, e945091. [Google Scholar] [CrossRef]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef]
- Maurice, T.; Goguadze, N. Sigma-1 (σ1) Receptor in Memory and Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2017, 244, 81–108. [Google Scholar] [CrossRef]
- Wu, N.H.; Ye, Y.; Wan, B.B.; Yu, Y.D.; Liu, C.; Chen, Q.J. Emerging Benefits: Pathophysiological Functions and Target Drugs of the Sigma-1 Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 5649–5666. [Google Scholar] [CrossRef]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [CrossRef]
- Su, T.P.; London, E.D.; Jaffe, J.H. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science 1988, 240, 219–221. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, X.; Bai, Y.; Zhu, X.; Wu, X.; Chao, J.; Duan, M.; Buch, S.; Chen, L.; Yao, H. Involvement of sigma-1 receptor in astrocyte activation induced by methamphetamine via up-regulation of its own expression. J Neuroinflammation 2015, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guo, Q.; Fang, L.P.; Yao, H.; Scheller, A.; Kirchhoff, F.; Huang, W. Specific detection and deletion of the sigma-1 receptor widely expressed in neurons and glial cells in vivo. J Neurochem. 2023, 164, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Meunier, J.; Hayashi, T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J. Pharmacol. Exp. Ther. 2010, 332, 388–397. [Google Scholar] [CrossRef]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef]
- Lahmy, V.; Meunier, J.; Malmström, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyper-phosphorylation and Aβ₁₋₄₂ generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ₁ receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef]
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/σ1 Agonist with Therapeutic Efficacy in Animal Models of Alzheimer’s Disease. Neurodegener. Dis. 2016, 16, 95–110. [Google Scholar] [CrossRef]
- Hall, H.; Iulita, M.F.; Gubert, P.; Flores Aguilar, L.; Ducatenzeiler, A.; Fisher, A.; Cuello, A.C. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 811–823. [Google Scholar] [CrossRef]
- Hayashi, T.; Maurice, T.; Su, T.P. Ca(2+) signaling via sigma(1)-receptors: Novel regulatory mechanism affecting intracellular Ca(2+) concentration. J. Pharmacol. Exp. Ther. 2000, 293, 788–798. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Pan, K.; Garaschuk, O. The role of intracellular calcium-store-mediated calcium signals in in vivo sensor and effector functions of microglia. J. Physiol. 2023, 601, 4203–4215. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; LeVault, K.R.; Brewer, G.J. Dual-energy precursor and nuclear erythroid-related factor 2 activator treatment additively improve redox glutathione levels and neuron survival in aging and Alzheimer mouse neurons upstream of reactive oxygen species. Neurobiol. Aging 2014, 35, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zuo, C.; Gu, Z.; Huang, Y.; Yang, Y.; Zhu, L.; Jiang, Y.; Wang, F. High frequency repetitive transcranial magnetic stimulation alleviates cognitive deficits in 3xTg-AD mice by modulating the PI3K/Akt/GLT-1 axis. Redox Biol. 2022, 54, 102354. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- García-Ayllón, M.S.; Silveyra, M.X.; Sáez-Valero, J. Association between acetylcholinesterase and beta-amyloid peptide in Alzheimer’s cerebrospinal fluid. Chem. Biol. Interact. 2008, 175, 209–215. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Villarreal-Silva, E.E.; González-Navarro, A.R.; Salazar-Ybarra, R.A.; Quiroga-García, O.; Cruz-Elizondo, M.A.J.; García-García, A.; Rodríguez-Rocha, H.; Morales-Gómez, J.A.; Quiroga-Garza, A.; Elizondo-Omaña, R.E.; et al. Aged rats learn Morris Water maze using non-spatial search strategies evidenced by a parameter-based algorithm. Transl. Neurosci. 2022, 13, 134–144. [Google Scholar] [CrossRef]
- Martínez-Orozco, H.; Reyes-Castro, L.A.; Lomas-Soria, C.; Sandoval-Salazar, C.; Ramírez-Emiliano, J.; Díaz-Cintra, S.; Solís-Ortiz, S. High-fat and combined high-fat-high-fructose diets impair episodic-like memory and decrease glutamate and glutamine in the hippocampus of adult mice. Nutr. Neurosci. 2022, 25, 2479–2489. [Google Scholar] [CrossRef]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of pro-tein-dye binding. Anal Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcho-linesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadegan, M.; Soodi, M. Comparison of Memory Impairment and Oxidative Stress Following Single or Repeated Doses Administration of Scopolamine in Rat Hippocampus. Basic Clin. Neurosci. 2018, 9, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.P.; Wolfgram, F. Spectrophotometric assay for choline acetyltransferase. Anal Biochem. 1972, 46, 114–118. [Google Scholar] [CrossRef] [PubMed]
- da Costa E Silva, L.D.; Pereira, P.; Regner, G.G.; Boaretto, F.B.M.; Hoffmann, C.; Pflüger, P.; da Silva, L.L.; Steffens, L.R.; Morás, A.M.; Moura, D.J.; et al. DNA damage and oxidative stress induced by seizures are decreased by anticonvulsant and neuroprotective effects of lobeline, a candidate to treat alcoholism. Metab. Brain Dis. 2018, 33, 53–61. [Google Scholar] [CrossRef]
- Reiniers, M.J.; van Golen, R.F.; Bonnet, S.; Broekgaarden, M.; van Gulik, T.M.; Egmond, M.R.; Heger, M. Preparation and Practical Applications of 2’,7’-Dichlorodihydrofluorescein in Redox Assays. Anal Chem. 2017, 89, 3853–3857. [Google Scholar] [CrossRef]
- Goswami, P.; Gupta, S.; Biswas, J.; Sharma, S.; Singh, S. Endoplasmic Reticulum Stress Instigates the Rotenone Induced Oxidative Apoptotic Neuronal Death: A Study in Rat Brain. Mol. Neurobiol. 2016, 53, 5384–5400. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [CrossRef]
- Spitz, D.R.; Oberley, L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989, 179, 8–18. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Orozco, H.; Bencomo-Martínez, A.; Maya-Arteaga, J.P.; Rubio-De Anda, P.F.; Sanabria-Romero, F.; Casas, Z.G.M.; Rodríguez-Vargas, I.; Hernández-Puga, A.G.; Sablón-Carrazana, M.; Menéndez-Soto del Valle, R.; et al. CNEURO-201, an Anti-amyloidogenic Agent and σ1-Receptor Agonist, Improves Cognition in the 3xTg Mouse Model of Alzheimer’s Disease by Multiple Actions in the Pathology. Int. J. Mol. Sci. 2025, 26, 1301. https://doi.org/10.3390/ijms26031301
Martínez-Orozco H, Bencomo-Martínez A, Maya-Arteaga JP, Rubio-De Anda PF, Sanabria-Romero F, Casas ZGM, Rodríguez-Vargas I, Hernández-Puga AG, Sablón-Carrazana M, Menéndez-Soto del Valle R, et al. CNEURO-201, an Anti-amyloidogenic Agent and σ1-Receptor Agonist, Improves Cognition in the 3xTg Mouse Model of Alzheimer’s Disease by Multiple Actions in the Pathology. International Journal of Molecular Sciences. 2025; 26(3):1301. https://doi.org/10.3390/ijms26031301
Chicago/Turabian StyleMartínez-Orozco, Humberto, Alberto Bencomo-Martínez, Juan Pablo Maya-Arteaga, Pedro Francisco Rubio-De Anda, Fausto Sanabria-Romero, Zyanya Gloria Mena Casas, Isaac Rodríguez-Vargas, Ana Gabriela Hernández-Puga, Marquiza Sablón-Carrazana, Roberto Menéndez-Soto del Valle, and et al. 2025. "CNEURO-201, an Anti-amyloidogenic Agent and σ1-Receptor Agonist, Improves Cognition in the 3xTg Mouse Model of Alzheimer’s Disease by Multiple Actions in the Pathology" International Journal of Molecular Sciences 26, no. 3: 1301. https://doi.org/10.3390/ijms26031301
APA StyleMartínez-Orozco, H., Bencomo-Martínez, A., Maya-Arteaga, J. P., Rubio-De Anda, P. F., Sanabria-Romero, F., Casas, Z. G. M., Rodríguez-Vargas, I., Hernández-Puga, A. G., Sablón-Carrazana, M., Menéndez-Soto del Valle, R., Rodríguez-Tanty, C., & Díaz-Cintra, S. (2025). CNEURO-201, an Anti-amyloidogenic Agent and σ1-Receptor Agonist, Improves Cognition in the 3xTg Mouse Model of Alzheimer’s Disease by Multiple Actions in the Pathology. International Journal of Molecular Sciences, 26(3), 1301. https://doi.org/10.3390/ijms26031301