
A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective


Abstract
:1. Introduction
2. Methodology
2.1. Aims and Research Planning
2.2. Search Strategy and Inclusion and Exclusion Criteria
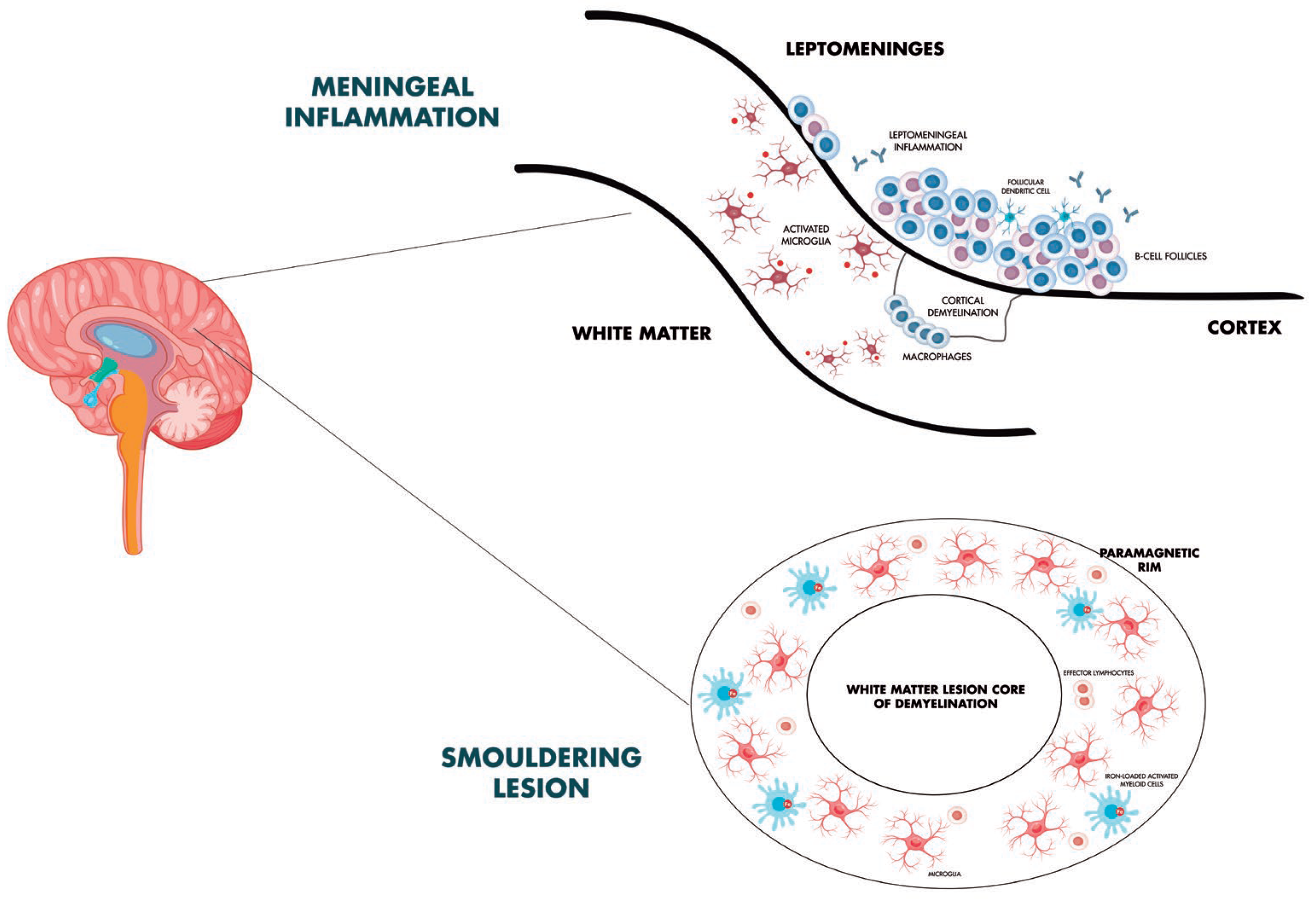
3. Smoldering Biology in Multiple Sclerosis
4. Radiological Expression of PIRA and Molecular Correlates
5. Meningeal Inflammation, Subpial Cortical Damage, and Focus on Microglia and Diffuse White Matter Pathology
6. Adaptive Immunity and PIRA: Role of T Cells and B Cells
7. Fluid Biomarkers and PIRA Phenomena
8. Therapies and PIRA

{kind=link}
| Authors | Study Design | Population | Interpretation of Results |
|---|---|---|---|
| Graf et al. [87] | Retrospective chart review study | 184 RRMS patients | Patients who are started on natalizumab early in the course of their disease, typically to treat an aggressive clinical presentation, are more likely to experience early confirmed progression independent of relapse activity. |
| Iaffaldano et al. [88] | Retrospective cohort study | 11,871 MS patients (BMSD) | DMTs should be commenced within 1.2 years from the disease onset to reduce the risk of disability accumulation over the long term. |
| Portaccio et al. [89] | Retrospective cohort study | 5169 MS patients (CIS, RRMS) (RISM) | Longer exposure to DMT is associated with a lower risk of both progression independent of relapse activity and relapse-associated worsening events. |
| Iaffaldano et al. [92] | Retrospective cohort study | Total population: 770 MS patients. Matched cohort: 195 patients treated with ocrelizumab, 195 with natalizumab (RISM) | Natalizumab and ocrelizumab strongly suppress RAW events and, in the short term, the risk of achieving PIRA events, EDSS 4.0 and 6.0 disability milestones is not significantly different. |
| Puthenparampil et al. [93] | Observational retrospective study | Total population: 160 MS patients. Matched cohort: 32 patients pediatric-onset MS and 64 with adult-onset MS | In naïve patients treated with natalizumab, PIRA was never observed in pediatric-onset MS, while a small percentage of adult-onset MS (12.5%) had PIRA events during the follow-up. |
| Chisari et al. [94] | Retrospective cohort study | Total population: 5321 SPMS patients. Matched cohort: 421 MS patients treated with natalizumab and 353 with interferon-beta 1b (RISM) | The proportion of patients who developed PIRA at 48 months is significantly higher in interferon beta-1b group compared to the natalizumab-treated cohort. Patients treated with IFNb-1b are 1.64 times more to likely to develop PIRA |
| Cross et al. [80] | Cohort study assessed data from 2 prospective MS cohorts | Test cohort: 131 MS patients Confirmation cohort: 68 MS patients. | Ocrelizumab reduced CSF measures of acute inflammation, including lymphocyte measures sTACI, sCD27, sBCMA, and chemokine/cytokine measures CXCL13 and CXCL10. Neuroaxonal injury measure NfH and glial measures sTREM2 and YKL-40 resulted modestly reduced. |
| Bajrami et al. [96] | Observational, prospective, longitudinal study | 95 RRMS | Compared to fingolimod, ocrelizumab-treated patients experience fewer new white matter lesions and lower deep grey matter volume loss, lower global cortical thickness change, and reduced cortical thinning/volume loss in several regions of interest. |
| Eisele et al. [97] | Retrospective study | 27 MS patients | Patients on fingolimod, dimethyl fumarate, and ocrelizumab have a considerably lower 2-year follow-up rate of T1/T2 ratio of iron rim lesions. than those not taking DMTs. |
| Elliott et al. [98] | PPMS study population of the ORATORIO trial | ITT population (n = 732); SEL analytical population (n = 555) | Ocrelizumab reduces longitudinal measures of chronic lesion activity such as T1 hypointense lesion volume accumulation and mean normalized T1 signal intensity decrease both in slowly expanding/evolving and non-slowly expanding/evolving lesions. |
| Maggi et al. [99] | Retrospective analysis and imaging, laboratory, and clinical data prospectively collected | 72 MS patients | Despite predicted effects on inflammatory networks related to microglia in CAL, anti-CD20 monoclonal antibodies failed to fully resolve paramagnetic rim lesions after a 2-year MRI follow-up |
| Preziosa et al. [100] | Single centre, prospective, longitudinal, open-label, non-randomized cohort study | 52 MS patients | Higher SEL number and volume is observed in the fingolimod vs. natalizumab group. Longitudinally, non-SEL MTR increased in both treatment groups. T1 signal intensity decreased in SELs with both treatments and increased in natalizumab non-SELs. |
| Montobbio et al. [102] | Retrospective study | 1405 MS patients | Across ages, patients diagnosed in more recent times had lower PIRA and RAW than those diagnosed in earlier periods. Patients diagnosed in later years had a significantly higher contribution of PIRA in EDSS progression. |
| Cortese et al. [105] | MRI data from the CLARITY study | Treatment group: 267 MS patients Placebo group: 265 MS patients | In the first six months of treatment, patients on cladribine experienced more GM and WM volume loss than those on placebo, most likely as a result of pseudoatrophy. Nonetheless, GM volume loss was considerably less in cladribine-treated patients than in placebo-treated group throughout the course of 6–24 months. |
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Jakimovski, D.; Bittner, S.; Zivadinov, R.; Morrow, S.A.; Benedict, R.H.; Zipp, F.; Weinstock-Guttman, B. Multiple sclerosis. Lancet 2024, 403, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Dalla Costa, G.; Moiola, L. Newly approved agents for relapsing remitting multiple sclerosis: How real-world evidence compares with randomized clinical trials? Expert Rev. Neurother. 2021, 21, 21–34. [Google Scholar] [CrossRef]
- Lublin, F.D.; Häring, D.A.; Ganjgahi, H.; Ocampo, A.; Hatami, F.; Čuklina, J.; Aarden, P.; Dahlke, F.; Arnold, D.L.; Wiendl, H.; et al. How patients with multiple sclerosis acquire disability. Brain 2022, 145, 3147–3161. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; Cohen, J.A.; Correale, J.; Graves, J.; Marrie, R.A.; Montalban, X.; Yong, V.W.; Thompson, A.J.; et al. Multiple sclerosis progression: Time for a new mechanism-driven framework. Lancet Neurol. 2023, 22, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Preziosa, P.; Scalfari, A.; Colato, E.; Marastoni, D.; Absinta, M.; Battaglini, M.; De Stefano, N.; Di Filippo, M.; Hametner, S.; et al. Determinants and biomarkers of progression independent of relapses in multiple sclerosis. Ann. Neurol. 2024, 96, 1–20. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Sellebjerg, F.; Hartung, H.P.; Montalban, X.; Comi, G.; Tintoré, M. The apparently milder course of multiple sclerosis: Changes in the diagnostic criteria, therapy and natural history. Brain 2020, 143, 2637–2652. [Google Scholar] [CrossRef]
- Portaccio, E.; Magyari, M.; Havrdova, E.K.; Ruet, A.; Brochet, B.; Scalfari, A.; Di Filippo, M.; Tur, C.; Montalban, X.; Amato, M.P. Multiple sclerosis: Emerging epidemiological trends and redefining the clinical course. Lancet Reg. Health Eur. 2024, 44, 100977. [Google Scholar] [CrossRef]
- Kappos, L.; Wolinsky, J.S.; Giovannoni, G.; Arnold, D.L.; Wang, Q.; Bernasconi, C.; Model, F.; Koendgen, H.; Manfrini, M.; Belachew, S.; et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020, 77, 1132–1140. [Google Scholar] [CrossRef]
- Giovannoni, G.; Popescu, V.; Wuerfel, J.; Hellwig, K.; Iacobaeus, E.; Jensen, M.B.; García-Domínguez, J.M.; Sousa, L.; De Rossi, N.; Hupperts, R.; et al. Smouldering multiple sclerosis: The ‘real MS’. Ther. Adv. Neurol. Disord. 2022, 15, 17562864211066751. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A.C. Treatment of multiple sclerosis: A review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Cagol, A.; Lorscheider, J.; Tsagkas, C.; Benkert, P.; Yaldizli, Ö.; Kuhle, J.; Derfuss, T.; Sormani, M.P.; Thompson, A.; et al. Harmonizing definitions for progression independent of relapse activity in multiple sclerosis: A systematic review. JAMA Neurol. 2023, 80, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, O.; Barkhof, F.; Calabrese, M.; De Stefano, N.; Eshaghi, A.; Filippi, M.; Gasperini, C.; Granziera, C.; Kappos, L.; Rocca, M.A.; et al. Using the Progression Independent of Relapse Activity Framework to Unveil the Pathobiological Foundations of Multiple Sclerosis. Neurology 2024, 103, e209444. [Google Scholar] [CrossRef] [PubMed]
- Sharrad, D.; Chugh, P.; Slee, M.; Bacchi, S. Defining progression independent of relapse activity (PIRA) in adult patients with relapsing multiple sclerosis: A systematic review. Mult. Scler. Relat. Disord. 2023, 78, 104899. [Google Scholar] [CrossRef]
- Iaffaldano, P.; Portaccio, E.; Lucisano, G.; Simone, M.; Manni, A.; Guerra, T.; Paolicelli, D.; Betti, M.; De Meo, E.; Pastò, L.; et al. Multiple Sclerosis Progression and Relapse Activity in Children. JAMA Neurol. 2024, 81, 50–58. [Google Scholar] [CrossRef]
- Portaccio, E.; Betti, M.; De Meo, E.; Addazio, I.; Pastò, L.; Razzolini, L.; Totaro, R.; Spitaleri, D.; Lugaresi, A.; Cocco, E.; et al. Progression independent of relapse activity in relapsing multiple sclerosis: Impact and relationship with secondary progression. J. Neurol. 2024, 271, 5074–5082. [Google Scholar] [CrossRef]
- Simone, M.; Lucisano, G.; Guerra, T.; Paolicelli, D.; Rocca, M.A.; Brescia Morra, V.; Patti, F.; Annovazzi, P.; Gasperini, C.; De Luca, G.; et al. Disability trajectories by progression independent of relapse activity status differ in pediatric, adult and late-onset multiple sclerosis. J. Neurol. 2024, 271, 6782–6790. [Google Scholar] [CrossRef]
- Prosperini, L.; Ruggieri, S.; Haggiag, S.; Tortorella, C.; Gasperini, C. Disability patterns in multiple sclerosis: A meta-analysis on RAW and PIRA in the real-world context. Mult. Scler. J. 2024, 30, 1309–1321. [Google Scholar] [CrossRef]
- Tur, C.; Carbonell-Mirabent, P.; Cobo-Calvo, Á.; Otero-Romero, S.; Arrambide, G.; Midaglia, L.; Castilló, J.; Vidal-Jordana, Á.; Rodríguez-Acevedo, B.; Zabalza, A.; et al. Association of Early Progression Independent of Relapse Activity with Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023, 80, 151–160. [Google Scholar] [CrossRef]
- Cagol, A.; Schaedelin, S.; Barakovic, M.; Benkert, P.; Todea, R.A.; Rahmanzadeh, R.; Galbusera, R.; Lu, P.J.; Weigel, M.; Melie-Garcia, L.; et al. Association of Brain Atrophy with Disease Progression Independent of Relapse Activity in Patients With Relapsing Multiple Sclerosis. JAMA Neurol. 2022, 79, 682–692. [Google Scholar] [CrossRef]
- Comi, G.; Dalla Costa, G.; Stankoff, B.; Hartung, H.P.; Soelberg Sørensen, P.; Vermersch, P.; Leocani, L. Assessing disease progression and treatment response in progressive multiple sclerosis. Nat. Rev. Neurol. 2024, 20, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Monaco, S.; Nicholas, R.; Reynolds, R.; Magliozzi, R. Intrathecal inflammation in progressive multiple sclerosis. Int. J. Mol. Sci. 2020, 21, 8217. [Google Scholar] [CrossRef] [PubMed]
- University of California, San Francisco MS-EPIC Team; Cree, B.A.C.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H.; et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar]
- Scalfari, A.; Traboulsee, A.; Oh, J.; Airas, L.; Bittner, S.; Calabrese, M.; Garcia Dominguez, J.M.; Granziera, C.; Greenberg, B.; Hellwig, K.; et al. Smouldering-Associated Worsening in Multiple Sclerosis: An International Consensus Statement on Definition, Biology, Clinical Implications, and Future Directions. Ann. Neurol. 2024, 96, 826–845. [Google Scholar] [CrossRef]
- Giovannoni, G.; Hawkes, C.H.; Lechner-Scott, J.; Levy, M.; Yeh, E.A. Smouldering-associated worsening or SAW: The next therapeutic challenge in managing multiple sclerosis. Mult. Scler. Relat. Disord. 2024, 30, 106194. [Google Scholar] [CrossRef]
- Di Filippo, M.; Gaetani, L.; Centonze, D.; Hegen, H.; Kuhle, J.; Teunissen, C.E.; Tintoré, M.; Villar, L.M.; Willemse, E.A.J.; Zetterberg, H.; et al. Fluid biomarkers in multiple sclerosis: From current to future applications. Lancet Reg. Health Eur. 2024, 44, 101009. [Google Scholar] [CrossRef]
- Hamzaoui, M.; Garcia, J.; Boffa, G.; Lazzarotto, A.; Absinta, M.; Ricigliano, V.A.G.; Soulier, T.; Tonietto, M.; Gervais, P.; Bissery, A.; et al. Positron Emission Tomography with [18F]-DPA-714 Unveils a Smoldering Component in Most Multiple Sclerosis Lesions which Drives Disease Progression. Ann. Neurol. 2023, 94, 366–383. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef]
- Trobisch, T.; Zulji, A.; Stevens, N.A.; Schwarz, S.; Wischnewski, S.; Öztürk, M.; Perales-Patón, J.; Haeussler, M.; Saez-Rodriguez, J.; Velmeshev, D.; et al. Cross-regional homeostatic and reactive glial signatures in multiple sclerosis. Acta Neuropathol. 2022, 144, 987–1003. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Nicholas, R.; Cruciani, C.; Castellaro, M.; Romualdi, C.; Rossi, S.; Pitteri, M.; Benedetti, M.D.; Gajofatto, A.; et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann. Neurol. 2018, 83, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, V.; Crescenzo, F.; Turano, E.; Guandalini, M.; Bertolazzo, M.; Ziccardi, S.; Virla, F.; Camera, V.; Marastoni, D.; Tamanti, A.; et al. The contribution of tumor necrosis factor to multiple sclerosis: A possible role in progression independent of relapse? J. Neuroinflammation 2024, 21, 209. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.P.; Guevara, C.; Olesen, M.A.; Orellana, J.A.; Quintanilla, R.A.; Ortiz, F.C. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants 2022, 11, 1146. [Google Scholar] [CrossRef] [PubMed]
- Husseini, L.; Geladaris, A.; Weber, M.S. Toward identifying key mechanisms of progression in multiple sclerosis. Trends Neurosci. 2024, 47, 58–70. [Google Scholar] [CrossRef]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Witte, M.E.; Bø, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.; De Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 2009, 219, 193–204. [Google Scholar] [CrossRef]
- Carotenuto, A.; Cacciaguerra, L.; Pagani, E.; Preziosa, P.; Filippi, M.; Rocca, M.A. Glymphatic system impairment in multiple sclerosis: Relation with brain damage and disability. Brain 2022, 145, 2785–2795. [Google Scholar] [CrossRef]
- Alghanimy, A.; Work, L.M.; Holmes, W.M. The glymphatic system and multiple sclerosis: An evolving connection. Mult. Scler. Relat. Disord. 2024, 83, 105456. [Google Scholar] [CrossRef]
- Aharoni, R.; Eilam, R.; Arnon, R. Astrocytes in Multiple Sclerosis-Essential Constituents with Diverse Multifaceted Functions. Int. J. Mol. Sci. 2021, 22, 5904. [Google Scholar] [CrossRef]
- Bagnato, F.; Sati, P.; Hemond, C.C.; Elliott, C.; Gauthier, S.A.; Harrison, D.M.; Mainero, C.; Oh, J.; Pitt, D.; Shinohara, R.T.; et al. Imaging chronic active lesions in multiple sclerosis: A consensus statement. Brain 2024, 147, 2913–2933. [Google Scholar] [CrossRef]
- Ludwig, N.; Cucinelli, S.; Hametner, S.; Muckenthaler, M.U.; Schirmer, L. Iron scavenging and myeloid cell polarization. Trends Immunol. 2024, 45, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.; Krajnc, N.; Dal-Bianco, A.; Riedl, C.J.; Zrzavy, T.; Lerma-Martin, C.; Kasprian, G.; Weber, C.E.; Pezzini, F.; Leutmezer, F.; et al. Myeloid cell iron uptake pathways and paramagnetic rim formation in multiple sclerosis. Acta Neuropathol. 2023, 146, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol. 2019, 76, 699–709. [Google Scholar] [CrossRef]
- Maggi, P.; Kuhle, J.; Schädelin, S.; van der Meer, F.; Weigel, M.; Galbusera, R.; Mathias, A.; Lu, P.J.; Rahmanzadeh, R.; Benkert, P.; et al. Chronic White Matter Inflammation and Serum Neurofilament Levels in Multiple Sclerosis. Neurology 2021, 97, e543–e553. [Google Scholar] [CrossRef]
- Wittayer, M.; Weber, C.E.; Platten, M.; Schirmer, L.; Gass, A.; Eisele, P. Spatial distribution of multiple sclerosis iron rim lesions and their impact on disability. Mult. Scler. Relat. Disord. 2022, 64, 103967. [Google Scholar] [CrossRef]
- Okar, S.V.; Dieckhaus, H.; Beck, E.S.; Gaitán, M.I.; Norato, G.; Pham, D.L.; Absinta, M.; Cortese, I.C.; Fletcher, A.; Jacobson, S.; et al. Highly Sensitive 3-Tesla Real Inversion Recovery MRI Detects Leptomeningeal Contrast Enhancement in Chronic Active Multiple Sclerosis. Investig. Radiol. 2024, 59, 243–251. [Google Scholar] [CrossRef]
- Preziosa, P.; Pagani, E.; Meani, A.; Moiola, L.; Rodegher, M.; Filippi, M.; Rocca, M.A. Slowly Expanding Lesions Predict 9-Year Multiple Sclerosis Disease Progression. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1139. [Google Scholar] [CrossRef]
- Cagol, A.; Benkert, P.; Melie-Garcia, L.; Schaedelin, S.A.; Leber, S.; Tsagkas, C.; Barakovic, M.; Galbusera, R.; Lu, P.J.; Weigel, M.; et al. Association of spinal cord atrophy and brain paramagnetic rim lesions with progression independent of relapse activity in people with MS. Neurology 2024, 102, e207768. [Google Scholar] [CrossRef]
- Wenzel, N.; Wittayer, M.; Weber, C.E.; Platten, M.; Gass, A.; Eisele, P. Multiple sclerosis iron rim lesions are linked to impaired cervical spinal cord integrity using the T1/T2-weighted ratio. J. Neuroimaging 2023, 33, 240–248. [Google Scholar] [CrossRef]
- Weber, C.E.; Kramer, J.; Wittayer, M.; Gregori, J.; Randoll, S.; Weiler, F.; Heldmann, S.; Roßmanith, C.; Platten, M.; Gass, A.; et al. Association of iron rim lesions with brain and cervical cord volume in relapsing multiple sclerosis. Eur. Radiol. 2021, 31, 6789–6902. [Google Scholar] [CrossRef]
- Herranz, E.; Treaba, C.A.; Barletta, V.T.; Mehndiratta, A.; Ouellette, R.; Sloane, J.A.; Ionete, C.; Babu, S.; Mastantuono, M.; Magon, S.; et al. Characterization of cortico-meningeal translocator protein expression in multiple sclerosis. Brain 2024, 147, 2566–2578. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, Y.; Hagiwara, A.; Hoshino, Y.; Nakaya, M.; Kamagata, K.; Cossu, D.; Yokoyama, K.; Aoki, S.; Hattori, N. The glymphatic system as a potential biomarker and therapeutic target in secondary progressive multiple sclerosis. Mult. Scler. Relat. Disord. 2024, 83, 105437. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Calabrese, M.; Reynolds, R. Meningeal inflammation as a driver of cortical grey matter pathology and clinical progression in multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signaling in cortical grey matter in multiple sclerosis. J. Neuroinflammation 2019, 16, 259. [Google Scholar] [CrossRef]
- Tham, M.; Frischer, J.M.; Weigand, S.D.; Fitz-Gibbon, P.D.; Webb, S.M.; Guo, Y.; Adiele, R.C.; Robinson, C.A.; Brück, W.; Lassmann, H.; et al. Iron heterogeneity in early active multiple sclerosis lesions. Ann. Neurol. 2021, 89, 498–510. [Google Scholar] [CrossRef]
- van den Bosch, A.M.R.; van der Poel, M.; Fransen, N.L.; Vincenten, M.C.; Bobeldijk, A.M.; Jongejan, A.; Engelenburg, H.J.; Moerland, P.D.; Smolders, J.; Huitinga, I.; et al. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat. Commun. 2024, 15, 1667. [Google Scholar] [CrossRef]
- Gallego-Delgado, P.; James, R.; Browne, E.; Meng, J.; Umashankar, S.; Tan, L.; Picon, C.; Mazarakis, N.D.; Faisal, A.A.; Howell, O.W.; et al. Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biol. 2020, 18, e3001008. [Google Scholar] [CrossRef]
- Preziosa, P.; Pagani, E.; Meani, A.; Storelli, L.; Margoni, M.; Yudin, Y.; Tedone, N.; Biondi, D.; Rubin, M.; Rocca, M.A.; et al. Chronic Active Lesions and Larger Choroid Plexus Explain Cognition and Fatigue in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2024, 11, e200205. [Google Scholar] [CrossRef]
- Zhan, J.; Kipp, M.; Han, W.; Kaddatz, H. Ectopic lymphoid follicles in progressive multiple sclerosis: From patients to animal models. Immunology 2021, 164, 450–466. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef]
- Reali, C.; Magliozzi, R.; Roncaroli, F.; Nicholas, R.; Howell, O.W.; Reynolds, R. B cell rich meningeal inflammation associates with increased spinal cord pathology in multiple sclerosis. Brain Pathol. 2020, 30, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; Lenhart, A.; Rosenwald, A.; Monoranu, C.M.; Berberich-Siebelt, F. Lymphoid aggregates in the CNS of progressive multiple sclerosis patients lack regulatory T cells. Front. Immunol. 2019, 10, 3090. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis—From targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Bar-Or, A.; Lassmann, H.; Uccelli, A.; Hartung, H.P.; Montalban, X.; Sørensen, P.S.; Hohlfeld, R.; Hauser, S.L.; Expert Panel of the 27th Annual Meeting of the European Charcot Foundation. Role of B Cells in Multiple Sclerosis and Related Disorders. Ann. Neurol. 2021, 89, 13–23. [Google Scholar] [CrossRef]
- Margoni, M.; Preziosa, P.; Filippi, M.; Rocca, M.A. Anti-CD20 therapies for multiple sclerosis: Current status and future perspectives. J. Neurol. 2022, 269, 1316–1334. [Google Scholar] [CrossRef]
- Krajnc, N.; Bsteh, G.; Berger, T.; Mares, J.; Hartung, H.P. Monoclonal Antibodies in the Treatment of Relapsing Multiple Sclerosis: An Overview with Emphasis on Pregnancy, Vaccination, and Risk Management. Neurotherapeutics 2022, 19, 753–773. [Google Scholar] [CrossRef]
- Afshar, B.; Khalifehzadeh-Esfahani, Z.; Seyfizadeh, N.; Rezaei Danbaran, G.; Hemmatzadeh, M.; Mohammadi, H. The role of immune regulatory molecules in multiple sclerosis. J. Neuroimmunol. 2019, 337, 577061. [Google Scholar] [CrossRef]
- Sarkar, S.K.; Willson, A.M.L.; Jordan, M.A. The Plasticity of Immune Cell Response Complicates Dissecting the Underlying Pathology of Multiple Sclerosis. J. Immunol. Res. 2024, 2024, 5383099. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Li, H.; Zheng, C.; Han, J.; Zhu, J.; Liu, S.; Jin, T. PD-1/PD-L1 Axis as a Potential Therapeutic Target for Multiple Sclerosis: A T Cell Perspective. Front. Cell. Neurosci. 2021, 15, 716747. [Google Scholar] [CrossRef]
- Planas, R.; Metz, I.; Martin, R.; Sospedra, M. Detailed Characterization of T Cell Receptor Repertoires in Multiple Sclerosis Brain Lesions. Front. Immunol. 2018, 9, 509. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Fransen, N.L.; Touil, H.; Michailidou, I.; Huitinga, I.; Gommerman, J.L.; Bar-Or, A.; Ramaglia, V. Accumulation of meningeal lymphocytes correlates with white matter lesion activity in progressive multiple sclerosis. JCI Insight. 2022, 7, e151683. [Google Scholar] [CrossRef] [PubMed]
- Fransen, N.L.; Hsiao, C.C.; van der Poel, M.; Engelenburg, H.J.; Verdaasdonk, K.; Vincenten, M.C.J.; Remmerswaal, E.B.M.; Kuhlmann, T.; Mason, M.R.J.; Hamann, J.; et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 2020, 143, 1714–1730. [Google Scholar] [CrossRef] [PubMed]
- Kosa, P.; Barbour, C.; Varosanec, M.; Wichman, A.; Sandford, M.; Greenwood, M.; Bielekova, B. Molecular models of multiple sclerosis severity identify heterogeneity of pathogenic mechanisms. Nat. Commun. 2022, 13, 7670. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Toscano, S.; Oteri, V.; Chisari, C.G.; Finocchiaro, C.; Lo Fermo, S.; Valentino, P.; Bertolotto, A.; Zappia, M.; Patti, F. Cerebrospinal fluid neurofilament light chains predict early disease activity in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2023, 80, 105131. [Google Scholar] [CrossRef]
- Abdelhak, A.; Benkert, P.; Schaedelin, S.; Boscardin, W.J.; Cordano, C.; Oechtering, J.; Ananth, K.; Granziera, C.; Melie-Garcia, L.; Montes, S.C.; et al. Neurofilament light chain elevation and disability progression in multiple sclerosis. JAMA Neurol. 2023, 80, 1317. [Google Scholar] [CrossRef]
- Magliozzi, R.; Pitteri, M.; Ziccardi, S.; Pisani, A.I.; Montibeller, L.; Marastoni, D.; Rossi, S.; Mazziotti, V.; Guandalini, M.; Dapor, C.; et al. CSF parvalbumin levels reflect interneuron loss linked with cortical pathology in multiple sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 534–547. [Google Scholar] [CrossRef]
- Ziccardi, S.; Tamanti, A.; Ruggieri, C.; Guandalini, M.; Marastoni, D.; Camera, V.; Montibeller, L.; Mazziotti, V.; Rossi, S.; Calderone, M.; et al. CSF Parvalbumin Levels at Multiple Sclerosis Diagnosis Predict Future Worse Cognition, Physical Disability, Fatigue, and Gray Matter Damage. Neurol. Neuroimmunol. Neuroinflamm. 2024, 11, e200301. [Google Scholar] [CrossRef]
- Cross, A.H.; Gelfand, J.M.; Thebault, S.; Bennett, J.L.; von Büdingen, H.C.; Cameron, B.; Carruthers, R.; Edwards, K.; Fallis, R.; Gerstein, R.; et al. Emerging Cerebrospinal Fluid Biomarkers of Disease Activity and Progression in Multiple Sclerosis. JAMA Neurol. 2024, 81, 373–383. [Google Scholar] [CrossRef]
- Meier, S.; Willemse, E.A.J.; Schaedelin, S.; Oechtering, J.; Lorscheider, J.; Melie-Garcia, L.; Cagol, A.; Barakovic, M.; Galbusera, R.; Subramaniam, S.; et al. Serum glial fibrillary acidic protein compared with neurofilament light chain as a biomarker for disease progression in multiple sclerosis. JAMA Neurol. 2023, 80, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, F.; Pisani, A.; Mazziotti, V.; Marastoni, D.; Tamanti, A.; Borroni, E.; Magon, S.; Zinnhardt, B.; Magliozzi, R.; Calabrese, M. Intrathecal versus Peripheral Inflammatory Protein Profile in MS Patients at Diagnosis: A Comprehensive Investigation on Serum and CSF. Int. J. Mol. Sci. 2023, 24, 3768. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Scalfari, A.; Pisani, A.I.; Ziccardi, S.; Marastoni, D.; Pizzini, F.B.; Bajrami, A.; Tamanti, A.; Guandalini, M.; Bonomi, S.; et al. The CSF Profile Linked to Cortical Damage Predicts Multiple Sclerosis Activity. Ann. Neurol. 2020, 88, 562–573. [Google Scholar] [CrossRef]
- Leurs, C.E.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Piehl, F.; Uitdehaag, B.M.; Killestein, J.; van Horssen, J.; et al. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult. Scler. 2018, 24, 472–480. [Google Scholar] [CrossRef]
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflammation 2016, 13, 36. [Google Scholar] [CrossRef]
- Gärtner, J.; Hauser, S.L.; Bar-Or, A.; Montalban, X.; Cohen, J.A.; Cross, A.H.; Deiva, K.; Ganjgahi, H.; Häring, D.A.; Li, B.; et al. Efficacy and safety of ofatumumab in recently diagnosed, treatment-naive patients with multiple sclerosis: Results from ASCLEPIOS I and II. Mult. Scler. 2022, 28, 1562–1575. [Google Scholar] [CrossRef]
- Graf, J.; Leussink, V.I.; Soncin, G.; Lepka, K.; Meinl, I.; Kümpfel, T.; Meuth, S.G.; Hartung, H.P.; Havla, J.; Aktas, O.; et al. Relapse-independent multiple sclerosis progression under natalizumab. Brain Commun. 2021, 3, fcab229. [Google Scholar] [CrossRef]
- Iaffaldano, P.; Lucisano, G.; Butzkueven, H.; Hillert, J.; Hyde, R.; Koch-Henriksen, N.; Magyari, M.; Pellegrini, F.; Spelman, T.; Sørensen, P.S.; et al. Early treatment delays long-term disability accrual in RRMS: Results from the BMSD network. Mult. Scler. 2021, 27, 1543–1555. [Google Scholar] [CrossRef]
- Portaccio, E.; Bellinvia, A.; Fonderico, M.; Pastò, L.; Razzolini, L.; Totaro, R.; Spitaleri, D.; Lugaresi, A.; Cocco, E.; Onofrj, M.; et al. Progression is independent of relapse activity in early multiple sclerosis: A real-life cohort study. Brain 2022, 145, 2796–2805. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Multiple sclerosis: Role of meningeal lymphoid aggregates in progression independent of relapse activity. Trends Immunol. 2023, 44, 266–275. [Google Scholar] [CrossRef]
- Mosconi, P.; Guerra, T.; Paletta, P.; D’Ettorre, A.; Ponzio, M.; Battaglia, M.A.; Amato, M.P.; Bergamaschi, R.; Capobianco, M.; Comi, G.; et al. Data monitoring roadmap. The experience of the Italian Multiple Sclerosis and Related Disorders Register. Neurol. Sci. 2023, 44, 4001–4011. [Google Scholar] [CrossRef] [PubMed]
- Iaffaldano, P.; Lucisano, G.; Guerra, T.; Paolicelli, D.; Portaccio, E.; Inglese, M.; Foschi, M.; Patti, F.; Granella, F.; Romano, S.; et al. A comparison of natalizumab and ocrelizumab on disease progression in multiple sclerosis. Ann. Clin. Transl. Neurol. 2024, 11, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Puthenparampil, M.; Gaggiola, M.; Ponzano, M.; Zanotelli, G.; Miscioscia, A.; Nosadini, M.; Di Paola, A.; Sartori, S.; Perini, P.; Rinaldi, F.; et al. High NEDA and No PIRA in Natalizumab-Treated Patients With Pediatric-Onset Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2024, 11, e200303. [Google Scholar] [CrossRef] [PubMed]
- Chisari, C.G.; Aguglia, U.; Amato, M.P.; Bergamaschi, R.; Bertolotto, A.; Bonavita, S.; Morra, V.B.; Cavalla, P.; Cocco, E.; Conte, A.; et al. Long-term effectiveness of natalizumab in secondary progressive multiple sclerosis: A propensity-matched study. Neurotherapeutics 2024, 21, e00363. [Google Scholar] [CrossRef]
- de Sèze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef]
- Bajrami, A.; Tamanti, A.; Peloso, A.; Ziccardi, S.; Guandalini, M.; Calderone, M.; Castellaro, M.; Pizzini, F.B.; Montemezzi, S.; Marastoni, D.; et al. Ocrelizumab reduces cortical and deep grey matter loss compared to the S1P-receptor modulator in multiple sclerosis. J. Neurol. 2024, 271, 2149–2158. [Google Scholar] [CrossRef]
- Eisele, P.; Wittayer, M.; Weber, C.E.; Platten, M.; Schirmer, L.; Gass, A. Impact of disease-modifying therapies on evolving tissue damage in iron rim multiple sclerosis lesions. Mult. Scler. 2022, 28, 2294–2298. [Google Scholar] [CrossRef]
- Elliott, C.; Belachew, S.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Fecker, J.; Model, F.; Wei, W.; et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain 2019, 142, 2787–2799. [Google Scholar] [CrossRef]
- Maggi, P.; Bulcke, C.V.; Pedrini, E.; Bugli, C.; Sellimi, A.; Wynen, M.; Stölting, A.; Mullins, W.A.; Kalaitzidis, G.; Lolli, V.; et al. B cell depletion therapy does not resolve chronic active multiple sclerosis lesions. EBioMedicine 2023, 94, 104701. [Google Scholar] [CrossRef]
- Preziosa, P.; Pagani, E.; Moiola, L.; Rodegher, M.; Filippi, M.; Rocca, M.A. Occurrence and microstructural features of slowly expanding lesions on fingolimod or natalizumab treatment in multiple sclerosis. Mult. Scler. 2021, 27, 1520–1532. [Google Scholar] [CrossRef]
- Spelman, T.; Glaser, A.; Hilert, J. Immediate high-efficacy treatment in multiple sclerosis is associated with long-term reduction in progression independent of relapse activity (PIRA) compared to low-moderate efficacy treatment—A Swedish MS Registry study. Mult. Scler. J. 2024, 30, 651, Poster ECTRIMS 2024. [Google Scholar]
- Montobbio, N.; Cordioli, C.; Signori, A.; Bovis, F.; Capra, R.; Sormani, M.P. Relapse-Associated and Relapse-Independent Contribution to Overall Expanded Disability Status Scale Progression in Multiple Sclerosis Patients Diagnosed in Different Eras. Ann. Neurol. 2024, 91, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Montalban, X.; Coyle, P.K.; Kappos, L.; Li, B.; Sfikas, N.; Willi, R.; Häring, D.A.; Merschhemke, M.; Hauser, S.L. Ofatumumab reduces disability progression independent of relapse activity in patients with relapsing multiple sclerosis. Neurology 2021, 96, 2299. [Google Scholar] [CrossRef]
- De Stefano, N.; Giorgio, A.; Battaglini, M.; De Leucio, A.; Hicking, C.; Dangond, F.; Giovannoni, G.; Sormani, M.P. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult. Scler. 2018, 24, 222–226. [Google Scholar] [CrossRef]
- Cortese, R.; Battaglini, M.; Sormani, M.P.; Luchetti, L.; Gentile, G.; Inderyas, M.; Alexandri, N.; De Stefano, N. Reduction in grey matter atrophy in patients with relapsing multiple sclerosis following treatment with cladribine tablets. Eur. J. Neurol. 2023, 30, 179–186. [Google Scholar] [CrossRef]
- Cortese, R.; Testa, G.; Assogna, F.; De Stefano, N. Magnetic Resonance Imaging Evidence Supporting the Efficacy of Cladribine Tablets in the Treatment of Relapsing-Remitting Multiple Sclerosis. CNS Drugs 2024, 38, 267–279. [Google Scholar] [CrossRef]
- Geladaris, A.; Torke, S.; Weber, M.S. Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis: Pioneering the Path Towards Treatment of Progression? CNS Drugs 2022, 36, 1019–1030. [Google Scholar] [CrossRef]
- García-Merino, A. Bruton’s Tyrosine Kinase Inhibitors: A New Generation of Promising Agents for Multiple Sclerosis Therapy. Cells 2021, 10, 2560. [Google Scholar] [CrossRef]
- Niedziela, N.; Kalinowska, A.; Kułakowska, A.; Mirowska-Guzel, D.; Rejdak, K.; Siger, M.; Stasiołek, M.; Adamczyk-Sowa, M. Clinical and therapeutic challenges of smouldering multiple sclerosis. Neurol. Neurochir. Pol. 2024, 58, 245–255. [Google Scholar] [CrossRef]
- Zurmati, B.M.; Khan, J.; Reich, D.S.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar]
- Gruber, R.; Blazier, A.; Lee, L.; Ryan, S.; Cheong, A.; Havari, E.; Turner, T.; Ofengeim, D. Evaluating the Effect of BTK Inhibitor Tolebrutinib in Human Tri-culture. Neurology 2022, 98, e140. [Google Scholar] [CrossRef]
- Bierhansl, L.; Hartung, H.P.; Aktas, O.; Ruck, T.; Roden, M.; Meuth, S.G. Thinking outside the box: Non-canonical targets in multiple sclerosis. Nat. Rev. Drug Discov. 2022, 21, 578–600. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Da Mesquita, S.; Kipnis, J. Lymphatics in Neurological Disorders: A Neuro-Lympho-Vascular Component of Multiple Sclerosis and Alzheimer’s Disease? Neuron 2016, 91, 957–973. [Google Scholar] [CrossRef]
- das Neves, S.P.; Delivanoglou, N.; Ren, Y.; Cucuzza, C.S.; Makuch, M.; Almeida, F.; Sanchez, G.; Barber, M.J.; Rego, S.; Schrader, R.; et al. Meningeal lymphatic function promotes oligodendrocyte survival and brain myelination. Immunity 2024, 57, 2328–2343.e8. [Google Scholar] [CrossRef]
- Tur, C.; Rocca, M.A. Progression Independent of Relapse Activity in Multiple Sclerosis: Closer to Solving the Pathologic Puzzle. Neurology 2024, 102, e207936. [Google Scholar] [CrossRef]
- Graves, J.S.; Krysko, K.M.; Hua, L.H.; Absinta, M.; Franklin, R.J.M.; Segal, B.M. Ageing and multiple sclerosis. Lancet Neurol. 2023, 22, 66–77. [Google Scholar] [CrossRef]
- Pukoli, D.; Vécsei, L. Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. Int. J. Mol. Sci. 2023, 24, 12631. [Google Scholar] [CrossRef]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef]
- Ananthavarathan, P.; Sahi, N.; Chard, D.T. An update on the role of magnetic resonance imaging in predicting and monitoring multiple sclerosis progression. Expert Rev. Neurother. 2024, 24, 201–216. [Google Scholar] [CrossRef]
- Montalban, X.; European Committee for Treatment and Research in Multiple Sclerosis. 2024 Revisions of the McDonald Criteria; ECTRIMS: New York, NY, USA, 2024. [Google Scholar]
- Trojano, M.; Kalincik, T.; Iaffaldano, P.; Amato, M.P. Interrogating large multiple sclerosis registries and databases: What information can be gained? Curr. Opin. Neurol. 2022, 35, 271–277. [Google Scholar] [CrossRef]
- Zaratin, P.; Samadzadeh, S.; Seferoğlu, M.; Ricigliano, V.; Dos Santos Silva, J.; Tunc, A.; Brichetto, G.; Coetzee, T.; Helme, A.; Khan, U.; et al. The global patient-reported outcomes for multiple sclerosis initiative: Bridging the gap between clinical research and care. Front. Neurol. 2024, 15, 1407257. [Google Scholar] [CrossRef] [PubMed]
- Tunç, A.; Seferoğlu, M.; Sivaci, Ö.; Köktürk, M.D.; Polat, A.K. Pediatric, adult, and late-onset multiple sclerosis patients: A unified analysis of clinical profiles and treatment responses. Mult. Scler. Relat. Disord. 2024, 24, e240789. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, T.; Iaffaldano, P. A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective. Int. J. Mol. Sci. 2025, 26, 884. https://doi.org/10.3390/ijms26030884
Guerra T, Iaffaldano P. A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective. International Journal of Molecular Sciences. 2025; 26(3):884. https://doi.org/10.3390/ijms26030884
Chicago/Turabian StyleGuerra, Tommaso, and Pietro Iaffaldano. 2025. "A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective" International Journal of Molecular Sciences 26, no. 3: 884. https://doi.org/10.3390/ijms26030884
APA StyleGuerra, T., & Iaffaldano, P. (2025). A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective. International Journal of Molecular Sciences, 26(3), 884. https://doi.org/10.3390/ijms26030884