

SMURF1-Induced Ubiquitination of FTH1 Disrupts Iron Homeostasis and Suppresses Myogenesis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
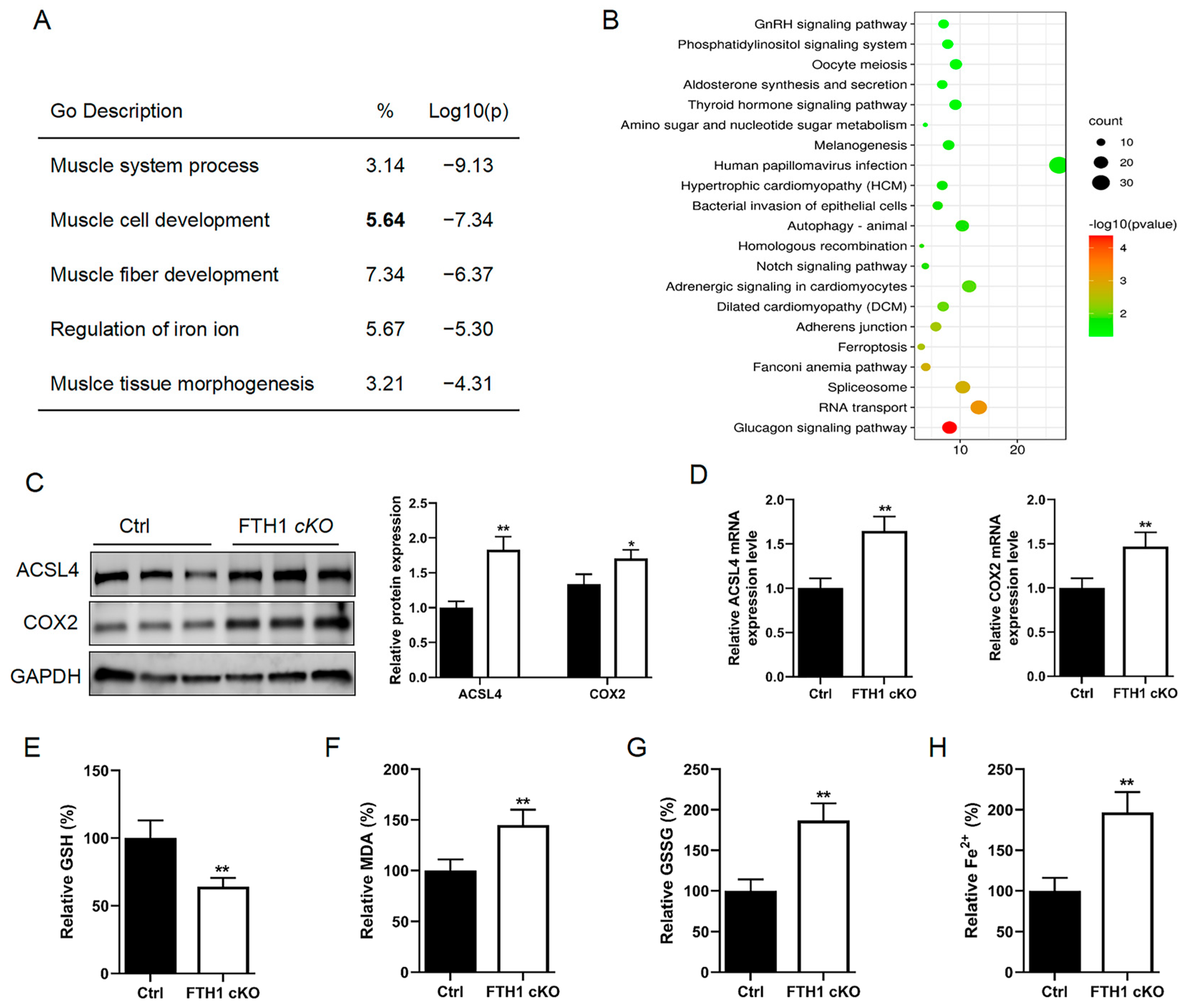
2.1. FTH1 Knockout Mice Result in Muscle Atrophy and Weakness
2.2. FTH1 Knockout Induces Activation of the Ferroptosis Pathway in Myoblasts
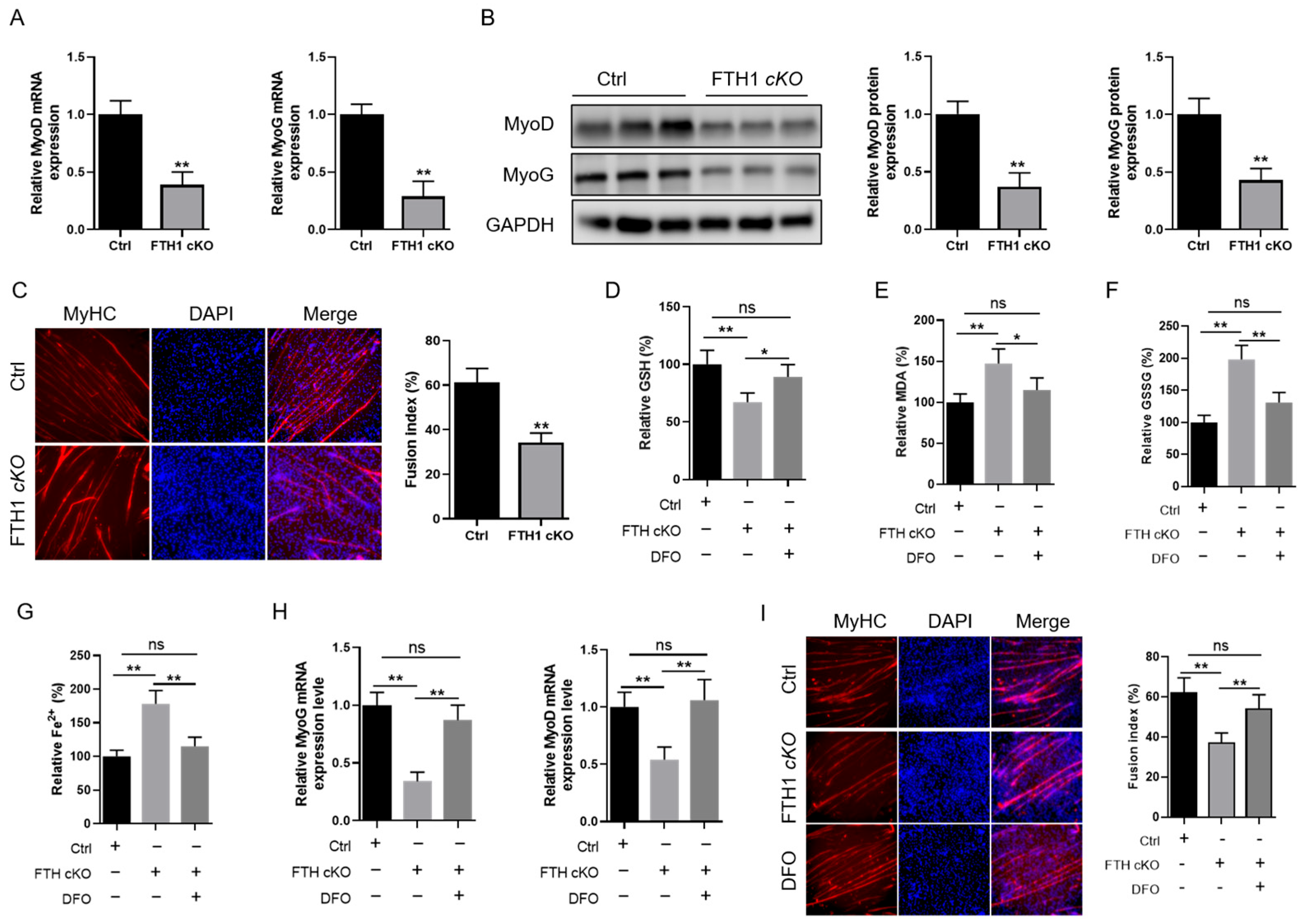
2.3. FTH1 Modulates Myoblast Differentiation into Myotubes via the Ferroptosis Pathway
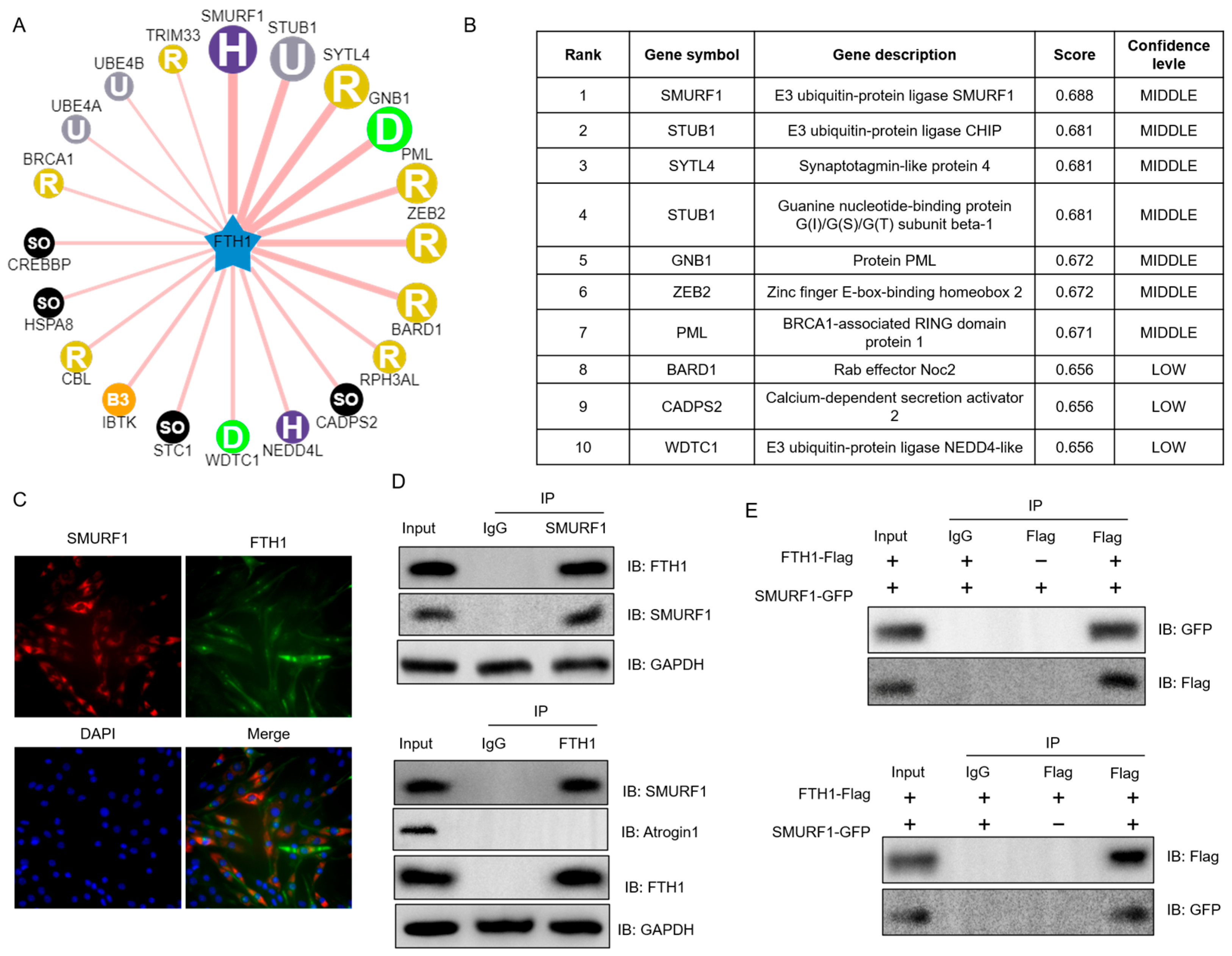
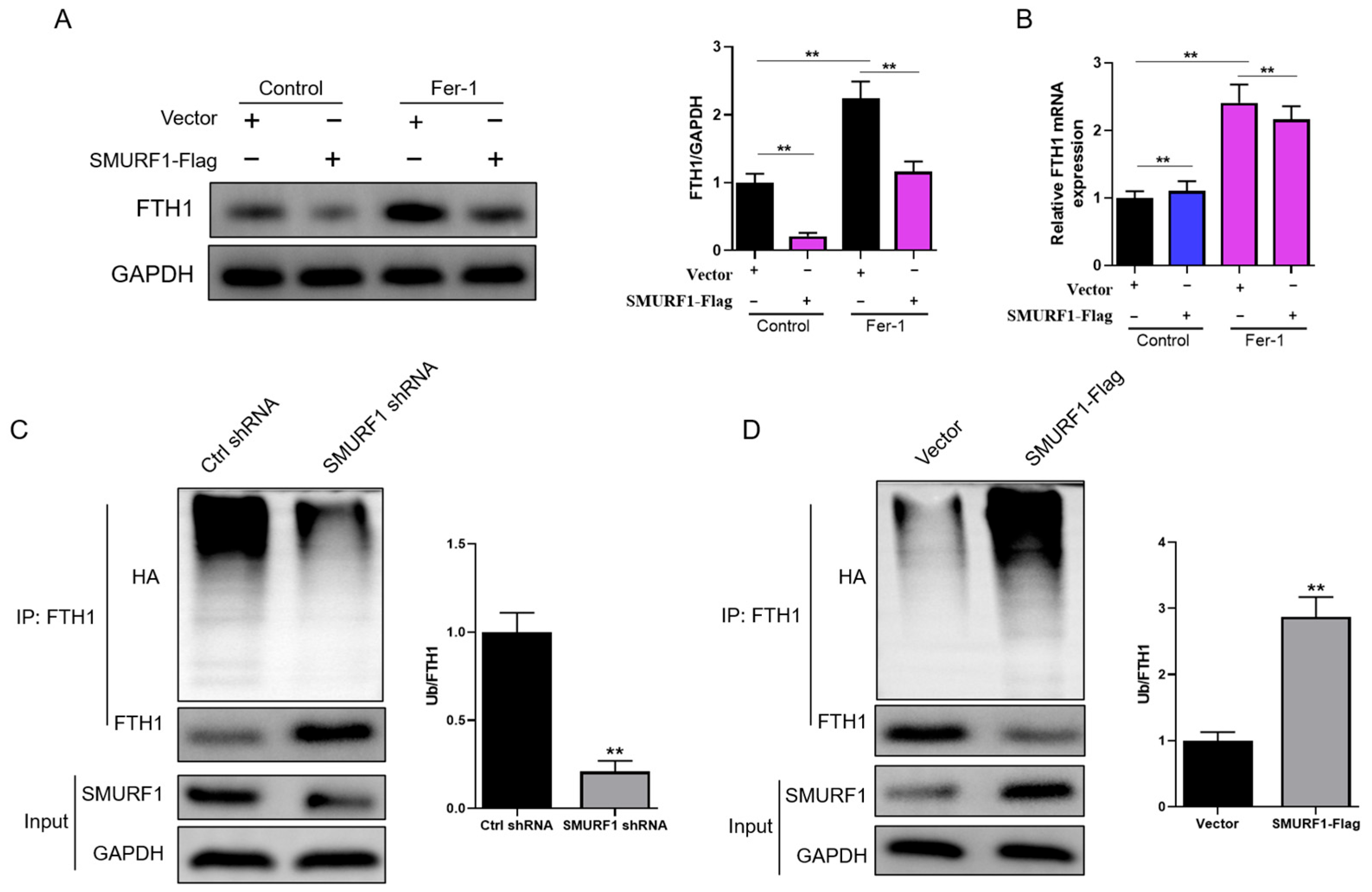
2.4. SMURF1 Directly Recognizes FTH1
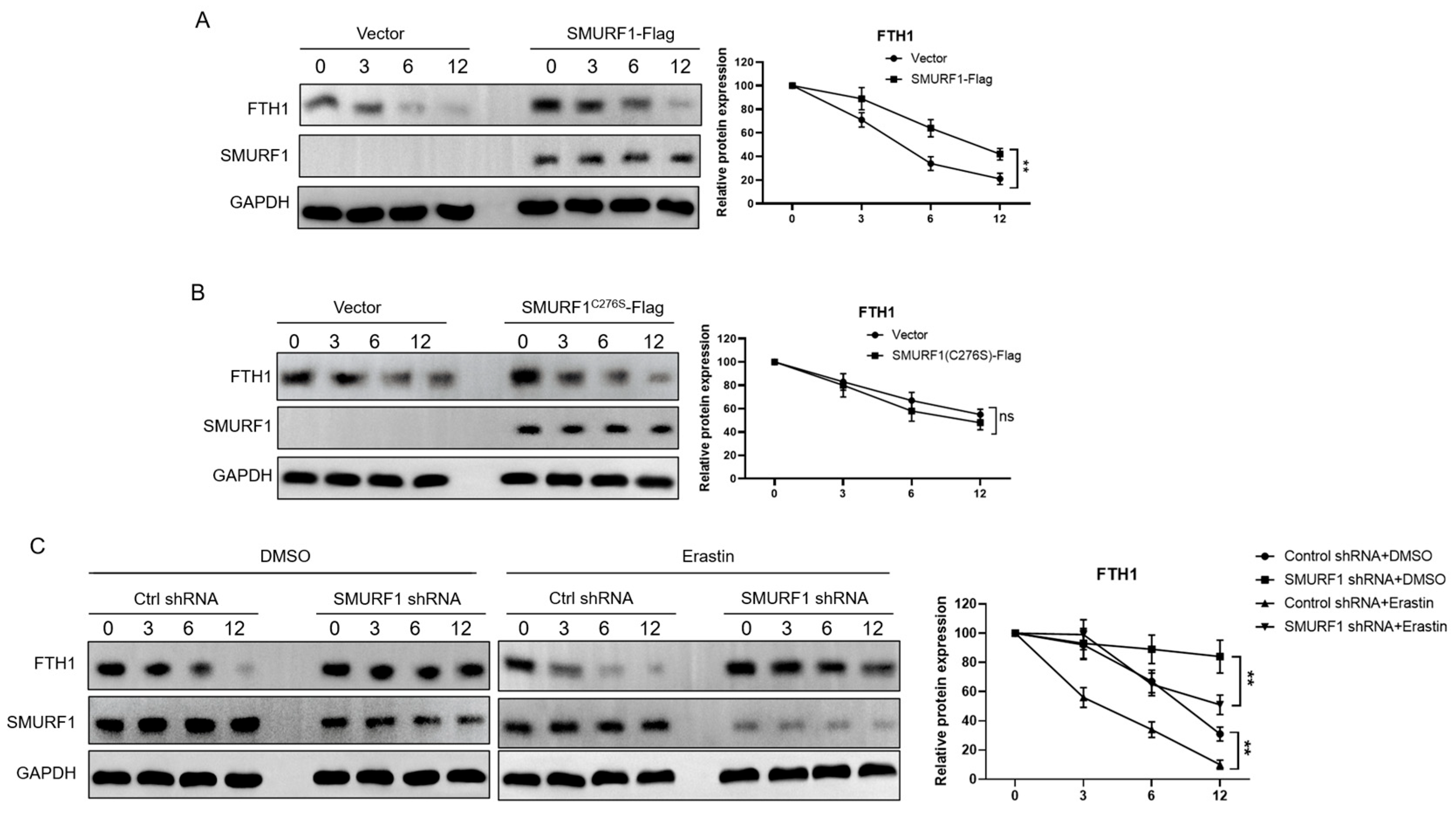
2.5. SMURF1 Ubiquitinates and Degrades FTH1
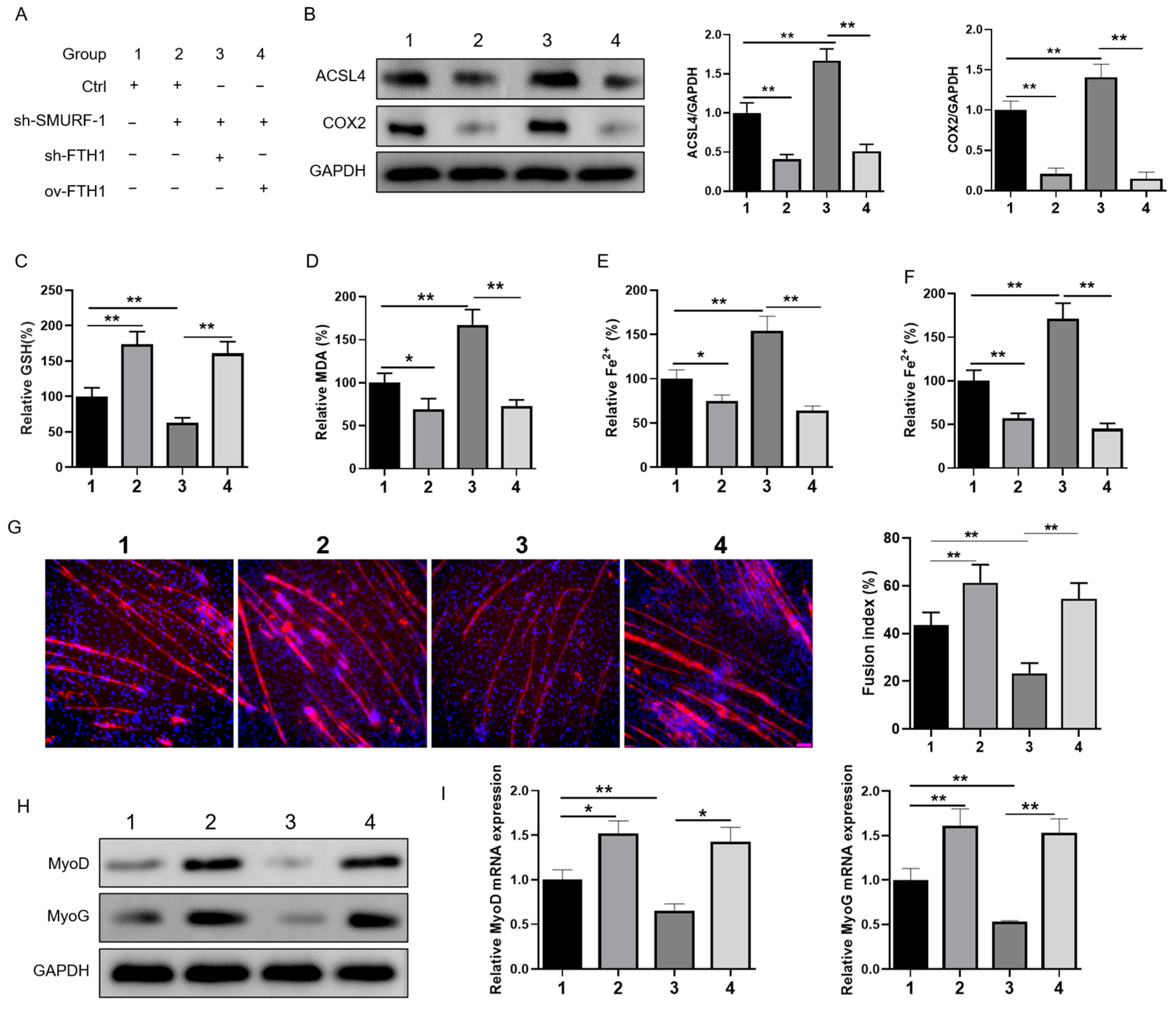
2.6. SMURF1 Regulates Ferroptosis via FTH1 to Affect Myoblast-to-Myotube Differentiation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. In Vitro Cell Culture and Differentiation
4.3. RNA Extraction and Real-Time PCR Analysis
4.4. Cell Transfection
4.5. Protein Extraction and Western Blot Analysis
4.6. Immunofluorescence Staining
4.7. Immunoprecipitation Assay
4.8. Assessment of Lipid Peroxidation, GSH Levels, and Iron Content
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Hayashi, K. Actin-related protein 5 functions as a novel modulator of MyoD and MyoG in skeletal muscle and in rhabdomyosarcoma. eLife 2022, 11, e77746. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Calabria, E. Skeletal muscle mass is controlled by the MRF4-MEF2 axis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 164–167. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, X.; Lei, H.; Lam, N.; Carter, S.; Yockey, O.; Xu, M.; Mendoza, A.; Hernandez, E.R.; Wei, J.S. CASZ1 induces skeletal muscle and rhabdomyosarcoma differentiation through a feed-forward loop with MYOD and MYOG. Nat. Commun. 2020, 11, 911. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Konishi, C.T.; Perez Carbajal, E.E.; Wang, T.T.; Walsh, R.A.; Gan, Q.; Wagner, M.W.; Rando, T.A. Mesenchymal Stromal Cells Are Required for Regeneration and Homeostatic Maintenance of Skeletal Muscle. Cell Rep. 2019, 27, 2029–2035.e5. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef]
- Reisz-Porszasz, S.; Bhasin, S.; Artaza, J.N.; Shen, R.; Sinha-Hikim, I.; Hogue, A.; Fielder, T.J.; Gonzalez-Cadavid, N.F. Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E876–E888. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.D.; Yu, B.; Yu, J.; Mao, X.B.; Zheng, P.; He, J.; Huang, Z.Q.; He, D.T.; Chen, D.W. Mitochondrial biogenesis is decreased in skeletal muscle of pig fetuses exposed to maternal high-energy diets. Anim. Int. J. Anim. Biosci. 2017, 11, 54–60. [Google Scholar] [CrossRef]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Di Sanzo, M.; Quaresima, B.; Biamonte, F.; Palmieri, C. FTH1 Pseudogenes in Cancer and Cell Metabolism. Cells 2020, 9, 2554. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Xie, J.; Pu, J.; Shen, Z.; Wang, A.; Li, T.; Wang, T.; Li, G.; Liu, Y.; et al. M(7)G modification of FTH1 and pri-miR-26a regulates ferroptosis and chemotherapy resistance in osteosarcoma. Oncogene 2024, 43, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Chen, X.; Feng, J.; Duan, T.; Liu, S.; Sun, X.; Chen, P.; Pan, T.; Yan, L.; Jin, T.; et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm. Sinica. B 2021, 11, 4045–4054. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, N.; Ji, Y.; Sheng, X.; Huo, J.; Zhu, Y.; Huang, M.; He, W.; Ma, J. Brusatol induces ferroptosis in oesophageal squamous cell carcinoma by repressing GSH synthesis and increasing the labile iron pool via inhibition of the NRF2 pathway. Biomed. Pharmacother. 2023, 167, 115567. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, C.; Yao, Y.; Tang, X.; Yin, E.; Lu, Z.; Sun, N.; He, J. A comprehensive pan-cancer analysis of prognostic value and potential clinical implications of FTH1 in cancer immunotherapy. Cancer Immunol. Immunother. 2024, 73, 37. [Google Scholar] [CrossRef]
- Alves, F.M.; Ayton, S. Age-Related Changes in Skeletal Muscle Iron Homeostasis. J. Gerontol. Ser. A 2023, 78, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Carbonara, R.; Ruggieri, R.; Passantino, A.; Scrutinio, D. Iron Deficiency: A New Target for Patients with Heart Failure. Front. Cardiovasc. Med. 2021, 8, 709872. [Google Scholar] [CrossRef]
- Fan, X.; Li, A. From Iron Metabolism to Ferroptosis: Pathologic Changes in Coronary Heart Disease. Oxid. Med. Cell. Longev. 2022, 2022, 6291889. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Jiao, W.; Wang, Y.; Wang, X.; Zhao, Y.; Fan, X.; Tian, L.; Li, X.; Mi, J. Ferroptosis and its role in skeletal muscle diseases. Front. Mol. Biosci. 2022, 9, 1051866. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B.; et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Nuclear Factor Erythroid 2-Related Factor 2 and Its Targets in Skeletal Muscle Repair and Regeneration. Antioxid. Redox Signal. 2023, 38, 619–642. [Google Scholar] [CrossRef]
- Cui, J.; Chen, Y.; Yang, Q.; Zhao, P.; Yang, M.; Wang, X.; Mang, G.; Yan, X.; Wang, D.; Tong, Z.; et al. Protosappanin A Protects DOX-Induced Myocardial Injury and Cardiac Dysfunction by Targeting ACSL4/FTH1 Axis-Dependent Ferroptosis. Adv. Sci. 2024, 11, e2310227. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell. Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef]
- Yi, W.; Zhang, J.; Huang, Y.; Zhan, Q.; Zou, M.; Cheng, X.; Zhang, X.; Yin, Z.; Tao, S.; Cheng, H.; et al. Ferritin-mediated mitochondrial iron homeostasis is essential for the survival of hematopoietic stem cells and leukemic stem cells. Leukemia 2024, 38, 1003–1018. [Google Scholar] [CrossRef]
- Holcomb, L.E.; Rowe, P.; O’Neill, C.C.; DeWitt, E.A.; Kolwicz, S.C., Jr. Sex differences in endurance exercise capacity and skeletal muscle lipid metabolism in mice. Physiol. Rep. 2022, 10, e15174. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Ferrucci, L.; Gonzalez-Freire, M.; Kosmac, K.; Leeuwenburgh, C.; Peterson, C.A.; Saini, S.; Sufit, R. Skeletal Muscle Pathology in Peripheral Artery Disease: A Brief Review. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Miura, M.; Kodama, J.; Ahmed, S.M.; Itokawa, Y. Role of iron in oxidative stress in skeletal muscle atrophied by immobilization. Pflügers Arch. Eur. J. Physiol. 1992, 421, 295–297. [Google Scholar] [CrossRef]
- Leermakers, P.A.; Remels, A.H.V.; Zonneveld, M.I.; Rouschop, K.M.A.; Schols, A.M.W.J.; Gosker, H.R. Iron deficiency-induced loss of skeletal muscle mitochondrial proteins and respiratory capacity; the role of mitophagy and secretion of mitochondria-containing vesicles. FASEB J. 2020, 34, 6703–6717. [Google Scholar] [CrossRef]
- Che, Y.; Li, J.; Wang, P.; Yu, W.; Lin, J.; Su, Z.; Ye, F.; Zhang, Z.; Xu, P.; Xie, Z.; et al. Iron deficiency–induced ferritinophagy impairs skeletal muscle regeneration through RNF20-mediated H2Bub1 modification. Sci. Adv. 2023, 9, eadf4345. [Google Scholar] [CrossRef]
- Fefelova, N.; Wongjaikam, S.; Pamarthi, S.H.; Siri-Angkul, N.; Comollo, T.; Kumari, A.; Garg, V.; Ivessa, A.; Chattipakorn, S.C.; Chattipakorn, N.; et al. Deficiency of mitochondrial calcium uniporter abrogates iron overload-induced cardiac dysfunction by reducing ferroptosis. Basic Res. Cardiol. 2023, 118, 1–14. [Google Scholar] [CrossRef]
- Polonifi, A.; Politou, M.; Kalotychou, V.; Xiromeritis, K.; Tsironi, M.; Berdoukas, V.; Vaiopoulos, G.; Aessopos, A. Iron metabolism gene expression in human skeletal muscle. Blood Cells Mol. Dis. 2010, 45, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.Z. Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar] [PubMed]
- Henderson, B.R. Iron regulatory proteins 1 and 2. BioEssays News Rev. Mol. Cell. Dev. Biol. 1996, 18, 739–746. [Google Scholar] [CrossRef]
- Xu, J.; Hwang, J.C.; Lees, H.A.; Wohlgemuth, S.E.; Knutson, M.D.; Judge, A.R.; Dupont-Versteegden, E.E.; Marzetti, E.; Leeuwenburgh, C. Long-term perturbation of muscle iron homeostasis following hindlimb suspension in old rats is associated with high levels of oxidative stress and impaired recovery from atrophy. Exp. Gerontol. 2012, 47, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Chen, A.; Li, L.; Liang, Q.; Wang, S.; Dong, Q.; Fu, M.; Lan, Z.; Li, Y.; Liu, X.; et al. Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int. 2022, 102, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Vulinović, M.P.; Turčić, P.; Micek, V.; Ljubojević, M. Light and heavy ferritin chain expression in the liver and kidneys of Wistar rats: Aging, sex differences, and impact of gonadectomy. Arch. Ind. Hyg. Toxicol. 2022, 73, 48–61. [Google Scholar] [CrossRef]
- Dang, Y.; He, Q.; Yang, S.; Sun, H.; Liu, Y.; Li, W.; Tang, Y.; Zheng, Y.; Wu, T. FTH1- and SAT1-Induced Astrocytic Ferroptosis Is Involved in Alzheimer’s Disease: Evidence from Single-Cell Transcriptomic Analysis. Pharmaceuticals 2022, 15, 1177. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Kong, B.; Fang, J.; Qin, T.; Dai, C.; Shuai, W.; Huang, H. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered 2021, 12, 9367–9376. [Google Scholar] [CrossRef] [PubMed]
- Patino, E.; Bhatia, D.; Vance, S.Z.; Antypiuk, A.; Uni, R.; Campbell, C.; Castillo, C.G.; Jaouni, S.; Vinchi, F.; Choi, M.E.; et al. Iron therapy mitigates chronic kidney disease progression by regulating intracellular iron status of kidney macrophages. J. Clin. Investig. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Li, W.; Wang, Y.; Zhao, M.; Liu, K.; Yang, Y.; Teng, S.; Zhang, N.; Min, L.; Li, P.; et al. M2 Macrophage-Derived Exosomal Ferritin Heavy Chain Promotes Colon Cancer Cell Proliferation. Biol. Trace Element Res. 2022, 201, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Zheng, H.; Li, Y.; Xu, W.; Wu, C.; Xu, J.; Li, S.; Zhang, L.; Dong, L. SMURF1 controls the PPP3/calcineurin complex and TFEB at a regulatory node for lysosomal biogenesis. Autophagy 2024, 20, 735–751. [Google Scholar] [CrossRef]
- Xia, Q.; Li, Y.; Han, D.; Dong, L. SMURF1, a promoter of tumor cell progression? Cancer Gene Ther. 2021, 28, 551–565. [Google Scholar] [CrossRef]
- Sangadala, S.; Metpally, R.P.; Reddy, B.V. Molecular interaction between Smurf1 WW2 domain and PPXY motifs of Smad1, Smad5, and Smad6—Modeling and analysis. J. Biomol. Struct. Dyn. 2007, 25, 11–23. [Google Scholar] [CrossRef]
- Wei, R.; Li, B.; Guo, J.; Li, M.; Zhu, R.; Yang, X.; Gao, R. Smurf1 targets Securin for ubiquitin-dependent degradation and regulates the metaphase-to-anaphase transition. Cell. Signal. 2017, 38, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chen, H.; Sun, J.; Chen, C.; Zhao, J.; Wang, Y.L.; Anderson, K.D.; Warburton, D. Overexpression of Smurf1 negatively regulates mouse embryonic lung branching morphogenesis by specifically reducing Smad1 and Smad5 proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L293-300. [Google Scholar] [CrossRef]
- Niu, H.; Bi, F.; Zhao, W.; Xu, Y.; Han, Q.; Guo, W. Smurf1 regulates ameloblast polarization by ubiquitination-mediated degradation of RhoA. Cell Prolif. 2023, 56, e13387. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhu, Z.; Zhang, H.; Peng, Y.; Liu, J.; Lu, H.; Li, J.; Liang, L.; Xia, S.; Wang, Q.; et al. GDF11 contributes to hepatic hepcidin (HAMP) inhibition through SMURF1-mediated BMP-SMAD signalling suppression. Br. J. Haematol. 2020, 188, 321–331. [Google Scholar] [CrossRef]
- Gu, X.; Song, Y.; Liu, X.; Cheng, Z.; Min, J.; Zhang, Y. METTL14-Mediated m6A Modification of TUG1 Represses Ferroptosis in Alzheimer’s Disease via Inhibiting GDF15 Ubiquitination. Front. Biosci. 2024, 29, 298. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Yamamoto, H.; Sakamaki, Y.; Saito, C. NCOA4 drives ferritin phase separation to facilitate macroferritinophagy and microferritinophagy. J. Cell Biol. 2022, 221, e202203102. [Google Scholar] [CrossRef] [PubMed]
- Koganti, P.; Levy-Cohen, G.; Blank, M. Smurfs in Protein Homeostasis, Signaling, and Cancer. Front. Oncol. 2018, 8, 295. [Google Scholar] [CrossRef]
- Xia, C.; Wang, Y.; Jiang, T.; Hu, Y.; Chen, Y.; Ma, X.; Zhang, X.; Gao, Y. Slo1 deficiency impaired skeletal muscle regeneration and slow-twitch fibre formation. J. Cachexia Sarcopenia Muscle 2023, 14, 1737–1752. [Google Scholar] [CrossRef]
- Shen, X.; Cui, C.; Tang, S.; Han, S.; Zhang, Y.; Xia, L.; Tan, B.; Ma, M.; Kang, H.; Yu, J.; et al. MyoG-enhanced circGPD2 regulates chicken skeletal muscle development by targeting miR-203a. Int. J. Biol. Macromol. 2022, 222, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, X.; Li, W.; Yu, C.; Qiu, M.; Zhang, Z.; Hu, C.; Zhu, S.; Yang, L.; Pen, H.; Song, X.; et al. SMURF1-Induced Ubiquitination of FTH1 Disrupts Iron Homeostasis and Suppresses Myogenesis. Int. J. Mol. Sci. 2025, 26, 1390. https://doi.org/10.3390/ijms26031390
Xiong X, Li W, Yu C, Qiu M, Zhang Z, Hu C, Zhu S, Yang L, Pen H, Song X, et al. SMURF1-Induced Ubiquitination of FTH1 Disrupts Iron Homeostasis and Suppresses Myogenesis. International Journal of Molecular Sciences. 2025; 26(3):1390. https://doi.org/10.3390/ijms26031390
Chicago/Turabian StyleXiong, Xia, Wen Li, Chunlin Yu, Mohan Qiu, Zengrong Zhang, Chenming Hu, Shiliang Zhu, Li Yang, Han Pen, Xiaoyan Song, and et al. 2025. "SMURF1-Induced Ubiquitination of FTH1 Disrupts Iron Homeostasis and Suppresses Myogenesis" International Journal of Molecular Sciences 26, no. 3: 1390. https://doi.org/10.3390/ijms26031390
APA StyleXiong, X., Li, W., Yu, C., Qiu, M., Zhang, Z., Hu, C., Zhu, S., Yang, L., Pen, H., Song, X., Chen, J., Xia, B., Han, S., & Yang, C. (2025). SMURF1-Induced Ubiquitination of FTH1 Disrupts Iron Homeostasis and Suppresses Myogenesis. International Journal of Molecular Sciences, 26(3), 1390. https://doi.org/10.3390/ijms26031390