
Signaling Pathways Driving MSC Osteogenesis: Mechanisms, Regulation, and Translational Applications


Abstract
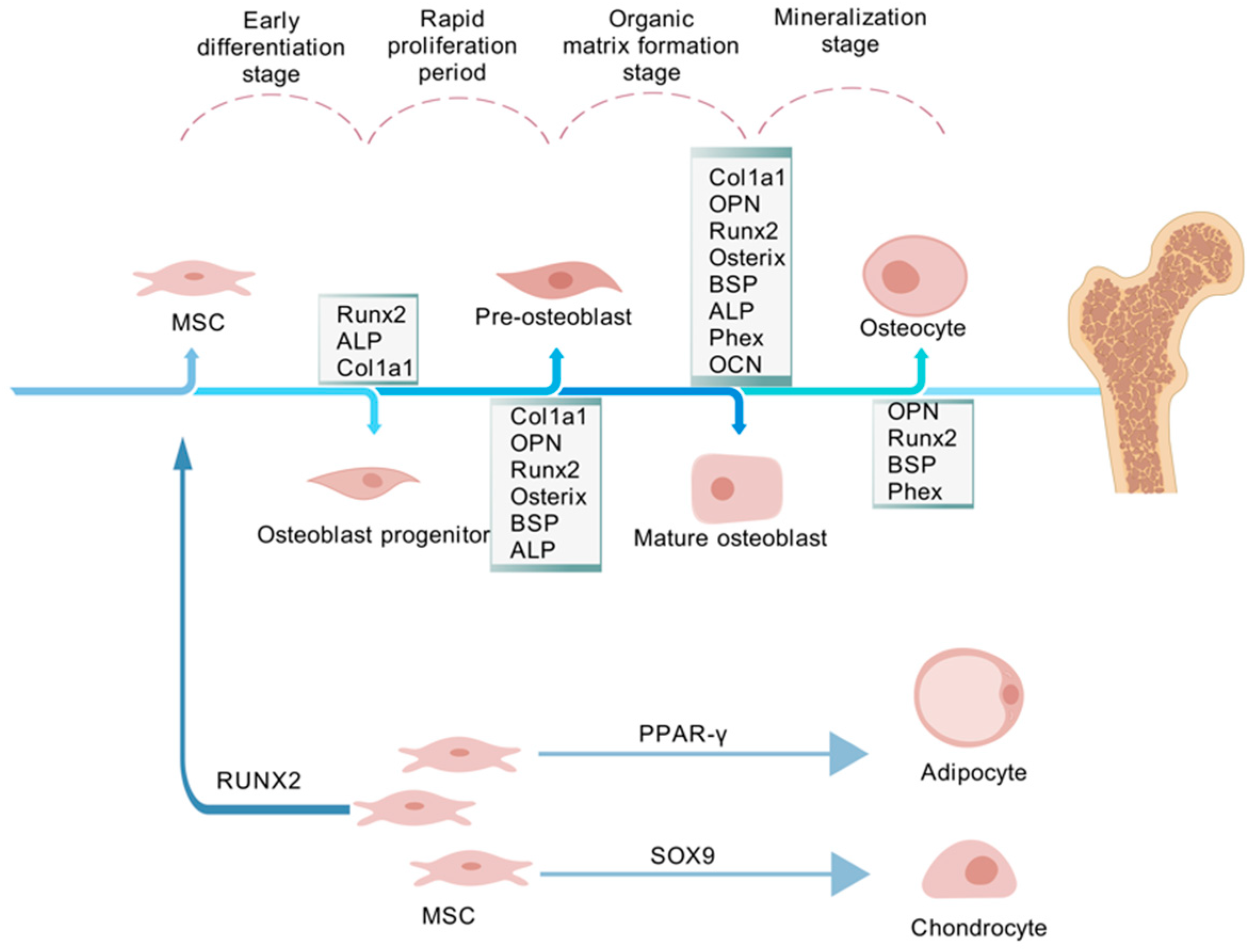
:1. Introduction
2. Wnt Signaling Pathway
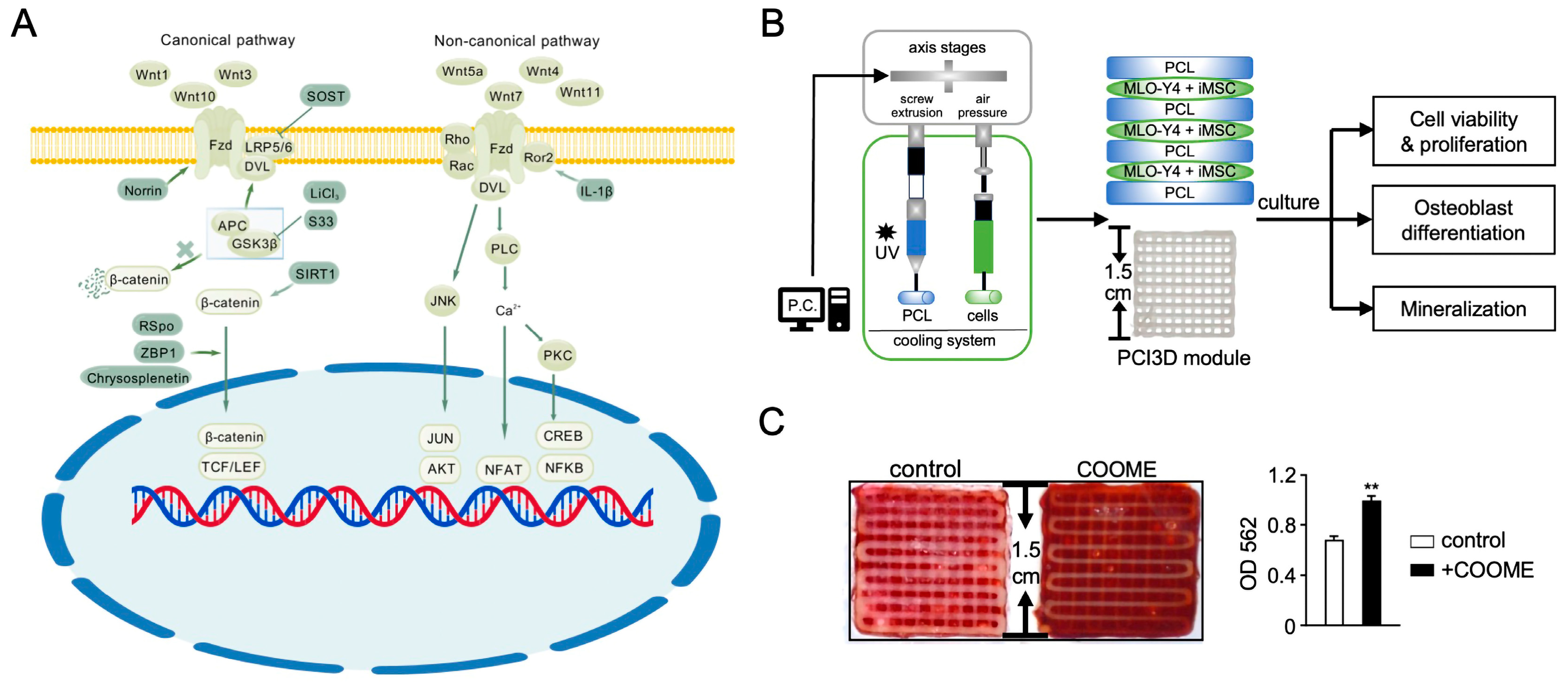
2.1. Overview of Wnt Signaling
2.2. The Role of Wnt Pathway Regulation in MSC Osteogenic Differentiation
2.3. Wnt Signaling in MSC Osteogenesis for Bone Regeneration
3. TGF-β/BMP Signaling Pathway
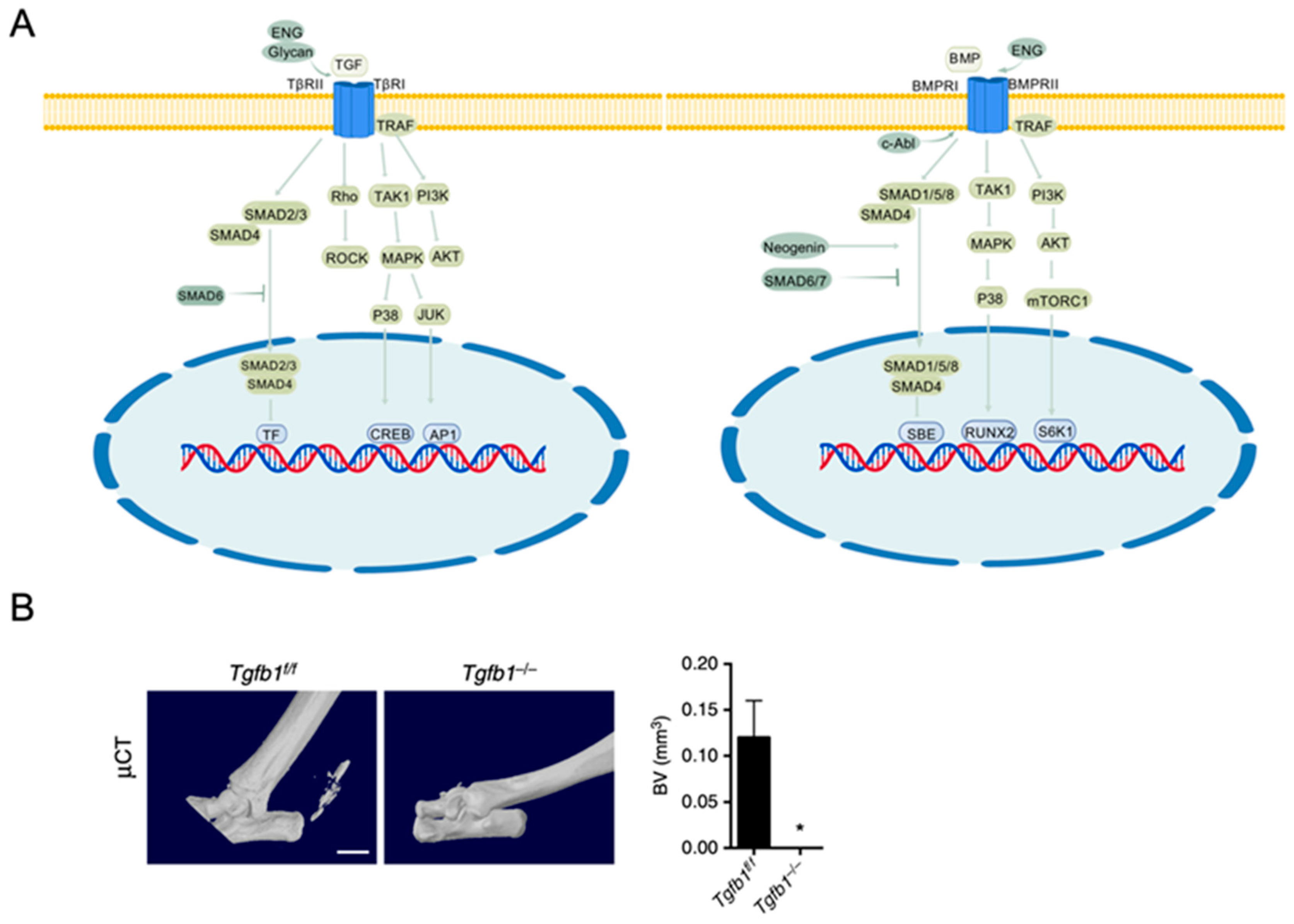
3.1. Overview of TGF-β/BMP Signaling
3.2. Effects of TGF-β/BMP Signaling on Osteogenesis
3.3. Applications of TGF-β and BMP Signaling Pathways in Bone Regeneration
4. PTH/PTHR Signaling Pathway
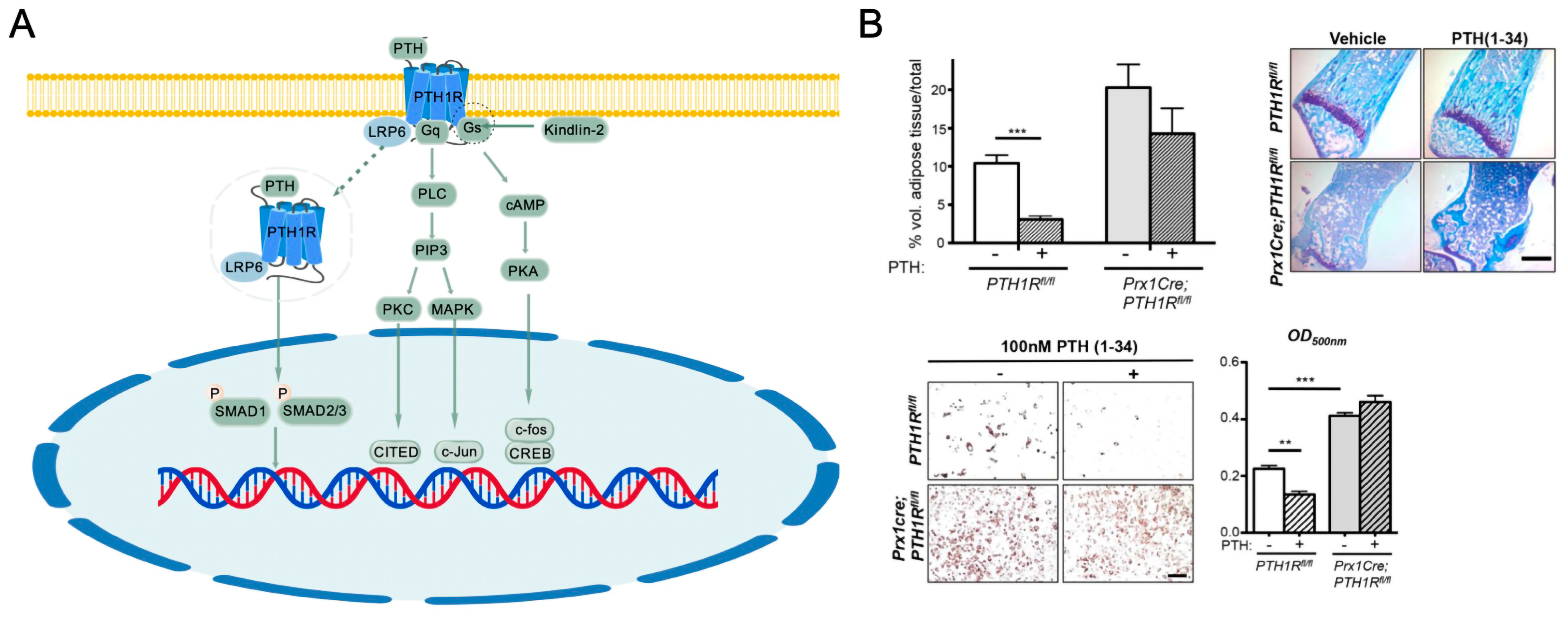
4.1. Overview of PTH/PTHR Signaling
4.2. Impact of PTH/PTHrP Signaling on Bone Homeostasis
4.3. Application of PTH/PTHrP Signaling Pathway in MSC-Based Bone Tissue Regeneration
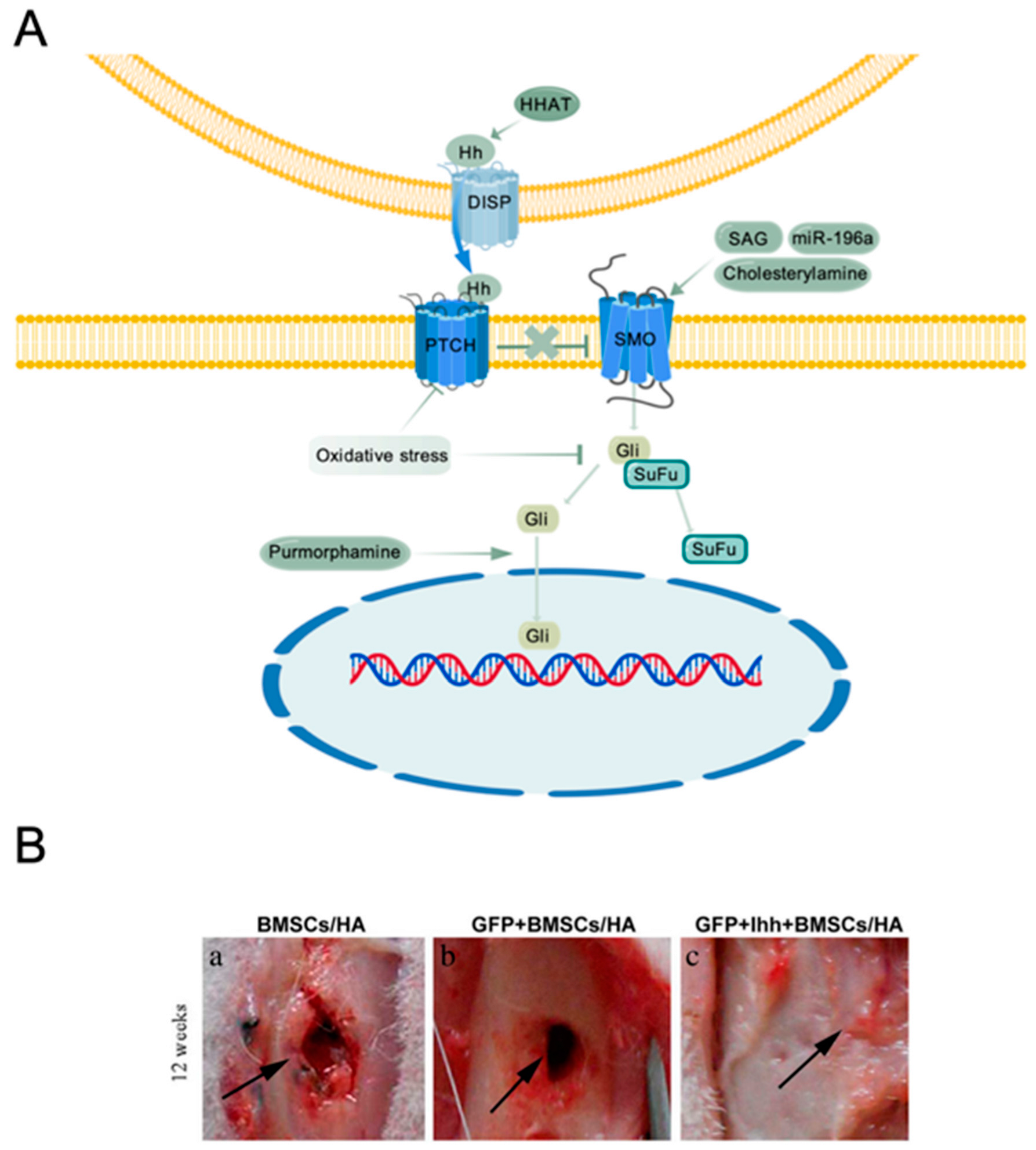
5. Hedgehog Signaling Pathway
5.1. Overview of Hedgehog Signaling
5.2. Hh Signaling Regulates Bone Tissue Development and Homeostasis
5.3. Hh Signaling in MSC-Mediated Bone Tissue Engineering
6. IGF Signaling Pathway
6.1. Overview of IGF Signaling
6.2. IGF Pathway in Bone Development and Homeostasis
6.3. IGF Pathway in Bone Tissue Engineering Applications
7. Innovative Approaches Targeting MSC Signaling Pathways
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Almalki, S.G.; Agrawal, D.K. Key Transcription Factors in the Differentiation of Mesenchymal Stem Cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Olsen, B.R. Bone Development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix Remodeling during Endochondral Ossification. Trends Cell Biol. 2004, 14, 86–93. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, H.; Zhang, Y.; Gao, S.; Liang, K.; Su, Y.; Wang, D.; Yang, Z.; Du, Y.; Xing, D.; et al. Enhanced Bone Regeneration via Endochondral Ossification Using Exendin-4-Modified Mesenchymal Stem Cells. Bioact. Mater. 2024, 34, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different Populations and Sources of Human Mesenchymal Stem Cells (MSC): A Comparison of Adult and Neonatal Tissue-Derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.; Ariyanti, A.D.; Luo, Q.; Song, G. Recent Progress in Engineering Mesenchymal Stem Cell Differentiation. Stem Cell Rev. Rep. 2020, 16, 661–674. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate Decision of Mesenchymal Stem Cells: Adipocytes or Osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.-P. Signaling and Transcriptional Regulation in Osteoblast Commitment and Differentiation. Front. Biosci. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Mareddy, S.; Crawford, R. Clonal Characterization of Bone Marrow Derived Stem Cells and Their Application for Bone Regeneration. Int. J. Oral Sci. 2010, 2, 127–135. [Google Scholar] [PubMed]
- Kim, H.D.; Amirthalingam, S.; Kim, S.L.; Lee, S.S.; Rangasamy, J.; Hwang, N.S. Biomimetic Materials and Fabrication Approaches for Bone Tissue Engineering. Adv. Healthc. Mater. 2017, 6, 1700612. [Google Scholar] [CrossRef]
- Barcena, A.J.R.; Mishra, A.; Bolinas, D.K.M.; Martin, B.M.; Melancon, M.P. Integration of Electrospun Scaffolds and Biological Polymers for Enhancing the Delivery and Efficacy of Mesenchymal Stem/Stromal Cell Therapies. Front. Biosci. 2024, 29, 228. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, S.; Tahmasebi Birgani, Z.; Habibovic, P. Biomaterial-Induced Pathway Modulation for Bone Regeneration. Biomaterials 2022, 283, 121431. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E. Wnt Signalling in Osteoporosis: Mechanisms and Novel Therapeutic Approaches. Nat. Rev. Endocrinol. 2013, 9, 575–583. [Google Scholar] [CrossRef]
- Hayat, R.; Manzoor, M.; Hussain, A. Wnt Signaling Pathway: A Comprehensive Review. Cell Biol. Int. 2022, 46, 863–877. [Google Scholar] [CrossRef]
- Hu, L.; Chen, W.; Qian, A.; Li, Y.-P. Wnt/β-Catenin Signaling Components and Mechanisms in Bone Formation, Homeostasis, and Disease. Bone Res. 2024, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Lories, R.J.; Corr, M.; Lane, N.E. To Wnt or Not to Wnt: The Bone and Joint Health Dilemma. Nat. Rev. Rheumatol. 2013, 9, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Mi, K.; Johnson, G.V.W. Role of the Intracellular Domains of LRP5 and LRP6 in Activating the Wnt Canonical Pathway. J. Cell. Biochem. 2005, 95, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Lerner, U.H.; Ohlsson, C. The WNT System: Background and Its Role in Bone. J. Intern. Med. 2015, 277, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast, Skeletal Development, and Bone Formation, Homeostasis and Disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT Signaling in Bone Homeostasis and Disease: From Human Mutations to Treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Appelman-Dijkstra, N.M.; Papapoulos, S.E. Clinical Advantages and Disadvantages of Anabolic Bone Therapies Targeting the WNT Pathway. Nat. Rev. Endocrinol. 2018, 14, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Loots, G.G.; Studer, A.; Keller, H.; Kneissel, M. Parathyroid Hormone (PTH)–Induced Bone Gain Is Blunted in SOST Overexpressing and Deficient Mice. J. Bone Miner. Res. 2010, 25, 178–189. [Google Scholar] [CrossRef]
- McClung, M.R. Sclerostin Antibodies in Osteoporosis: Latest Evidence and Therapeutic Potential. Ther. Adv. Musculoskelet. 2017, 9, 263–270. [Google Scholar] [CrossRef]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP Signaling and Other Molecular Events: Regulation of Osteoblastogenesis and Bone Formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP Signalling in Skeletal Development, Disease and Repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Martin, T.J. Parathyroid Hormone-Related Protein, Its Regulation of Cartilage and Bone Development, and Role in Treating Bone Diseases. Physiol. Rev. 2016, 96, 831–871. [Google Scholar] [CrossRef]
- Kuo, S.-W.; Rimando, M.; Liu, Y.-S.; Lee, O. Intermittent Administration of Parathyroid Hormone 1–34 Enhances Osteogenesis of Human Mesenchymal Stem Cells by Regulating Protein Kinase Cδ. Int. J. Mol. Sci. 2017, 18, 2221. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cho, H.-Y.; Bui, H.T.T.; Kang, D. The Osteogenic or Adipogenic Lineage Commitment of Human Mesenchymal Stem Cells Is Determined by Protein Kinase C Delta. BMC Cell Biol. 2014, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; He, X.; Shen, Y.; Chen, X.; Yang, F.; Yang, P.; Pang, F.; Han, X.; He, W.; Wei, Q. Chrysosplenetin Promotes Osteoblastogenesis of Bone Marrow Stromal Cells via Wnt/β-Catenin Pathway and Enhances Osteogenesis in Estrogen Deficiency-Induced Bone Loss. Stem Cell Res. Ther. 2019, 10, 277. [Google Scholar] [CrossRef]
- Zhao, X.; Xie, L.; Wang, Z.; Wang, J.; Xu, H.; Han, X.; Bai, D.; Deng, P. ZBP1 (DAI/DLM-1) Promotes Osteogenic Differentiation While Inhibiting Adipogenic Differentiation in Mesenchymal Stem Cells through a Positive Feedback Loop of Wnt/β-Catenin Signaling. Bone Res. 2020, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- James, A.W. Review of Signaling Pathways Governing MSC Osteogenic and Adipogenic Differentiation. Scientifica 2013, 2013, 684736. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef]
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch Signaling Maintains Bone Marrow Mesenchymal Progenitors by Suppressing Osteoblast Differentiation. Nat. Med. 2008, 14, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Remark, L.H.; Josephson, A.M.; Leclerc, K.; Lopez, E.M.; Kirby, D.J.; Mehta, D.; Litwa, H.P.; Wong, M.Z.; Shin, S.Y.; et al. Notch-Wnt Signal Crosstalk Regulates Proliferation and Differentiation of Osteoprogenitor Cells during Intramembranous Bone Healing. NPJ Regen. Med. 2021, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Joeng, K.S.; Nakayama, K.I.; Nakayama, K.; Rajagopal, J.; Carroll, T.J.; McMahon, A.P.; Long, F. Noncanonical Wnt Signaling through G Protein-Linked PKCδ Activation Promotes Bone Formation. Dev. Cell 2007, 12, 113–127. [Google Scholar] [CrossRef]
- Baron, R.; Saito, H.; Gori, F. Bone Cells Crosstalk: Noncanonical Roring in the Wnt. Cell Metab. 2012, 15, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Sonomoto, K.; Yamaoka, K.; Oshita, K.; Fukuyo, S.; Zhang, X.; Nakano, K.; Okada, Y.; Tanaka, Y. Interleukin-1β Induces Differentiation of Human Mesenchymal Stem Cells into Osteoblasts via the Wnt-5a/Receptor Tyrosine Kinase-like Orphan Receptor 2 Pathway. Arthritis Rheum. 2012, 64, 3355–3363. [Google Scholar] [CrossRef]
- Bolzoni, M.; Donofrio, G.; Storti, P.; Guasco, D.; Toscani, D.; Lazzaretti, M.; Bonomini, S.; Agnelli, L.; Capocefalo, A.; Dalla Palma, B.; et al. Myeloma Cells Inhibit Non-Canonical Wnt Co-Receptor Ror2 Expression in Human Bone Marrow Osteoprogenitor Cells: Effect of Wnt5a/Ror2 Pathway Activation on the Osteogenic Differentiation Impairment Induced by Myeloma Cells. Leukemia 2013, 27, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Hang, K.; Ye, C.; Xu, J.; Chen, E.; Wang, C.; Zhang, W.; Ni, L.; Kuang, Z.; Ying, L.; Xue, D.; et al. Apelin Enhances the Osteogenic Differentiation of Human Bone Marrow Mesenchymal Stem Cells Partly through Wnt/β-Catenin Signaling Pathway. Stem Cell Res. Ther. 2019, 10, 189. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, G.; Weng, J.; Jia, G.; Fang, C.; Xu, H.; Xiong, A.; Qin, H.; Qi, T.; Yang, Q.; et al. Repair of Osteoporotic Bone Defects in Rats via the Sirtuin 1-Wnt/β-Catenin Signaling Pathway by Novel Icariin/Porous Magnesium Alloy Scaffolds. Biomater. Res. 2024, 28, 0090. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Chen, J.; Gong, W.; Tu, X. The Osteocyte with SB216763-Activated Canonical Wnt Signaling Constructs a Multifunctional 4D Intelligent Osteogenic Module. Biomolecules 2024, 14, 354. [Google Scholar] [CrossRef]
- Wang, X.; Ma, Y.; Chen, J.; Liu, Y.; Liu, G.; Wang, P.; Wang, B.; Taketo, M.M.; Bellido, T.; Tu, X. A Novel Decellularized Matrix of Wnt Signaling-Activated Osteocytes Accelerates the Repair of Critical-Sized Parietal Bone Defects with Osteoclastogenesis, Angiogenesis, and Neurogenesis. Bioact. Mater. 2023, 21, 110–128. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, J.; Bu, Q.; Zhang, J.; Ruan, M.; Chen, X.; Zhao, M.; Tu, X.; Zhao, C. Establishing Stable and Highly Osteogenic hiPSC-Derived MSCs for 3D-Printed Bone Graft through Microenvironment Modulation by CHIR99021-Treated Osteocytes. Mater. Today Bio 2024, 26, 101111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ruan, X.; Li, J.; Wang, B.; Chen, J.; Wang, X.; Wang, P.; Tu, X. The Osteocyte Stimulated by Wnt Agonist SKL2001 Is a Safe Osteogenic Niche Improving Bioactivities in a Polycaprolactone and Cell Integrated 3D Module. Cells 2022, 11, 831. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β Signaling in Health, Disease and Therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, S.; Chen, W.; Li, Y.-P. The Roles and Regulatory Mechanisms of TGF-β and BMP Signaling in Bone and Cartilage Development, Homeostasis and Disease. Cell Res. 2024, 34, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; Ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming Growth Factor-Β1 to the Bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef]
- Kang, Q.; Song, W.-X.; Luo, Q.; Tang, N.; Luo, J.; Luo, X.; Chen, J.; Bi, Y.; He, B.-C.; Park, J.K.; et al. A Comprehensive Analysis of the Dual Roles of BMPs in Regulating Adipogenic and Osteogenic Differentiation of Mesenchymal Progenitor Cells. Stem Cells Dev. 2009, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, X. BMP Signaling and Skeletogenesis. Ann. N. Y. Acad. Sci. 2006, 1068, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Li, C.; Zhen, G.; Jiao, K.; He, W.; Jia, X.; Wang, W.; Shi, C.; Xing, Q.; Chen, Y.; et al. Injury-Activated Transforming Growth Factor β Controls Mobilization of Mesenchymal Stem Cells for Tissue Remodeling. Stem Cells 2012, 30, 2498–2511. [Google Scholar] [CrossRef]
- Crane, J.L.; Cao, X. Bone Marrow Mesenchymal Stem Cells and TGF-β Signaling in Bone Remodeling. J. Clin. Investig. 2014, 124, 466–472. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-Β1–Induced Migration of Bone Mesenchymal Stem Cells Couples Bone Resorption with Formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, F.; Xie, L.; Crane, J.; Zhen, G.; Mishina, Y.; Deng, R.; Gao, B.; Chen, H.; Liu, S.; et al. Inhibition of Overactive TGF-β Attenuates Progression of Heterotopic Ossification in Mice. Nat. Commun. 2018, 9, 551. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.W.; Rosen, V. The BMP Pathway and Its Inhibitors in the Skeleton. Physiol. Rev. 2018, 98, 2431–2452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xie, J.; Lee, D.; Liu, Y.; Jung, J.; Zhou, L.; Xiong, S.; Mei, L.; Xiong, W.-C. Neogenin Regulation of BMP-Induced Canonical Smad Signaling and Endochondral Bone Formation. Dev. Cell 2010, 19, 90–102. [Google Scholar] [CrossRef]
- Kua, H.-Y.; Liu, H.; Leong, W.F.; Li, L.; Jia, D.; Ma, G.; Hu, Y.; Wang, X.; Chau, J.F.L.; Chen, Y.-G.; et al. C-Abl Promotes Osteoblast Expansion by Differentially Regulating Canonical and Non-Canonical BMP Pathways and p16INK4a Expression. Nat. Cell Biol. 2012, 14, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.; Iacovino, M.; Mayerhofer, T.; Koyano-Nakagawa, N.; Baik, J.; Garry, D.J.; Kyba, M.; Letarte, M.; Perlingeiro, R.C.R. A Critical Role for Endoglin in the Emergence of Blood during Embryonic Development. Blood 2012, 119, 5417–5428. [Google Scholar] [CrossRef]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin Is an Accessory Protein That Interacts with the Signaling Receptor Complex of Multiple Members of the Transforming Growth Factor-β Superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Fei, W.; Gao, W.; Fan, C.; Li, Y.; Hong, Y.; Cui, R. SOD3 Regulates FLT1 to Affect Bone Metabolism by Promoting Osteogenesis and Inhibiting Adipogenesis through PI3K/AKT and MAPK Pathways. Free Radic. Biol. Med. 2024, 212, 65–79. [Google Scholar] [CrossRef]
- Lim, J.; Shi, Y.; Karner, C.M.; Lee, S.-Y.; Lee, W.-C.; He, G.; Long, F. Dual Function of Bmpr1a Signaling in Restricting Preosteoblast Proliferation and Stimulating Osteoblast Activity in the Mouse. Development 2015, 143, 339–347. [Google Scholar] [CrossRef]
- Melzer, M.; Schubert, S.; Müller, S.F.; Geyer, J.; Hagen, A.; Niebert, S.; Burk, J. Rho/ROCK Inhibition Promotes TGF-Β3-Induced Tenogenic Differentiation in Mesenchymal Stromal Cells. Stem Cells Int. 2021, 2021, 8284690. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.; Niebert, S.; Heimann, M.; Ullm, F.; Pompe, T.; Scheiner-Bobis, G.; Burk, J. Differential Smad2/3 Linker Phosphorylation Is a Crosstalk Mechanism of Rho/ROCK and Canonical TGF-Β3 Signaling in Tenogenic Differentiation. Sci. Rep. 2024, 14, 10393. [Google Scholar] [CrossRef] [PubMed]
- Gou, Y.; Huang, Y.; Luo, W.; Li, Y.; Zhao, P.; Zhong, J.; Dong, X.; Guo, M.; Li, A.; Hao, A.; et al. Adipose-Derived Mesenchymal Stem Cells (MSCs) Are a Superior Cell Source for Bone Tissue Engineering. Bioact. Mater. 2024, 34, 51–63. [Google Scholar] [CrossRef]
- Zouani, O.F.; Chollet, C.; Guillotin, B.; Durrieu, M.-C. Differentiation of Pre-Osteoblast Cells on Poly(Ethylene Terephthalate) Grafted with RGD and/or BMPs Mimetic Peptides. Biomaterials 2010, 31, 8245–8253. [Google Scholar] [CrossRef]
- Lo, K.W.-H.; Ulery, B.D.; Ashe, K.M.; Laurencin, C.T. Studies of Bone Morphogenetic Protein-Based Surgical Repair. Adv. Drug Deliv. Rev. 2012, 64, 1277–1291. [Google Scholar] [CrossRef]
- Gardella, T.J.; Vilardaga, J.-P.; Ohlstein, E.H. International Union of Basic and Clinical Pharmacology. XCIII. The Parathyroid Hormone Receptors—Family B G Protein–Coupled Receptors. Pharmacol. Rev. 2015, 67, 310–337. [Google Scholar] [CrossRef]
- Martin, T.J.; Sims, N.A.; Seeman, E. Physiological and Pharmacological Roles of PTH and PTHrP in Bone Using Their Shared Receptor, PTH1R. Endocr. Rev. 2021, 42, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Esen, E.; Lee, S.-Y.; Wice, B.M.; Long, F. PTH Promotes Bone Anabolism by Stimulating Aerobic Glycolysis via IGF Signaling. J. Bone Miner. Res. 2015, 30, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Atfi, A.; Baron, R. PTH Battles TGF-β in Bone. Nat. Cell Biol. 2010, 12, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.; Tsuda, M.C.; Usdin, T.B.; Dobolyi, A. Behavioural Actions of Tuberoinfundibular Peptide 39 (Parathyroid Hormone 2). J. Neuroendocrinol. 2022, 34, e13130. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Mefford, S.; Bambino, T.; Nissenson, R.A. Transmembrane Residues Together with the Amino Terminus Limit the Response of the Parathyroid Hormone (PTH) 2 Receptor to PTH-Related Peptide. J. Biol. Chem. 1998, 273, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hanai, J.; Le, P.T.; Bi, R.; Maridas, D.; DeMambro, V.; Figueroa, C.A.; Kir, S.; Zhou, X.; Mannstadt, M.; et al. Parathyroid Hormone Directs Bone Marrow Mesenchymal Cell Fate. Cell Metab. 2017, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Sims, N.A.; Hunzelman, J.L.; Knight, M.C.; Giovannetti, A.; Saxton, J.M.; Kronenberg, H.M.; Baron, R.; Schipani, E. Activated Parathyroid Hormone/Parathyroid Hormone–Related Protein Receptor in Osteoblastic Cells Differentially Affects Cortical and Trabecular Bone. J. Clin. Investig. 2001, 107, 277–286. [Google Scholar] [CrossRef]
- Hoogendam, J.; Farih-Sips, H.; Wÿnaendts, L.C.; Löwik, C.W.G.M.; Wit, J.M.; Karperien, M. Novel Mutations in the Parathyroid Hormone (PTH)/PTH-Related Peptide Receptor Type 1 Causing Blomstrand Osteochondrodysplasia Types I and II. J. Clin. Endocrinol. Metab. 2007, 92, 1088–1095. [Google Scholar] [CrossRef]
- Fu, X.; Zhou, B.; Yan, Q.; Tao, C.; Qin, L.; Wu, X.; Lin, S.; Chen, S.; Lai, Y.; Zou, X.; et al. Kindlin-2 Regulates Skeletal Homeostasis by Modulating PTH1R in Mice. Signal Transduct. Target. Ther. 2020, 5, 297. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wada, A.; Le, I.; Le, P.T.; Lee, A.W.; Zhou, J.; Gori, F.; Baron, R.; Rosen, C.J. PTH Regulates Osteogenesis and Suppresses Adipogenesis through Zfp467 in a Feed-Forward, PTH1R-Cyclic AMP-Dependent Manner. eLife 2023, 12, e83345. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, L.; Rosen, C.J. PTH and the Regulation of Mesenchymal Cells within the Bone Marrow Niche. Cells 2024, 13, 406. [Google Scholar] [CrossRef]
- Wan, M.; Yang, C.; Li, J.; Wu, X.; Yuan, H.; Ma, H.; He, X.; Nie, S.; Chang, C.; Cao, X. Parathyroid Hormone Signaling through Low-Density Lipoprotein-Related Protein 6. Genes Dev. 2008, 22, 2968–2979. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-β Type II Receptor Phosphorylates PTH Receptor to Integrate Bone Remodelling Signalling. Nat. Cell Biol. 2010, 12, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Shapiro, G.; Tawackoli, W.; Jun, D.S.; Koh, Y.; Kang, K.B.; Su, S.; Da, X.; Ben-David, S.; Bez, M.; et al. PTH Induces Systemically Administered Mesenchymal Stem Cells to Migrate to and Regenerate Spine Injuries. Mol. Ther. 2016, 24, 318–330. [Google Scholar] [CrossRef]
- Xia, H.; Tian, Y.; Lin, Y.; Huang, Q.; Xue, Y. Evaluating Osteogenic Differentiation of Osteoblastic Precursors Upon Intermittent Administration of PTH/IGFBP7. Front. Pharmacol. 2022, 13, 839035. [Google Scholar] [CrossRef] [PubMed]
- Takahata, M.; Awad, H.A.; O’Keefe, R.J.; Bukata, S.V.; Schwarz, E.M. Endogenous Tissue Engineering: PTH Therapy for Skeletal Repair. Cell Tissue Res. 2012, 347, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.E.; Cochran, D.L.; Domken, O.; Seibl, R.; Jones, A.A.; Buser, D.; Hammerle, C.H.F. The Effect of Matrix Bound Parathyroid Hormone on Bone Regeneration. Clin. Oral Implant. Res. 2007, 18, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, I.; Mark, S.; Alvisi, M.; Von Rechenberg, B.; Hubbell, J.A.; Schense, J.C. Bone Healing Induced by Local Delivery of an Engineered Parathyroid Hormone Prodrug. Biomaterials 2009, 30, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, S.T.; Liu, S.; Chareunsouk, A.; Serowoky, M.; Mariani, F.V. On the Horizon: Hedgehog Signaling to Heal Broken Bones. Bone Res. 2022, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Qiao, M.; Harris, S.E.; Chen, D.; Oyajobi, B.O.; Mundy, G.R. The Zinc Finger Transcription Factor Gli2 Mediates Bone Morphogenetic Protein 2 Expression in Osteoblasts in Response to Hedgehog Signaling. Mol. Cell. Biol. 2006, 26, 6197–6208. [Google Scholar] [CrossRef] [PubMed]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; Spohr, T.C.L.D.S.E. A Highlight on Sonic Hedgehog Pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Zhong, L.; Zhang, Y.; Li, H.; Fu, H.; Lv, C.; Jia, X. Overexpressed miR-196a Accelerates Osteogenic Differentiation in Osteoporotic Mice via GNAS-dependent Hedgehog Signaling Pathway. J. Cell. Biochem. 2019, 120, 19422–19431. [Google Scholar] [CrossRef]
- Cobourne, M.T.; Xavier, G.M.; Depew, M.; Hagan, L.; Sealby, J.; Webster, Z.; Sharpe, P.T. Sonic Hedgehog Signalling Inhibits Palatogenesis and Arrests Tooth Development in a Mouse Model of the Nevoid Basal Cell Carcinoma Syndrome. Dev. Biol. 2009, 331, 38–49. [Google Scholar] [CrossRef]
- Kitamura, A.; Kawasaki, M.; Kawasaki, K.; Meguro, F.; Yamada, A.; Nagai, T.; Kodama, Y.; Trakanant, S.; Sharpe, P.T.; Maeda, T.; et al. Ift88 Is Involved in Mandibular Development. J. Anat. 2020, 236, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and Defective Axial Patterning in Mice Lacking Sonic Hedgehog Gene Function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.P.; Mitchell, J. Indian Hedgehog: Its Roles and Regulation in Endochondral Bone Development. J. Cell. Biochem. 2005, 96, 1163–1173. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian Hedgehog Signaling Regulates Proliferation and Differentiation of Chondrocytes and Is Essential for Bone Formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Coucke, P.J.; Giedion, A.; Paepe, A.D.; Kramer, P.; Beemer, F.; Mortier, G.R. Homozygous Mutations in IHH Cause Acrocapitofemoral Dysplasia, an Autosomal Recessive Disorder with Cone-Shaped Epiphyses in Hands and Hips. Am. J. Hum. Genet. 2003, 72, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. Indian Hedgehog Stimulates Periarticular Chondrocyte Differentiation to Regulate Growth Plate Length Independently of PTHrP. J. Clin. Investig. 2005, 115, 1734–1742. [Google Scholar] [CrossRef]
- Dennis, J.F.; Kurosaka, H.; Iulianella, A.; Pace, J.; Thomas, N.; Beckham, S.; Williams, T.; Trainor, P.A. Mutations in Hedgehog Acyltransferase (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects. PLoS Genet. 2012, 8, e1002927. [Google Scholar] [CrossRef] [PubMed]
- Ghuloum, F.I.; Johnson, C.A.; Riobo-Del Galdo, N.A.; Amer, M.H. From Mesenchymal Niches to Engineered in Vitro Model Systems: Exploring and Exploiting Biomechanical Regulation of Vertebrate Hedgehog Signalling. Mater. Today Bio 2022, 17, 100502. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, L.; Chen, Y.; Zhu, C.; Chen, F.; Li, A. Research Progress on the Hedgehog Signalling Pathway in Regulating Bone Formation and Homeostasis. Cell Prolif. 2022, 55, e13162. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Chen, J.K. Purmorphamine Activates the Hedgehog Pathway by Targeting Smoothened. Nat. Chem. Biol. 2006, 2, 29–30. [Google Scholar] [CrossRef]
- Kim, W.-K.; Meliton, V.; Amantea, C.M.; Hahn, T.J.; Parhami, F. 20(S)-Hydroxycholesterol Inhibits PPARγ Expression and Adipogenic Differentiation of Bone Marrow Stromal Cells Through a Hedgehog-Dependent Mechanism. J. Bone Miner. Res. 2007, 22, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shin, D.Y.; Eiseman, M.; Yallowitz, A.R.; Li, N.; Lalani, S.; Li, Z.; Cung, M.; Bok, S.; Debnath, S.; et al. SLITRK5 Is a Negative Regulator of Hedgehog Signaling in Osteoblasts. Nat. Commun. 2021, 12, 4611. [Google Scholar] [CrossRef] [PubMed]
- Beak, J.Y.; Kang, H.S.; Kim, Y.-S.; Jetten, A.M. Krüppel-Like Zinc Finger Protein Glis3 Promotes Osteoblast Differentiation by Regulating FGF18 Expression. J. Bone Miner. Res. 2007, 22, 1234–1244. [Google Scholar] [CrossRef]
- Zhang, L.; Fu, X.; Ni, L.; Liu, C.; Zheng, Y.; You, H.; Li, M.; Xiu, C.; Zhang, L.; Gong, T.; et al. Hedgehog Signaling Controls Bone Homeostasis by Regulating Osteogenic/Adipogenic Fate of Skeletal Stem/Progenitor Cells in Mice. J. Bone Miner. Res. 2020, 37, 559–576. [Google Scholar] [CrossRef]
- Kim, W.; Meliton, V.; Bourquard, N.; Hahn, T.J.; Parhami, F. Hedgehog Signaling and Osteogenic Differentiation in Multipotent Bone Marrow Stromal Cells Are Inhibited by Oxidative Stress. J. Cell. Biochem. 2010, 111, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, Y.; Fu, Q.; Dong, Y. Osterix Is Required for Sonic Hedgehog-Induced Osteoblastic MC3T3-E1 Cell Differentiation. Cell Biochem. Biophys. 2012, 64, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Tevlin, R.; Seo, E.Y.; Marecic, O.; McArdle, A.; Tong, X.; Zimdahl, B.; Malkovskiy, A.; Sinha, R.; Gulati, G.; Li, X.; et al. Pharmacological Rescue of Diabetic Skeletal Stem Cell Niches. Sci. Transl. Med. 2017, 9, eaag2809. [Google Scholar] [CrossRef] [PubMed]
- Breathwaite, E.; Weaver, J.; Odanga, J.; Dela Pena-Ponce, M.; Lee, J.B. 3D Bioprinted Osteogenic Tissue Models for In Vitro Drug Screening. Molecules 2020, 25, 3442. [Google Scholar] [CrossRef]
- Huang, C.; Tang, M.; Yehling, E.; Zhang, X. Overexpressing Sonic Hedgehog Peptide Restores Periosteal Bone Formation in a Murine Bone Allograft Transplantation Model. Mol. Ther. 2014, 22, 430–439. [Google Scholar] [CrossRef]
- Zou, S.; Chen, T.; Wang, Y.; Tian, R.; Zhang, L.; Song, P.; Yang, S.; Zhu, Y.; Guo, X.; Huang, Y.; et al. Mesenchymal Stem Cells Overexpressing Ihh Promote Bone Repair. J. Orthop. Surg. Res. 2014, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Aghaloo, T.L.; Amantea, C.M.; Cowan, C.M.; Richardson, J.A.; Wu, B.M.; Parhami, F.; Tetradis, S. Oxysterols Enhance Osteoblast Differentiation in Vitro and Bone Healing in Vivo. J. Orthop. Res. 2007, 25, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, J. Insulin-like Growth factor-II and Bioactive Proteins Containing a Part of the E-domain of Pro-insulin-like Growth factor-II. BioFactors 2020, 46, 563–578. [Google Scholar] [CrossRef]
- Foulstone, E.; Prince, S.; Zaccheo, O.; Burns, J.; Harper, J.; Jacobs, C.; Church, D.; Hassan, A. Insulin-like Growth Factor Ligands, Receptors, and Binding Proteins in Cancer. J. Pathol. 2005, 205, 145–153. [Google Scholar] [CrossRef]
- Wang, Y.; Bikle, D.D.; Chang, W. Autocrine and Paracrine Actions of IGF-I Signaling in Skeletal Development. Bone Res. 2013, 1, 249–259. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, X.; Chen, X.; Wang, Z.; Zheng, S.; Cheng, Y.; Liu, S.; Hao, L. The Role of Insulin-like Growth Factor-1 in Bone Remodeling: A Review. Int. J. Biol. Macromol. 2023, 238, 124125. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yuan, K.; Mao, X.; Miano, J.M.; Wu, H.; Chen, Y. Serum Response Factor Regulates Bone Formation via IGF-1 and Runx2 Signals. J. Bone Miner. Res. 2012, 27, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. Signaling Pathways of the Insulin-like Growth Factor Binding Proteins. Endocr. Rev. 2023, 44, 753–778. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth Hormone, Insulin-Like Growth Factors, and the Skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice Carrying Null Mutations of the Genes Encoding Insulin-like Growth Factor I (Igf-1) and Type 1 IGF Receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, W.; Wang, H.; Liu, Y.; Li, Z.; Yi, C.; Shi, Y.; Ma, T.; Chen, J. Downregulation of Insulin-Like Growth Factor-1 Receptor Mediates Chondrocyte Death and Matrix Degradation in Kashin-Beck Disease. Cartilage 2021, 13, 809S–817S. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yuan, K.; Liu, Y.; Wang, Q.; Lin, Y.; Yang, S.; Huang, K.; Kan, T.; Zhang, Y.; Xu, M.; et al. Constitutively Activated AMPKα1 Protects against Skeletal Aging in Mice by Promoting Bone-derived IGF-1 Secretion. Cell Prolif. 2023, 56, e13476. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; Chikazu, D.; Kubota, N.; Terauchi, Y.; Tobe, K.; Azuma, Y.; Ohta, T.; Kadowaki, T.; Nakamura, K.; Kawaguchi, H. Insulin Receptor Substrate-1 in Osteoblast Is Indispensable for Maintaining Bone Turnover. J. Clin. Investig. 2000, 105, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Akune, T.; Ogata, N.; Hoshi, K.; Kubota, N.; Terauchi, Y.; Tobe, K.; Takagi, H.; Azuma, Y.; Kadowaki, T.; Nakamura, K.; et al. Insulin Receptor Substrate-2 Maintains Predominance of Anabolic Function over Catabolic Function of Osteoblasts. J. Cell Biol. 2002, 159, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Wai, C.; Rosen, C.J.; Clemmons, D.R. A Peptide Containing the Receptor Binding Site of Insulin-like Growth Factor Binding Protein-2 Enhances Bone Mass in Ovariectomized Rats. Bone Res. 2018, 6, 23. [Google Scholar] [CrossRef]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 Maintains Bone Mass by Activation of mTOR in Mesenchymal Stem Cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef]
- Conover, C.A.; Oxvig, C. The Pregnancy-Associated Plasma Protein-A (PAPP-A) Story. Endocr. Rev. 2023, 44, 1012–1028. [Google Scholar] [CrossRef]
- Ning, Y.; Schuller, A.G.P.; Bradshaw, S.; Rotwein, P.; Ludwig, T.; Frystyk, J.; Pintar, J.E. Diminished Growth and Enhanced Glucose Metabolism in Triple Knockout Mice Containing Mutations of Insulin-Like Growth Factor Binding Protein-3, -4, and -5. Mol. Endocrinol. 2006, 20, 2173–2186. [Google Scholar] [CrossRef]
- Zhang, M.; Faugere, M.-C.; Malluche, H.; Rosen, C.J.; Chernausek, S.D.; Clemens, T.L. Paracrine Overexpression of IGFBP-4 in Osteoblasts of Transgenic Mice Decreases Bone Turnover and Causes Global Growth Retardation. J. Bone Miner. Res. 2003, 18, 836–843. [Google Scholar] [CrossRef]
- Miyakoshi, N.; Richman, C.; Kasukawa, Y.; Linkhart, T.A.; Baylink, D.J.; Mohan, S. Evidence That IGF-Binding Protein-5 Functions as a Growth Factor. J. Clin. Investig. 2001, 107, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Bauss, F.; Lang, K.; Dony, C.; Kling, L. The Complex of Recombinant Human Insulin-like Growth Factor-I (rhIGF-I) and Its Binding Protein-5 (IGFBP-5) Induces Local Bone Formation in Murine Calvariae and in Rat Cortical Bone after Local or Systemic Administration. Growth Horm. IGF Res. 2001, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Zhu, C.; Peng, T.; Huang, D.; Ma, X.; Pan, X.; Wu, C. Release Mechanisms of Bovine Serum Albumin Loaded–PLGA Microspheres Prepared by Ultra-Fine Particle Processing System. Drug Deliv. Transl. Res. 2020, 10, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, L.; Gong, Y.; Wang, M.; Li, S.; Chen, C.; Yu, B. Long-Term Delivery of rhIGF-1 from Biodegradable Poly(Lactic Acid)/hydroxyapatite@Eudragit Double-Layer Microspheres for Prevention of Bone Loss and Articular Degeneration in C57BL/6 Mice. J. Mater. Chem. B 2018, 6, 3085–3095. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, Q.; Cao, D.; Peng, Q.; Zhang, X.; Li, C.; Zhang, C.; Zhou, B.O.; Yue, R. Bone Marrow-Derived IGF-1 Orchestrates Maintenance and Regeneration of the Adult Skeleton. Proc. Natl. Acad. Sci. USA 2023, 120, e2203779120. [Google Scholar] [CrossRef]
- Busilacchi, A.; Gigante, A.; Mattioli-Belmonte, M.; Manzotti, S.; Muzzarelli, R.A.A. Chitosan Stabilizes Platelet Growth Factors and Modulates Stem Cell Differentiation toward Tissue Regeneration. Carbohydr. Polym. 2013, 98, 665–676. [Google Scholar] [CrossRef]
- Deng, M.; Luo, K.; Hou, T.; Luo, F.; Xie, Z.; Zhang, Z.; Yang, A.; Yu, B.; Yi, S.; Tan, J.; et al. IGFBP3 Deposited in the Human Umbilical Cord Mesenchymal Stem Cell-secreted Extracellular Matrix Promotes Bone Formation. J. Cell. Physiol. 2018, 233, 5792–5804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/Tricalcium Phosphate Combination Scaffolds Can Enhance Bone Regeneration by Activating the PI3K/Akt Signaling Pathway. Stem Cell Res. Ther. 2016, 7, 136. [Google Scholar] [CrossRef]
- Wu, F.; Wu, Z.; Ye, Z.; Niu, G.; Ma, Z.; Zhang, P. PLGA/BGP/Nef Porous Composite Restrains Osteoclasts by Inhibiting the NF-κB Pathway, Enhances IGF-1-Mediated Osteogenic Differentiation and Promotes Bone Regeneration. J. Biol. Eng. 2023, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Zhang, N.; Cheng, G.; Zhang, Q.; He, Y.; Shen, Y.; Zhang, Q.; Zhu, B.; Zhang, Q.; Qin, L. Rehmannia glutinosa Libosch Extracts Prevent Bone Loss and Architectural Deterioration and Enhance Osteoblastic Bone Formation by Regulating the IGF-1/PI3K/mTOR Pathway in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 3964. [Google Scholar] [CrossRef] [PubMed]
- Rieunier, G.; Wu, X.; Macaulay, V.M.; Lee, A.V.; Weyer-Czernilofsky, U.; Bogenrieder, T. Bad to the Bone: The Role of the Insulin-Like Growth Factor Axis in Osseous Metastasis. Clin. Cancer Res. 2019, 25, 3479–3485. [Google Scholar] [CrossRef]
- Khodabandehloo, F.; Taleahmad, S.; Aflatoonian, R.; Rajaei, F.; Zandieh, Z.; Nassiri-Asl, M.; Eslaminejad, M.B. Microarray Analysis Identification of Key Pathways and Interaction Network of Differential Gene Expressions during Osteogenic Differentiation. Hum. Genom. 2020, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, H.; Li, X.; Wu, P.; Liu, J.; Wang, T.; Zhou, X.; Xiong, J.; Li, W. Bioinformatics Analysis on the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts and Adipocytes. Mol. Med. Rep. 2017, 15, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Blayney, J.W.; Francis, H.; Rampasekova, A.; Camellato, B.; Mitchell, L.; Stolper, R.; Cornell, L.; Babbs, C.; Boeke, J.D.; Higgs, D.R.; et al. Super-Enhancers Include Classical Enhancers and Facilitators to Fully Activate Gene Expression. Cell 2023, 186, 5826–5839.e18. [Google Scholar] [CrossRef]
- Cheng, H.; Chawla, A.; Yang, Y.; Li, Y.; Zhang, J.; Jang, H.L.; Khademhosseini, A. Development of Nanomaterials for Bone-Targeted Drug Delivery. Drug Discov. Today 2017, 22, 1336–1350. [Google Scholar] [CrossRef]
- Zhou, H.; Qian, J.; Wang, J.; Yao, W.; Liu, C.; Chen, J.; Cao, X. Enhanced Bioactivity of Bone Morphogenetic Protein-2 with Low Dose of 2-N, 6-O-Sulfated Chitosan in Vitro and in Vivo. Biomaterials 2009, 30, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Werkmeister, J.A.; Wang, J.; Glattauer, V.; McLean, K.M.; Liu, C. Bone Regeneration Using Photocrosslinked Hydrogel Incorporating rhBMP-2 Loaded 2-N, 6-O-Sulfated Chitosan Nanoparticles. Biomaterials 2014, 35, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Huang, B.J.; Kaltz, S.R.; Sur, S.; Newcomb, C.J.; Stock, S.R.; Shah, R.N.; Stupp, S.I. Bone Regeneration with Low Dose BMP-2 Amplified by Biomimetic Supramolecular Nanofibers within Collagen Scaffolds. Biomaterials 2013, 34, 452–459. [Google Scholar] [CrossRef]
- Feng, K.; Xie, N.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Modular Design of Poly(Norbornenes) for Organelle-Specific Imaging in Tumor Cells. Biomacromolecules 2016, 17, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, B.; Goodman, J.T.; Vela Ramirez, J.E. Rational Design of Targeted Next-Generation Carriers for Drug and Vaccine Delivery. Annu. Rev. Biomed. Eng. 2016, 18, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, J.; Qiu, T.; Zhang, H.; Liao, L.; Su, X. Epigenetic Therapy Targeting Bone Marrow Mesenchymal Stem Cells for Age-Related Bone Diseases. Stem Cell Res. Ther. 2022, 13, 201. [Google Scholar] [CrossRef] [PubMed]
- Zha, K.; Tian, Y.; Panayi, A.C.; Mi, B.; Liu, G. Recent Advances in Enhancement Strategies for Osteogenic Differentiation of Mesenchymal Stem Cells in Bone Tissue Engineering. Front. Cell Dev. Biol. 2022, 10, 824812. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | Drug | Brand Name | Target Disease | Therapeutic Methods | Mechanisms of Action | Limitations and Risks |
|---|---|---|---|---|---|---|
| Wnt | Monoclonal antibody of SOST (Romosozumab) | Evenity (Manufacturer: Amgen Inc., Thousand Oaks, CA, USA; UCB Pharma, Brussels, Belgium) | Osteoporosis | Subcutaneous injection | Blocking sclerostin increases osteoblast activity and simultaneously decreases osteoclast activity. | Increased risk of cardiovascular events and bone-related side effects [24,26] |
| Wnt | Monoclonal antibody of SOST (Romosozumab) | APC+® (Manufacturer: Amgen Inc., Thousand Oaks, CA, USA) | Osteoporosis | Local injection or implantation | Blocking sclerostin increases osteoblast activity and simultaneously decreases osteoclast activity. | Injection site reactions, risk of osteonecrosis of the jaw, and possible cardiovascular concerns [24,26] |
| Wnt | Fully human antibody of SOST (Setrusumab) | BPS804 (Manufacturer: Novartis, Basel, Switzerland) | OI | Subcutaneous injection | Blocking sclerostin improves bone density and reduces fracture risk in patients with OI. | Incomplete efficacy across all OI subtypes, injection site reactions, and mild flu-like symptoms [24,26] |
| Wnt | Monoclonal antibody of SOST (Blosozumab) | Blosozumab (Manufacturer: Eli Lilly and Company, Indianapolis, IN, USA) | Osteoporosis | Subcutaneous injection | Blocking sclerostin, promoting osteoblast activity and bone formation. | Potential risks include long-term safety and efficacy, injection site reactions, and possible cardiovascular events [24,26] |
| TGF-β/BMP | rhBMP-2 | InFuse™ (Manufacturer: Medtronic, Minneapolis, MN, USA) | Spinal fusion and other bone repair procedures | Implantation with biomaterials | Activating the Smad1/5/8 pathway, promoting osteoblast differentiation, bone matrix production, and new bone formation. | Easily degraded by enzymes and rapidly cleared. Inflammatory responses, ectopic bone formation, and cancer risk at the implantation site [22,27,28] |
| TGF-β/BMP | rhBMP-7 | OP-1® (Manufacturer: Stryker Corporation, Kalamazoo, MI, USA) | OI, bone fractures and other bone defects | Local injection or implantation | Activating the Smad1/5/8 pathway stimulates bone formation by activating osteoblast differentiation and bone matrix production. | Swelling, pain, and infection at the implantation site, and the risk of ectopic bone formation [22,27,28] |
| PTH/PTHrP | PTH (1–34) (Teriparatide) | Forteo® (Manufacturer: Eli Lilly and Company, Indianapolis, IN, USA) | Osteoporosis | Intermittent subcutaneous injection | Stimulating osteoblasts to increase bone formation while also reducing bone resorption. | Hypercalcemia, orthostatic hypotension, dizziness, and an increased risk of osteosarcoma in animal studies, making long-term use a concern [29,30,31] |
| PTH/PTHrP | PTHrP analog (Abaloparatide) | TYMLOS® (Manufacturer: Radius Health, Inc., Boston, MA, USA) | Osteoporosis | Subcutaneous injection | Activates the parathyroid hormone receptor to stimulate osteoblast activity, thereby promoting bone formation and increasing bone mineral density. | Hypercalcemia, dizziness, headache, and an increased risk of osteosarcoma in animal studies. Long-term use should be limited [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Ruan, M.; Bu, Q.; Zhao, C. Signaling Pathways Driving MSC Osteogenesis: Mechanisms, Regulation, and Translational Applications. Int. J. Mol. Sci. 2025, 26, 1311. https://doi.org/10.3390/ijms26031311
Wang L, Ruan M, Bu Q, Zhao C. Signaling Pathways Driving MSC Osteogenesis: Mechanisms, Regulation, and Translational Applications. International Journal of Molecular Sciences. 2025; 26(3):1311. https://doi.org/10.3390/ijms26031311
Chicago/Turabian StyleWang, Liuqing, Minjie Ruan, Qiqi Bu, and Chengzhu Zhao. 2025. "Signaling Pathways Driving MSC Osteogenesis: Mechanisms, Regulation, and Translational Applications" International Journal of Molecular Sciences 26, no. 3: 1311. https://doi.org/10.3390/ijms26031311
APA StyleWang, L., Ruan, M., Bu, Q., & Zhao, C. (2025). Signaling Pathways Driving MSC Osteogenesis: Mechanisms, Regulation, and Translational Applications. International Journal of Molecular Sciences, 26(3), 1311. https://doi.org/10.3390/ijms26031311