
Decoding Local Adaptation in the Exploited Native Marine Mussel Mytilus chilensis: Genomic Evidence from a Reciprocal Transplant Experiment




Abstract
:1. Introduction
2. Results
2.1. Environmental Characterization
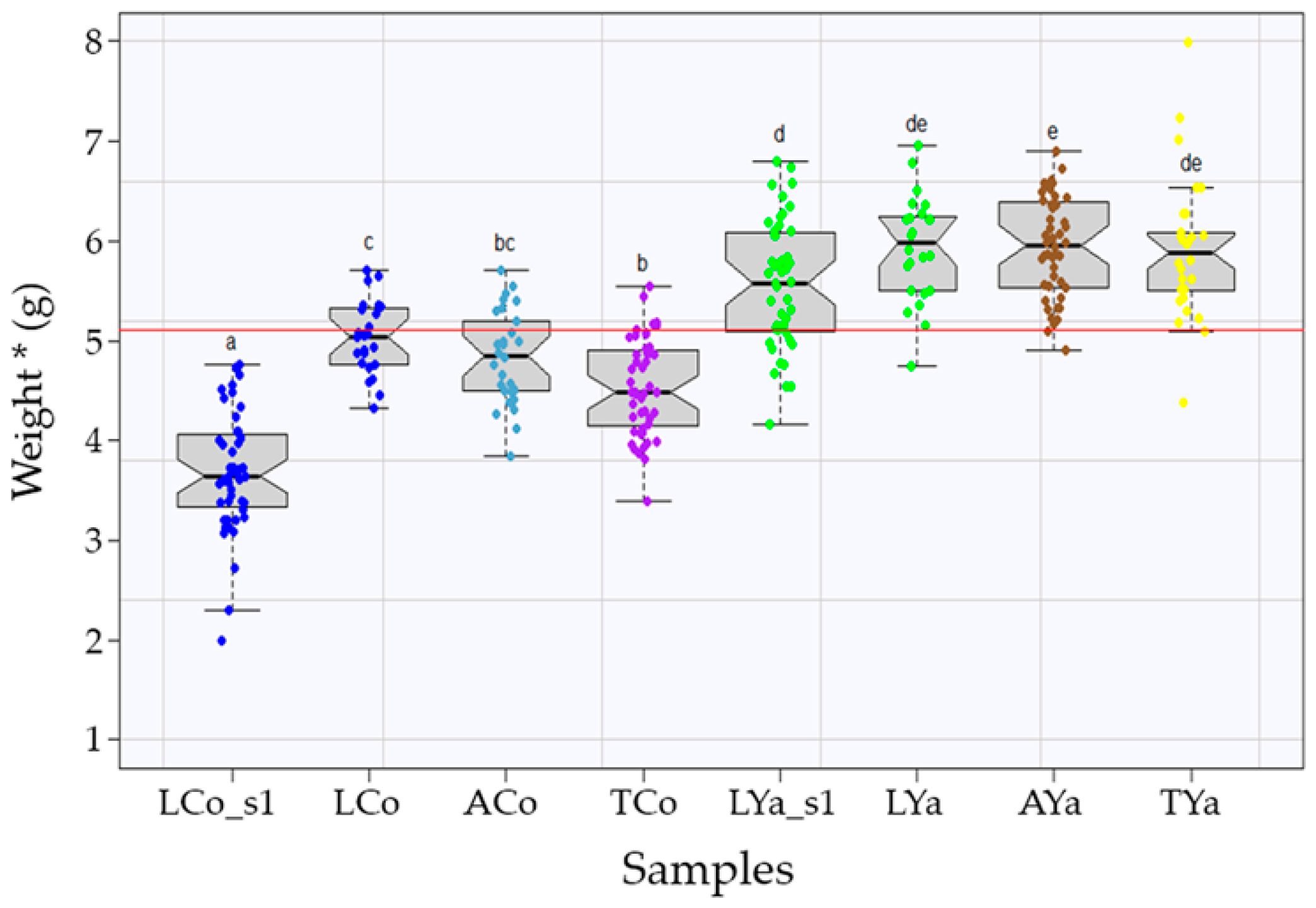
2.2. Mean Size Comparisons
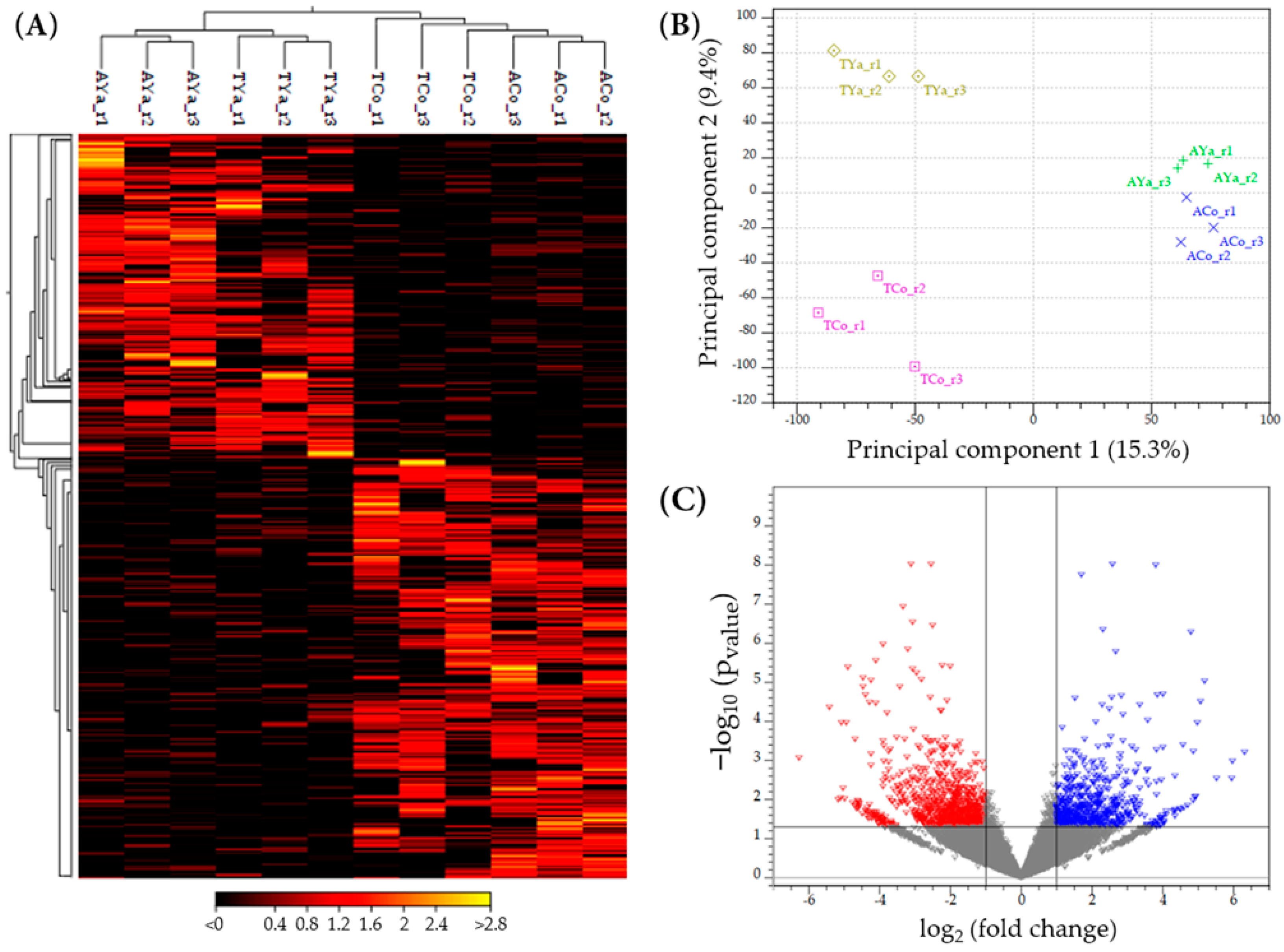
2.3. Mappings of Clean Reads
2.4. Differential Expression Analysis
2.5. Number of Differentially Expressed Genes (DEGs) by Location
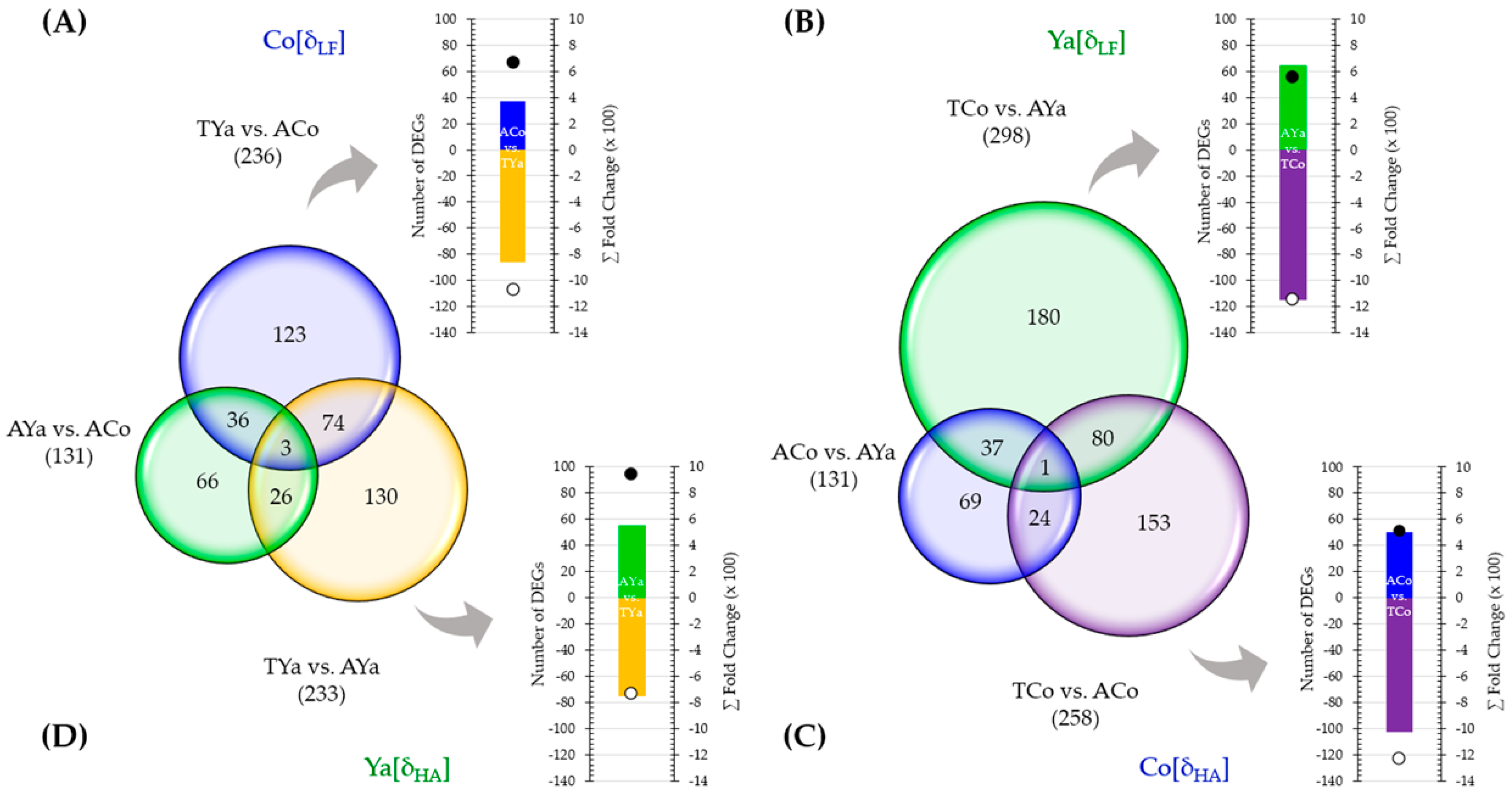
2.6. Venn Diagrams and DEG Selection
2.7. Annotation of DEGs
2.7.1. Local vs. Foreign (δLF) Criterion Comparison
2.7.2. Home vs. Away (δHA) Criterion Comparison
2.8. Functional Categorization of DEGs
2.8.1. KEGG Categorization
2.8.2. Gene Ontology (GO) Categorization
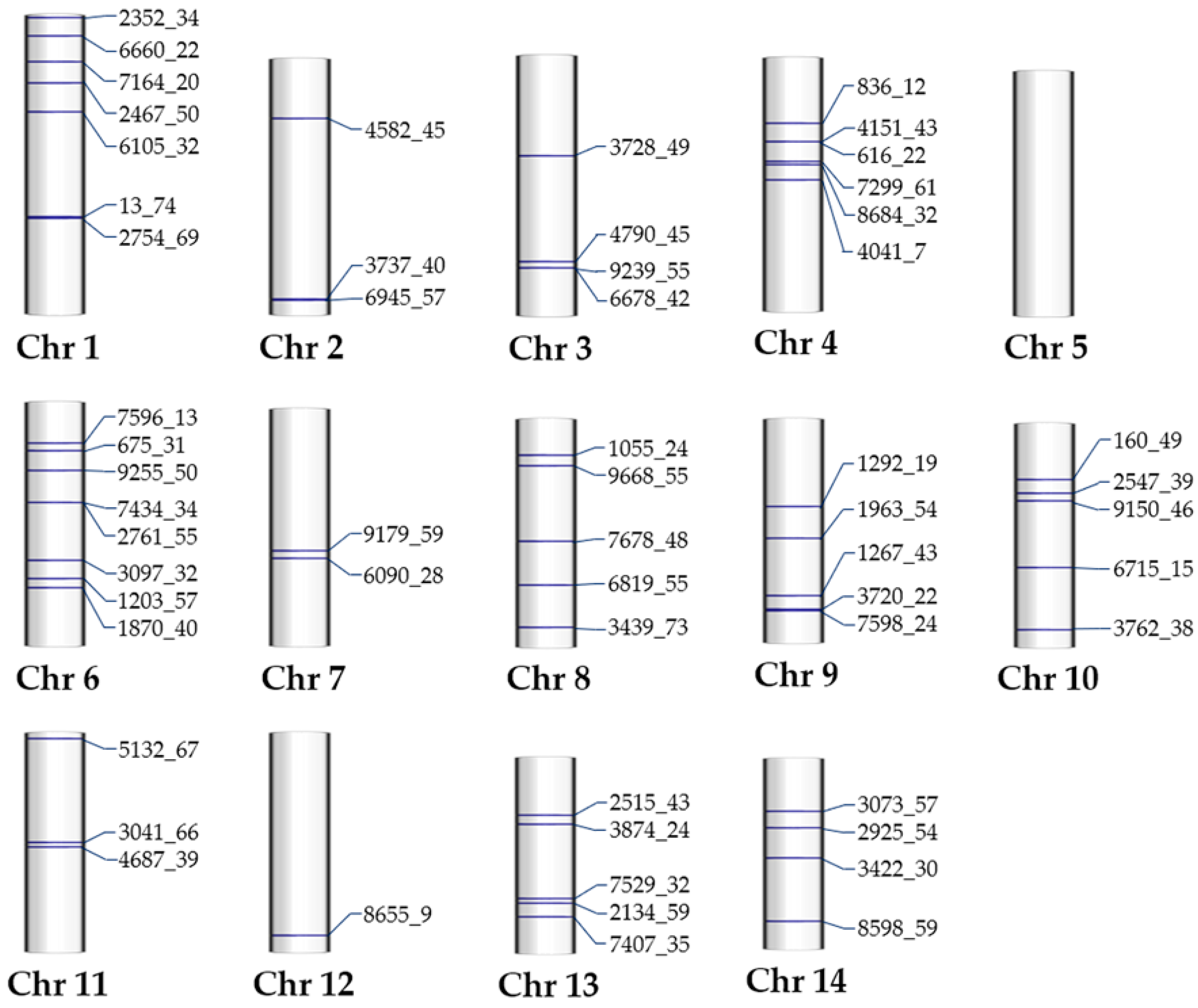
2.9. Genome Mapping of Outlier SNPs and Their Neighboring Genes
2.9.1. Mapping of the Outlier SNPs
2.9.2. Annotations of Outlier SNP-Neighboring Genes
2.9.3. GO Categorization of Outlier SNP-Neighboring Genes
3. Discussion
3.1. Environmental Barriers as Drivers of Local Adaptation in Mytilus chilensis
3.2. Growth Rate as Indicator of Local Adaptation in Mytilus chilensis
3.3. Differential Transcription Across the Mytilus chilensis Genome
3.4. Genomic Differentiation and Local Adaptation in Mytilus chilensis
3.5. Identifying Candidate Adaptive DEGs in Mytilus chilensis for Local Adaptation
3.6. Mapping of Outlier SNP-Neighbor Genes in the Mytilus chilensis Genome
4. Materials and Methods
4.1. Study Sites and Sampling
4.2. Reciprocal Transplant Experiment
4.3. Taxonomic Affiliation
4.4. RNA Extraction and Sequencing
4.5. RNA-Seq and Differential Expression Analysis
4.6. DEG Annotations and Functional Categorization
4.7. Outlier SNP Genome Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawecki, T.J.; Ebert, D. Conceptual Issues in Local Adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef]
- Savolainen, O.; Lascoux, M.; Merilä, J. Ecological Genomics of Local Adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Laikre, L.; Hoban, S.; Bruford, M.W.; Segelbacher, G.; Allendorf, F.W.; Gajardo, G.; Rodríguez, A.G.; Hedrick, P.W.; Heuertz, M.; Hohenlohe, P.A.; et al. Post-2020 Goals Overlook Genetic Diversity. Science 2020, 367, 1083–1085. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Bruford, M.; D’Urban Jackson, J.; Lopes-Fernandes, M.; Heuertz, M.; Hohenlohe, P.A.; Paz-Vinas, I.; Sjögren-Gulve, P.; Segelbacher, G.; Vernesi, C.; et al. Genetic Diversity Targets and Indicators in the CBD Post-2020 Global Biodiversity Framework Must Be Improved. Biol. Conserv. 2020, 248, 108654. [Google Scholar] [CrossRef]
- Sanford, E.; Kelly, M.W. Local Adaptation in Marine Invertebrates. Annu. Rev. Mar. Sci. 2011, 3, 509–535. [Google Scholar] [CrossRef]
- Blanquart, F.; Gandon, S.; Nuismer, S.L. The Effects of Migration and Drift on Local Adaptation to a Heterogeneous Environment. J. Evol. Biol. 2012, 25, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, M.C. Modern Approaches to Local Adaptation. Am. Nat. 2015, 186, S1–S4. [Google Scholar] [CrossRef]
- Frankham, R. Where Are We in Conservation Genetics and Where Do We Need to Go? Conserv. Genet. 2010, 11, 661–663. [Google Scholar] [CrossRef]
- Waldvogel, A.-M.; Schreiber, D.; Pfenninger, M.; Feldmeyer, B. Climate Change Genomics Calls for Standardized Data Reporting. Front. Ecol. Evol. 2020, 8, 242. [Google Scholar] [CrossRef]
- Astorga, M.P.; Vargas, J.; Valenzuela, A.; Molinet, C.; Marín, S.L. Population Genetic Structure and Differential Selection in Mussel Mytilus chilensis. Aquac. Res. 2018, 49, 919–927. [Google Scholar] [CrossRef]
- Astorga, M.P.; Cárdenas, L.; Pérez, M.; Toro, J.E.; Martínez, V.; Farías, A.; Uriarte, I. Complex Spatial Genetic Connectivity of Mussels Mytilus chilensis Along the Southeastern Pacific Coast and Its Importance for Resource Management. J. Shellfish. Res. 2020, 39, 77. [Google Scholar] [CrossRef]
- Blanquart, F.; Kaltz, O.; Nuismer, S.L.; Gandon, S. A Practical Guide to Measuring Local Adaptation. Ecol. Lett. 2013, 16, 1195–1205. [Google Scholar] [CrossRef]
- Burford, M.; Scarpa, J.; Cook, B.; Hare, M. Local Adaptation of a Marine Invertebrate with a High Dispersal Potential: Evidence from a Reciprocal Transplant Experiment of the Eastern Oyster Crassostrea virginica. Mar. Ecol. Prog. Ser. 2014, 505, 161–175. [Google Scholar] [CrossRef]
- Yanick, J.F.; Heath, J.W.; Heath, D.D. Survival and Growth of Local and Transplanted Blue Mussels (Mytilus trossulus, Lamark): Variation in Survival and Growth of Blue Mussels. Aquac. Res. 2003, 34, 869–875. [Google Scholar] [CrossRef]
- Jahnsen-Guzmán, N.; Lagos, N.A.; Lardies, M.A.; Vargas, C.A.; Fernández, C.; San Martín, V.A.; Saavedra, L.; Cuevas, L.A.; Quijón, P.A.; Duarte, C. Environmental Refuges Increase Performance of Juvenile Mussels Mytilus chilensis: Implications for Mussel Seedling and Farming Strategies. Sci. Total Environ. 2021, 751, 141723. [Google Scholar] [CrossRef] [PubMed]
- Schulte, P.M. Changes in Gene Expression as Biochemical Adaptations to Environmental Change: A Tribute to Peter Hochachka. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2004, 139, 519–529. [Google Scholar] [CrossRef]
- Barrett, R.D.H.; Hoekstra, H.E. Molecular Spandrels: Tests of Adaptation at the Genetic Level. Nat. Rev. Genet. 2011, 12, 767–780. [Google Scholar] [CrossRef]
- Sork, V.L.; Aitken, S.N.; Dyer, R.J.; Eckert, A.J.; Legendre, P.; Neale, D.B. Putting the Landscape into the Genomics of Trees: Approaches for Understanding Local Adaptation and Population Responses to Changing Climate. Tree Genet. Genomes 2013, 9, 901–911. [Google Scholar] [CrossRef]
- Weissenbach, J. The Rise of Genomics. Comptes Rendus. Biol. 2016, 339, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, J.F.; Ravago-Gotanco, R. Transcriptome-Derived SNP Markers for Population Assignment of Sandfish, Holothuria (Metriatyla) scabra. Aquaculture 2024, 579, 740261. [Google Scholar] [CrossRef]
- Pespeni, M.H.; Sanford, E.; Gaylord, B.; Hill, T.M.; Hosfelt, J.D.; Jaris, H.K.; LaVigne, M.; Lenz, E.A.; Russell, A.D.; Young, M.K.; et al. Evolutionary Change during Experimental Ocean Acidification. Proc. Natl. Acad. Sci. USA 2013, 110, 6937–6942. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.; Hansen, M.M.; Normandeau, E.; Mensberg, K.-L.D.; Frydenberg, J.; Larsen, P.F.; Bekkevold, D.; Bernatchez, L. Local Adaptation at the Transcriptome Level in Brown Trout: Evidence from Early Life History Temperature Genomic Reaction Norms. PLoS ONE 2014, 9, e85171. [Google Scholar] [CrossRef]
- Lockwood, B.L.; Connor, K.M.; Gracey, A.Y. The Environmentally Tuned Transcriptomes of Mytilus Mussels. J. Exp. Biol. 2015, 218, 1822–1833. [Google Scholar] [CrossRef]
- Pastenes, L.; Valdivieso, C.; Di Genova, A.; Travisany, D.; Hart, A.; Montecino, M.; Orellana, A.; Gonzalez, M.; Gutiérrez, R.A.; Allende, M.L.; et al. Global Gene Expression Analysis Provides Insight into Local Adaptation to Geothermal Streams in Tadpoles of the Andean Toad Rhinella spinulosa. Sci. Rep. 2017, 7, 1966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, A.; She, Z.; Wang, X.; Jia, Z.; Wang, W.; Zhang, G.; Li, L. Adaptive Divergence and Underlying Mechanisms in Response to Salinity Gradients between Two Crassostrea Oysters Revealed by Phenotypic and Transcriptomic Analyses. Evol. Appl. 2023, 16, 234–249. [Google Scholar] [CrossRef]
- Gu, H.; Qi, X.; Jia, Y.; Zhang, Z.; Nie, C.; Li, X.; Li, J.; Jiang, Z.; Wang, Q.; Qu, L. Inheritance Patterns of the Transcriptome in Hybrid Chickens and Their Parents Revealed by Expression Analysis. Sci. Rep. 2019, 9, 5750. [Google Scholar] [CrossRef]
- DeBiasse, M.B.; Kelly, M.W. Plastic and Evolved Responses to Global Change: What Can We Learn from Comparative Transcriptomics? J. Hered. 2016, 107, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Talbert, P.B.; Meers, M.P.; Henikoff, S. Old Cogs, New Tricks: The Evolution of Gene Expression in a Chromatin Context. Nat. Rev. Genet. 2019, 20, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Escárate, C.; Valenzuela-Muñoz, V.; Nuñez-Acuña, G.; Valenzuela-Miranda, D.; Tapia, F.J.; Yévenes, M.; Gajardo, G.; Toro, J.E.; Oyarzún, P.A.; Arriagada, G.; et al. Chromosome-Level Genome Assembly of the Blue Mussel Mytilus chilensis Reveals Molecular Signatures Facing the Marine Environment. Genes 2023, 14, 876. [Google Scholar] [CrossRef] [PubMed]
- Molinet, C.A.; Gomez, M.A.D.; Muñoz, C.B.A.; Pérez, L.E.C.; Arribas, S.L.M.; Opazo, M.P.A.; Huaquin, E.J.E.N. Spatial Distribution Pattern of Mytilus chilensis Beds in the Reloncaví Fjord: Hypothesis on Associated Processes. Rev. Chil. Hist. Nat. 2015, 88, 11. [Google Scholar] [CrossRef]
- Curelovich, J.; Lovrich, G.A.; Calcagno, J.A. The Role of the Predator Trophon geversianus in an Intertidal Population of Mytilus chilensis in a Rocky Shore of the Beagle Channel, Tierra Del Fuego, Argentina. Mar. Biol. Res. 2016, 12, 1053–1063. [Google Scholar] [CrossRef]
- Duarte, C.; Navarro, J.M.; Quijón, P.A.; Loncon, D.; Torres, R.; Manríquez, P.H.; Lardies, M.A.; Vargas, C.A.; Lagos, N.A. The Energetic Physiology of Juvenile Mussels, Mytilus chilensis (Hupe): The Prevalent Role of Salinity under Current and Predicted pCO2 Scenarios. Environ. Pollut. 2018, 242, 156–163. [Google Scholar] [CrossRef]
- Yévenes, M.; Núñez-Acuña, G.; Gallardo-Escárate, C.; Gajardo, G. Adaptive Differences in Gene Expression in Farm-Impacted Seedbeds of the Native Blue Mussel Mytilus chilensis. Front. Genet. 2021, 12, 666539. [Google Scholar] [CrossRef]
- Yévenes, M.; Núñez-Acuña, G.; Gallardo-Escárate, C.; Gajardo, G. Adaptive Mitochondrial Genome Functioning in Ecologically Different Farm-Impacted Natural Seedbeds of the Endemic Blue Mussel Mytilus chilensis. Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 42, 100955. [Google Scholar] [CrossRef]
- Yévenes, M.; Gallardo-Escárate, C.; Gajardo, G. Epigenetic Variation Mediated by lncRNAs Accounts for Adaptive Genomic Differentiation of the Endemic Blue Mussel Mytilus chilensis. Heliyon 2024, 10, e23695. [Google Scholar] [CrossRef]
- Toro, J.E.; Alcapan, A.C.; Vergara, A.M.; Ojeda, J.A. Heritability Estimates of Larval and Spat Shell Height in the Chilean Blue Mussel (Mytilus chilensis Hupe 1854) Produced under Controlled Laboratory Conditions. Aquac Res. 2004, 35, 56–61. [Google Scholar] [CrossRef]
- Ruiz, M.; Tarifeño, E.; Llanos-Rivera, A.; Padget, C.; Campos, B. Efecto de la temperatura en el desarrollo embrionario y larval del mejillón, Mytilus galloprovincialis (Lamarck, 1819). Rev. Biol. Mar. Oceanogr. 2008, 43, 51–61. [Google Scholar] [CrossRef]
- Barría, A.; Gebauer, P.; Molinet, C. Variabilidad espacial y temporal del suministro larval de mitílidos en el Seno de Reloncaví, sur de Chile. Rev. Biol. Mar. Oceanogr. 2012, 47, 461–473. [Google Scholar] [CrossRef]
- Toro, J.E.; Castro, G.C.; Ojeda, J.A.; Vergara, A.M. Allozymic Variation and Differentiation in the Chilean Blue Mussel, Mytilus chilensis, along Its Natural Distribution. Genet. Mol. Biol. 2006, 29, 174–179. [Google Scholar] [CrossRef]
- Larraín, M.A.; Díaz, N.F.; Lamas, C.; Uribe, C.; Araneda, C. Traceability of Mussel (Mytilus chilensis) in Southern Chile Using Microsatellite Molecular Markers and Assignment Algorithms. Exploratory Survey. Food Res. Int. 2014, 62, 104–110. [Google Scholar] [CrossRef]
- Díaz-Puente, B.; Pita, A.; Uribe, J.; Cuéllar-Pinzón, J.; Guiñez, R.; Presa, P. A Biogeography-based Management for Mytilus chilensis: The Genetic Hodgepodge of Los Lagos versus the Pristine Hybrid Zone of the Magellanic Ecotone. Aquat. Conserv. Mar. Freshw. Ecosyst. 2020, 30, 412–425. [Google Scholar] [CrossRef]
- Segovia, N.I.; Coral-Santacruz, D.; Haye, P.A. Genetic Homogeneity and Weak Signatures of Local Adaptation in the Marine Mussel Mytilus chilensis. Sci. Rep. 2024, 14, 21081. [Google Scholar] [CrossRef] [PubMed]
- Araneda, C.; Larraín, M.A.; Hecht, B.; Narum, S. Adaptive Genetic Variation Distinguishes Chilean Blue Mussels (Mytilus chilensis) from Different Marine Environments. Ecol. Evol. 2016, 6, 3632–3644. [Google Scholar] [CrossRef]
- Dokan, K.; Kawamura, S.; Teshima, K.M. Effects of Single Nucleotide Polymorphism Ascertainment on Population Structure Inferences. G3 Genes|Genomes|Genetics 2021, 11, jkab128. [Google Scholar] [CrossRef]
- Lachance, J.; Tishkoff, S.A. SNP Ascertainment Bias in Population Genetic Analyses: Why It Is Important, and How to Correct It. BioEssays 2013, 35, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Poblete, E.; Hurtado, C.; Rojo, C.; Norambuena, R. Blue Mussel Aquaculture in Chile: Small or Large Scale Industry? Aquaculture 2018, 493, 113–122. [Google Scholar] [CrossRef]
- Martínez, V.; Lara, C.; Silva, N.; Gudiño, V.; Montecino, V. Variability of Environmental Heterogeneity in Northern Patagonia, Chile: Effects on the Spatial Distribution, Size Structure and Abundance of Chlorophyll-a. Rev. Biol. Mar. Oceanogr. 2015, 50, 39–52. [Google Scholar] [CrossRef]
- Lara, C.; Saldías, G.S.; Tapia, F.J.; Iriarte, J.L.; Broitman, B.R. Interannual Variability in Temporal Patterns of Chlorophyll–a and Their Potential Influence on the Supply of Mussel Larvae to Inner Waters in Northern Patagonia (41–44°S). J. Mar. Syst. 2016, 155, 11–18. [Google Scholar] [CrossRef]
- Ottenburghs, J. The Genic View of Hybridization in the Anthropocene. Evol. Appl. 2021, 14, 2342–2360. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.P.; Lucas, I.A.N.; Seed, R.; Richardson, C.A. Mytilus edulis chilensis Infested with Coccomyxa parasitica (Chlorococcales, Coccomyxaceae). J. Molluscan Stud. 1999, 65, 289–294. [Google Scholar] [CrossRef]
- Aranda, C.P.; Yévenes, M.; Rodriguez-Benito, C.; Godoy, F.A.; Ruiz, M.; Cachicas, V. Distribution and Growth of Vibrio parahaemolyticus in Southern Chilean Clams (Venus antiqua) and Blue Mussels (Mytilus chilensis). Foodborne Pathog. Dis. 2015, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Détrée, C.; Gallardo-Escárate, C. Polyethylene Microbeads Induce Transcriptional Responses with Tissue-Dependent Patterns in the Mussel Mytilus galloprovincialis. J. Molluscan Stud. 2017, 83, 220–225. [Google Scholar] [CrossRef]
- Blanc, J.M.; Molinet, C.; Subiabre, R.; Díaz, P.A. Cadmium Determination in Chilean Blue Mussels Mytilus chilensis: Implications for Environmental and Agronomic Interest. Mar. Pollut. Bull. 2018, 129, 913–917. [Google Scholar] [CrossRef]
- Harvell, C.D.; Mitchell, C.E.; Ward, J.R.; Altizer, S.; Dobson, A.P.; Ostfeld, R.S.; Samuel, M.D. Climate Warming and Disease Risks for Terrestrial and Marine Biota. Science 2002, 296, 2158–2162. [Google Scholar] [CrossRef] [PubMed]
- Hüning, A.K.; Melzner, F.; Thomsen, J.; Gutowska, M.A.; Krämer, L.; Frickenhaus, S.; Rosenstiel, P.; Pörtner, H.-O.; Philipp, E.E.R.; Lucassen, M. Impacts of Seawater Acidification on Mantle Gene Expression Patterns of the Baltic Sea Blue Mussel: Implications for Shell Formation and Energy Metabolism. Mar. Biol. 2013, 160, 1845–1861. [Google Scholar] [CrossRef]
- Vihtakari, M.; Hendriks, I.; Holding, J.; Renaud, P.; Duarte, C.; Havenhand, J. Effects of Ocean Acidification and Warming on Sperm Activity and Early Life Stages of the Mediterranean Mussel (Mytilus galloprovincialis). Water 2013, 5, 1890–1915. [Google Scholar] [CrossRef]
- Castillo, N.; Saavedra, L.M.; Vargas, C.A.; Gallardo-Escárate, C.; Détrée, C. Ocean Acidification and Pathogen Exposure Modulate the Immune Response of the Edible Mussel Mytilus chilensis. Fish Shellfish. Immunol. 2017, 70, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Díaz, R.; Lardies, M.A.; Tapia, F.J.; Tarifeño, E.; Vargas, C.A. Transgenerational Effects of pCO2-Driven Ocean Acidification on Adult Mussels Mytilus chilensis Modulate Physiological Response to Multiple Stressors in Larvae. Front. Physiol. 2018, 9, 1349. [Google Scholar] [CrossRef] [PubMed]
- Malachowicz, M.; Wenne, R. Mantle Transcriptome Sequencing of Mytilus spp. and Identification of Putative Biomineralization Genes. PeerJ 2019, 6, e6245. [Google Scholar] [CrossRef] [PubMed]
- Mlouka, R.; Cachot, J.; Sforzini, S.; Oliveri, C.; Boukadida, K.; Clerandeau, C.; Pacchioni, B.; Millino, C.; Viarengo, A.; Banni, M. Molecular Mechanisms Underlying the Effects of Temperature Increase on Mytilus sp. and Their Hybrids at Early Larval Stages. Sci. Total Environ. 2020, 708, 135200. [Google Scholar] [CrossRef] [PubMed]
- Haye, P.A.; Segovia, N.I. Shedding Light on Variation in Reproductive Success through Studies of Population Genetic Structure in a Southeast Pacific Coast Mussel. Heredity 2023, 130, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.; Navarro, J.M.; Acuña, K.; Torres, R.; Manríquez, P.H.; Lardies, M.A.; Vargas, C.A.; Lagos, N.A.; Aguilera, V. Combined Effects of Temperature and Ocean Acidification on the Juvenile Individuals of the Mussel Mytilus chilensis. J. Sea Res. 2014, 85, 308–314. [Google Scholar] [CrossRef]
- Navarro, J.M.; Duarte, C.; Manríquez, P.H.; Lardies, M.A.; Torres, R.; Acuña, K.; Vargas, C.A.; Lagos, N.A. Ocean Warming and Elevated Carbon Dioxide: Multiple Stressor Impacts on Juvenile Mussels from Southern Chile. ICES J. Mar. Sci. 2016, 73, 764–771. [Google Scholar] [CrossRef]
- Mellado, C.; Chaparro, O.R.; Duarte, C.; Villanueva, P.A.; Ortiz, A.; Valdivia, N.; Torres, R.; Navarro, J.M. Ocean Acidification Exacerbates the Effects of Paralytic Shellfish Toxins on the Fitness of the Edible Mussel Mytilus chilensis. Sci. Total Environ. 2019, 653, 455–464. [Google Scholar] [CrossRef]
- Núñez-Acuña, G.; Aballay, A.E.; Hégaret, H.; Astuya, A.P.; Gallardo-Escárate, C. Transcriptional Responses of Mytilus chilensis Exposed in Vivo to Saxitoxin (STX). J. Molluscan Stud. 2013, 79, 323–331. [Google Scholar] [CrossRef]
- Détrée, C.; Núñez-Acuña, G.; Roberts, S.; Gallardo-Escárate, C. Uncovering the Complex Transcriptome Response of Mytilus chilensis against Saxitoxin: Implications of Harmful Algal Blooms on Mussel Populations. PLoS ONE 2016, 11, e0165231. [Google Scholar] [CrossRef]
- León-Muñoz, J.; Urbina, M.A.; Garreaud, R.; Iriarte, J.L. Hydroclimatic Conditions Trigger Record Harmful Algal Bloom in Western Patagonia (Summer 2016). Sci. Rep. 2018, 8, 1330. [Google Scholar] [CrossRef]
- Riccialdelli, L.; Newsome, S.D.; Fogel, M.L.; Fernández, D.A. Trophic Interactions and Food Web Structure of a Subantarctic Marine Food Web in Beagle Channel: Bahía Lapataia, Argentina. Polar Biol. 2017, 40, 807–821. [Google Scholar] [CrossRef]
- Uzkiaga, N.; Gebauer, P.; Niklitschek, E.; Montory, J.; Paschke, K.; Garcés, C.; De Lázaro-López, O. Predation of the Crab Acanthocyclus albatrossis on Seeds of the Bivalve Mytilus chilensis under Different Environmental Conditions: Importance of Prey and Predator Size. J. Exp. Mar. Biol. Ecol. 2022, 551, 151730. [Google Scholar] [CrossRef]
- Edmonds, D.; Dreslik, M.J.; Lovich, J.E.; Wilson, T.P.; Ernst, C.H. Growing as Slow as a Turtle: Unexpected Maturational Differences in a Small, Long-Lived Species. PLoS ONE 2021, 16, e0259978. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Hascup, E.; Hascup, K.; Bartke, A. Relationships among Development, Growth, Body Size, Reproduction, Aging, and Longevity—Trade-Offs and Pace-Of-Life. Biochem. Mosc. 2023, 88, 1692–1703. [Google Scholar] [CrossRef]
- Krapivka, S.; Toro, J.E.; Alcapán, A.C.; Astorga, M.; Presa, P.; Pérez, M.; Guiñez, R. Shell-Shape Variation along the Latitudinal Range of the Chilean Blue Mussel Mytilus chilensis (Hupe 1854). Aquac. Res. 2007, 38, 1770–1777. [Google Scholar] [CrossRef]
- Valladares, A.; Manríquez, G.; Suárez-Isla, B.A. Shell Shape Variation in Populations of Mytilus chilensis (Hupe 1854) from Southern Chile: A Geometric Morphometric Approach. Mar. Biol. 2010, 157, 2731–2738. [Google Scholar] [CrossRef]
- Fan, K.; Pfister, E.; Weng, Z. Toward a Comprehensive Catalog of Regulatory Elements. Hum. Genet. 2023, 142, 1091–1111. [Google Scholar] [CrossRef]
- Innes, P.A.; Goebl, A.M.; Smith, C.C.R.; Rosenberger, K.; Kane, N.C. Gene Expression and Alternative Splicing Contribute to Adaptive Divergence of Ecotypes. Heredity 2024, 132, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive Heterochromatin Formation and Transcription in Mammals. Epigenetics Chromatin 2015, 8, 3. [Google Scholar] [CrossRef]
- Penagos-Puig, A.; Furlan-Magaril, M. Heterochromatin as an Important Driver of Genome Organization. Front. Cell Dev. Biol. 2020, 8, 579137. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative Splicing and Evolution: Diversification, Exon Definition and Function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Singh, P.; Ahi, E.P. The Importance of Alternative Splicing in Adaptive Evolution. Mol. Ecol. 2022, 31, 1928–1938. [Google Scholar] [CrossRef]
- Lan, W.; Zhou, Y.; Hazra, R. Editorial: The Evolution in RNA: 2023. Front. Genet. 2024, 15, 1457242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yang, M.; Hu, Z.; Shi, P.; Li, Y.; Guo, Y.; Zhang, T.; Song, H. Molecular Evidence for the Adaptive Evolution in Euryhaline Bivalves. Mar. Environ. Res. 2023, 192, 106240. [Google Scholar] [CrossRef]
- Kouser, L.; Madhukaran, S.P.; Shastri, A.; Saraon, A.; Ferluga, J.; Al-Mozaini, M.; Kishore, U. Emerging and Novel Functions of Complement Protein C1q. Front. Immunol. 2015, 6, 317. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Kishore, U. C1 Complex: An Adaptable Proteolytic Module for Complement and Non-Complement Functions. Front. Immunol. 2017, 8, 592. [Google Scholar] [CrossRef]
- So, E.C.; Zhou, H.; Greenwell, A.; Burch, E.E.; Ji, Y.; Mérigeon, E.Y.; Olsen, H.S.; Bentzen, S.M.; Block, D.S.; Zhang, X.; et al. Complement Component C1q Is an Immunological Rheostat That Regulates Fc:FcgR Interactions. Immunogenetics 2023, 75, 369–383. [Google Scholar] [CrossRef]
- Calcino, A.D.; De Oliveira, A.L.; Simakov, O.; Schwaha, T.; Zieger, E.; Wollesen, T.; Wanninger, A. The Quagga Mussel Genome and the Evolution of Freshwater Tolerance. DNA Res. 2019, 26, 411–422. [Google Scholar] [CrossRef]
- Schoville, S.D.; Barreto, F.S.; Moy, G.W.; Wolff, A.; Burton, R.S. Investigating the Molecular Basis of Local Adaptation to Thermal Stress: Population Differences in Gene Expression across the Transcriptome of the Copepod Tigriopus californicus. BMC Evol. Biol. 2012, 12, 170. [Google Scholar] [CrossRef]
- Jianfang, W.; Raza, S.H.A.; Pant, S.D.; Juan, Z.; Prakash, A.; Abdelnour, S.A.; Aloufi, B.H.; Mahasneh, Z.M.H.; Amin, A.A.; Shokrollahi, B.; et al. Exploring Epigenetic and Genetic Modulation in Animal Responses to Thermal Stress. Mol. Biotechnol. 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Montúfar-Romero, M.; Valenzuela-Muñoz, V.; Valenzuela-Miranda, D.; Gallardo-Escárate, C. Hypoxia in the Blue Mussel Mytilus chilensis Induces a Transcriptome Shift Associated with Endoplasmic Reticulum Stress, Metabolism, and Immune Response. Genes 2024, 15, 658. [Google Scholar] [CrossRef]
- Pinilla, E.; Castillo, M.I.; Pérez-Santos, I.; Venegas, O.; Valle-Levinson, A. Water Age Variability in a Patagonian Fjord. J. Mar. Syst. 2020, 210, 103376. [Google Scholar] [CrossRef]
- Pandey, D.M.; Kim, S.-R. Identification and Expression Analysis of Hypoxia Stress Inducible CCCH-Type Zinc Finger Protein Genes in Rice. J. Plant Biol. 2012, 55, 489–497. [Google Scholar] [CrossRef]
- Nascimento-Schulze, J.C.; Bean, T.P.; Peñaloza, C.; Paris, J.R.; Whiting, J.R.; Simon, A.; Fraser, B.A.; Houston, R.D.; Bierne, N.; Ellis, R.P. SNP Discovery and Genetic Structure in Blue Mussel Species Using Low Coverage Sequencing and a Medium Density 60 K SNP-array. Evol. Appl. 2023, 16, 1044–1060. [Google Scholar] [CrossRef]
- Fernández-Tajes, J.; Longa, A.; García-Gil, J.; Chiu, Y.-W.; Huang, Y.-S.; Méndez, J.; Lee, R.-S. Alternative PCR–RFLP Methods for Mussel Mytilus Species Identification. Eur. Food Res. Technol. 2011, 233, 791–796. [Google Scholar] [CrossRef]
- Larraín, M.A.; Diaz, N.F.; Lamas, C.; Vargas, C.; Araneda, C. Genetic Composition of Mytilus Species in Mussel Populations from Southern Chile. Lat. Am. J. Aquat. Res. 2012, 40, 1077–1084. [Google Scholar] [CrossRef]
- Oyarzún, P.A.; Toro, J.E.; Cañete, J.I.; Gardner, J.P.A. Bioinvasion Threatens the Genetic Integrity of Native Diversity and a Natural Hybrid Zone: Smooth-Shelled Blue Mussels (Mytilus spp.) in the Strait of Magellan. Biol. J. Linn. Soc. 2016, 117, 574–585. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2016 Update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | COCHAMÓ | ||||||
| Self-Transplanted | Cross-Transplanted | ||||||
| Parameter | Location | Media | SD | % | Media | SD | % |
| Total of reads | 54,260,671 | 4,114,336 | 100.00 | 33,079,882 | 1,061,904 | 100.00 | |
| Reads mapped in unique fragments | Intron | 4,341,568 | 330,607 | 8.00 | 2,836,805 | 102,368 | 8.58 |
| Exon | 809,780 | 57,644 | 1.49 | 498,607 | 13,532 | 1.51 | |
| Exon–exon | 24,695 | 2,749 | 0.05 | 16,996 | 516 | 0.05 | |
| Intergenic | 37,866,782 | 3,219,598 | 69.79 | 24,642,892 | 724,681 | 74.50 | |
| Reads not mapped | 11,217,845 | 644,857 | 20.67 | 5,084,581 | 333,123 | 15.37 | |
| (B) | YALDAD | ||||||
| Self-Transplanted | Cross-Transplanted | ||||||
| Parameter | Location | Media | SD | % | Media | SD | % |
| Total of reads | 55,553,941 | 677,061 | 100.00 | 32,849,423 | 2,932,648 | 100.00 | |
| Reads mapped in unique fragments | Intron | 4,465,683 | 119,094 | 8.04 | 2,800,303 | 200,794 | 8.52 |
| Exon | 831,013 | 20,365 | 1.50 | 483,324 | 41,619 | 1.47 | |
| Exon–exon | 24,864 | 1,313 | 0.04 | 17,286 | 1,209 | 0.05 | |
| Intergenic | 38,862,256 | 809,785 | 69.95 | 24,531,770 | 2,111,543 | 74.68 | |
| Reads not mapped | 11,370,125 | 280,989 | 20.47 | 5,016,740 | 613,594 | 15.27 | |
| Comparison | Up-Reg | DEG ID | FCvalue | Database | Database ID | Description | |
|---|---|---|---|---|---|---|---|
| (A) | TYa vs. ACo | ACo | MCH017805.1 | 216.33 | SwissProt | A0A1S3IRL5_LINUN | T-cell-specific GTP nucleotide protein |
| MCH006397.1 | 91.16 | pfam | PF00098.22 | Zinc knuckle | |||
| MCH020394.1 | 43.56 | SwissProt | K1QDR7_CRAGI | Metallo-beta-lactamase domain | |||
| MCH006871.1 | 39.41 | BLAST NR | XP_021346654.1 | Uncharacterized protein LOC110446034 | |||
| MCH029407.1 | 30.48 | - | - | - | |||
| MCH018328.1 | 29.46 | - | - | - | |||
| MCH027090.1 | 14.57 | eggNOG | 7739.XP_002593795.1 | Ribonuclease H protein | |||
| MCH020558.1 | 13.57 | - | - | - | |||
| MCH016839.1 | 12.61 | - | - | - | |||
| MCH009573.1 | 11.80 | pfam | PF13833.5 | EF-hand domain pair | |||
| TYa | MCH017771.1 | −73.63 | SwissProt | A0A210QJ59_MIZYE | Solute carrier family 46 member 3 | ||
| MCH033811.1 | −55.39 | BLAST NR | OPL20729.1 | Hypothetical protein AM593_09521 | |||
| MCH019286.1 | −48.22 | - | - | - | |||
| MCH000986.1 | −48.01 | pfam | PF00386.20 | C1q domain | |||
| MCH023306.1 | −37.26 | BLAST NR | XP_011445537.1 | Late secretory pathway protein AVL9 | |||
| MCH032954.1 | −34.72 | SwissProt | A0A210PU25_MIZYE | Carboxylic ester hydrolase | |||
| MCH026984.1 | −33.34 | BLAST NR | XP_022326523.1 | Uncharacterized protein LOC111126294 | |||
| MCH025896.1 | −31.24 | - | - | - | |||
| MCH032866.1 | −29.60 | BLAST NR | OPL20628.1 | Collagen chain alpha-1, partial | |||
| MCH024575.1 | −25.32 | - | - | - | |||
| (B) | TCo vs. AYa | AYa | MCH024186.1 | 44.93 | eggNOG | 126957.SMAR011084 | Intracellular signal transduction |
| MCH023948.1 | 39.73 | eggNOG | 144197.XP_008303365.1 | Caprin family member 2 | |||
| MCH018636.1 | 20.41 | eggNOG | 6412.HelroP185617 | Chymotrypsin-like elastase family | |||
| MCH017912.1 | 19.65 | BLAST NR | XP_022336752.1 | Glycolipid transfer protein-like | |||
| MCH024803.1 | 18.35 | eggNOG | 6500.XP_005107509.1 | Zinc ion binding | |||
| MCH028910.1 | 15.34 | - | - | - | |||
| MCH001759.1 | 13.44 | BLAST NR | XP_021343198.1 | Proton myo-inositol cotransporter | |||
| MCH023151.1 | 13.32 | BLAST NR | XP_021339691.1 | LARGE xylosyl glucuronyltransferase | |||
| MCH018876.1 | 12.92 | - | - | - | |||
| MCH019654.1 | 11.15 | BLAST NR | XP_006817019.1 | Uncharacterized protein LOC102808054 | |||
| TCo | MCH003374.1 | −58.66 | pfam | PF00643.23 | B-box zinc finger | ||
| MCH017866.1 | −54.50 | BLAST NR | XP_021372906.1 | Uncharacterized protein LOC110462954 | |||
| MCH009251.1 | −44.54 | BLAST NR | XP_002585874.1 | Hypothetical BRAFLDRAFT_110974 | |||
| MCH004849.1 | −39.00 | eggNOG | 6412.HelroP193608 | HEPN domain | |||
| MCH033594.1 | −37.98 | eggNOG | 6500.XP_005107641.1 | Peptidase activity | |||
| MCH000114.1 | −30.53 | eggNOG | 136037.KDR22711 | Interleukin-like EMT inducer | |||
| MCH003442.1 | −26.55 | pfam | PF17517.1 | IgGFc binding protein | |||
| MCH003165.1 | −24.15 | pfam | PF01885.15 | RNA 2′-phosphotransferase family | |||
| MCH019043.1 | −23.81 | pfam | PF00098.22 | Zinc knuckle | |||
| MCH008976.1 | −23.63 | SwissProt | U5PYN6_CRAGI | Toll-like receptor 3 |
| Comparison | Up-Reg | DEG ID | FCvalue | Database | Database ID | Description | |
|---|---|---|---|---|---|---|---|
| (A) | TCo vs. ACo | ACo | MCH015136.1 | 137.36 | - | - | - |
| MCH016307.1 | 39.17 | eggNOG | 10224.XP_006822956.1 | Coagulation factor C-terminal domain | |||
| MCH029407.1 | 30.40 | - | - | - | |||
| MCH014593.1 | 22.93 | BLAST NR | XP_011447428.1 | Complement C1q-like protein 4 | |||
| MCH027699.1 | 17.12 | eggNOG | 7955.ENSDARP00124702 | Zinc ion binding | |||
| MCH025331.1 | 14.51 | BLAST NR | XP_022325980.1 | NF-kappa-B-repressing factor-like | |||
| MCH013756.1 | 12.28 | pfam | PF00643.23 | B-box zinc finger | |||
| MCH027090.1 | 12.11 | eggNOG | 7739.XP_002593795.1 | Ribonuclease H protein | |||
| MCH009101.1 | 9.61 | BLAST NR | AAQ83892.1 | Interferon gamma-inducible protein 30 | |||
| MCH029447.1 | 9.17 | SwissProt | K1PVI1_CRAGI | Iporin | |||
| TCo | MCH017771.1 | −108.62 | SwissProt | A0A210QJ59_MIZYE | Solute carrier family 46 member 3 | ||
| MCH033811.1 | −96.51 | BLAST NR | OPL20729.1 | Hypothetical protein AM593_09521 | |||
| MCH014167.1 | −43.74 | pfam | PF00160.20 | Cyclophilin peptidyl-prolyl isomerase | |||
| MCH019531.1 | −43.29 | SwissProt | A0A210Q3C1_MIZYE | Cyclic GMP-AMP synthase | |||
| MCH008293.1 | −34.14 | eggNOG | 59894.ENSFALP014062 | Protein tyrosine phosphatase receptor | |||
| MCH029089.1 | −28.80 | BLAST NR | OWF44506.1 | GTPase IMAP family member 7 | |||
| MCH022781.1 | −28.78 | - | - | - | |||
| MCH034174.1 | −28.71 | BLAST NR | OPL33663.1 | Hypothetical protein AM593_02374 | |||
| MCH014606.1 | −24.68 | BLAST NR | XP_013385709.1 | Periodic tryptophan protein 2 homolog | |||
| MCH023306.1 | −22.61 | BLAST NR | XP_011445537.1 | Late secretory pathway protein | |||
| (B) | TYa vs. AYa | AYa | MCH023206.1 | 198.83 | pfam | PF04505.11 | Interferon-induced transmembrane |
| MCH018071.1 | 157.70 | pfam | PF00098.22 | Zinc knuckle | |||
| MCH029089.1 | 100.05 | BLAST NR | OWF44506.1 | GTPase IMAP family member 7 | |||
| MCH033168.1 | 76.37 | eggNOG | 209285.XP_6694433.1 | Cell wall integrity and stress response | |||
| MCH002581.1 | 38.25 | - | - | - | |||
| MCH011954.1 | 22.44 | SwissProt | A0A210Q5N4_MIZYE | PR domain zinc finger protein 2 | |||
| MCH034109.1 | 16.39 | BLAST NR | OPL33917.1 | Hypothetical protein AM593_04500 | |||
| MCH020918.1 | 15.13 | pfam | PF12349.7 | Sterol-sensing cleavage activation | |||
| MCH028910.1 | 14.26 | - | - | - | |||
| MCH024145.1 | 11.40 | SwissProt | A0A210QZX4_MIZYE | Beta-hexosaminidase | |||
| TYa | MCH006622.1 | −48.58 | SwissProt | K1QES1_CRAGI | Peroxisomal NADH pyrophosphatase | ||
| MCH025739.1 | −24.57 | BLAST NR | EKC32884.1 | Aquaporin-2 | |||
| MCH012314.1 | −24.02 | BLAST NR | OPL32994.1 | Hypothetical protein AM593_08485 | |||
| MCH008238.1 | −23.74 | BLAST NR | XP_022294261.1 | Protocadherin Fat 4-like | |||
| MCH029124.1 | −23.07 | - | - | - | |||
| MCH012803.1 | −20.50 | pfam | PF00023.29 | Ankyrin repeat | |||
| MCH022944.1 | −19.55 | BLAST NR | OPL33555.1 | Hypothetical protein AM593_04533 | |||
| MCH015808.1 | −18.05 | eggNOG | 10224.XP_002740032.1 | NACHT domain | |||
| MCH020824.1 | −17.64 | eggNOG | 10160.XP_004627003.1 | Regulation of T-helper differentiation | |||
| MCH032590.1 | −15.26 | pfam | PF00169.28 | PH domain |
| Comparison | Up-Reg | DEG ID | KEGG ID | FDR pvalue | Description | |
|---|---|---|---|---|---|---|
| (A) | TYa vs. ACo | ACo | - | - | - | - |
| TYa | MCH026485.1; MCH003699.1 | crg00340 | 2.63 × 10−3 | Histidine metabolism | ||
| MCH026485.1; MCH003699.1 | crg00380 | 4.21 × 10−3 | Tryptophan metabolism | |||
| MCH026485.1; MCH003699.1 | crg00330 | 4.39 × 10−3 | Arginine and proline metabolism | |||
| MCH015372.1 | crg03450 | 3.06 × 10−2 | Non-homologous end joining | |||
| MCH013290.1 | crg00500 | 3.66 × 10−2 | Starch and sucrose metabolism | |||
| MCH026485.1; MCH003699.1; MCH002453.1; MCH013290.1 | crg01100 | 3.66 × 10−2 | Metabolic pathways | |||
| MCH015372.1 | crg03440 | 3.66 × 10−2 | Homologous recombination | |||
| MCH002453.1 | crg00601 | 3.66 × 10−2 | Glycosphingolipid biosynthesis | |||
| (B) | TCo vs. AYa | AYa | MCH015895.1 | crg03060 | 9.86 × 10−2 | Protein export |
| MCH011254.1 | crg04512 | 9.86 × 10−2 | ECM–receptor interaction | |||
| MCH011254.1 | crg04145 | 1.41 × 10−1 | Phagosome | |||
| MCH000888.1 | crg04144 | 1.54 × 10−1 | Endocytosis | |||
| TCo | MCH032590.1 | crg00592 | 1.39 × 10−1 | alpha-Linolenic acid metabolism | ||
| MCH032590.1 | crg00591 | 1.39 × 10−1 | Linoleic acid metabolism | |||
| MCH032166.1 | crg00340 | 1.39 × 10−1 | Histidine metabolism | |||
| MCH032590.1 | crg00565 | 1.39 × 10−1 | Ether lipid metabolism | |||
| MCH032166.1 | crg00380 | 1.39 × 10−1 | Tryptophan metabolism | |||
| MCH032590.1 | crg00590 | 1.39 × 10−1 | Arachidonic acid metabolism | |||
| MCH032166.1 | crg00330 | 1.41 × 10−1 | Arginine and proline metabolism | |||
| MCH032590.1 | crg00564 | 1.41 × 10−1 | Glycerophospholipid metabolism | |||
| MCH000692.1 | crg04141 | 1.95 × 10−1 | Protein processing in ER | |||
| MCH007384.1 | crg03040 | 1.95 × 10−1 | Spliceosome | |||
| MCH032166.1; MCH032590.1 | crg01100 | 5.86 × 10−1 | Metabolic pathways |
| Comparison | Up-Reg | DEG ID | KEGG ID | FDR pvalue | Description | |
|---|---|---|---|---|---|---|
| (A) | TCo vs. ACo | ACo | - | - | - | - |
| TCo | MCH029783.1; MCH029782.1 | crg00750 | 4.31 × 10−4 | Vitamin B6 metabolism | ||
| MCH029783.1; MCH029782.1 | crg00260 | 7.35 × 10−3 | Gly, Ser, and Thre metabolism | |||
| MCH029783.1; MCH029782.1 | crg00270 | 7.35 × 10−3 | Cysteine Methionine metabolism | |||
| MCH029783.1; MCH029782.1 | crg01230 | 1.07 × 10−2 | Biosynthesis of amino acids | |||
| MCH029783.1; MCH029782.1 | crg01200 | 2.15 × 10−2 | Carbon metabolism | |||
| MCH003699.1 | crg00340 | 6.91 × 10−2 | Histidine metabolism | |||
| MCH003699.1 | crg00380 | 9.72 × 10−2 | Tryptophan metabolism | |||
| MCH002447.1 | crg00590 | 9.72 × 10−2 | Arachidonic acid metabolism | |||
| MCH003699.1 | crg00330 | 1.03 × 10−1 | Arginine and proline metabolism | |||
| MCH002447.1; MCH029782.1; MCH003699.1; MCH029783.1 | crg01100 | 1.17 × 10−1 | Metabolic pathways | |||
| MCH017106.1 | crg04144 | 2.06 × 10−1 | Endocytosis | |||
| (B) | TYa vs. AYa | AYa | MCH029782.1 | crg00750 | 3.67 × 10−2 | Vitamin B6 metabolism |
| MCH015895.1 | crg03060 | 6.96 × 10−2 | Protein export | |||
| MCH029782.1 | crg00260 | 6.96 × 10−2 | Glycine, Ser, and Thre metabolism | |||
| MCH029782.1 | crg00270 | 6.96 × 10−2 | Cysteine Methionine metabolism | |||
| MCH029782.1 | crg01230 | 7.77 × 10−2 | Biosynthesis of amino acids | |||
| MCH029782.1 | crg01200 | 1.03 × 10−1 | Carbon metabolism | |||
| MCH029782.1 | crg01100 | 6.87 × 10−1 | Metabolic pathways | |||
| TYa | MCH032590.1 | crg00592 | 7.36 × 10−2 | alpha-Linolenic acid metabolism | ||
| MCH032590.1 | crg00591 | 7.36 × 10−2 | Linoleic acid metabolism | |||
| MCH032590.1 | crg00565 | 7.36 × 10−2 | Ether lipid metabolism | |||
| MCH032590.1 | crg00590 | 8.15 × 10−2 | Arachidonic acid metabolism | |||
| MCH032590.1 | crg00564 | 8.90 × 10−2 | Glycerophospholipid metabolism | |||
| MCH012803.1 | crg04142 | 1.51 × 10−1 | Lysosome | |||
| MCH032590.1 | crg01100 | 7.01 × 10−1 | Metabolic pathways |
| Chromosome | Outlier SNP ID | Neighbor Gene ID | Database | Database ID | Description |
|---|---|---|---|---|---|
| Chr 1 | 2352_34 | MCH000058.1 | BLAST NR | XP_021343742.1 | uncharacterized protein C19orf44-like |
| Chr 1 | 6660_22 | MCH000252.1 | Swissprot | A0A210R0A2 | protein phosphatase 1 regulatory subunit 42 |
| Chr 1 | 6660_22 | MCH000253.1 | BLAST NR | XP_022314097.1 | zinc finger SWIM domain-containing protein |
| Chr 1 | 7164_20 | MCH000584.1 | Pfam | PF13650.5 | aspartyl protease |
| Chr 1 | 2467_50 | MCH000860.1 | BLAST NR | OWF55543.1 | hypothetical protein KP79_PYT08876 |
| Chr 1 | 2467_50 | MCH000861.1 | Swissprot | A0A1S3JHP6 | probable rRNA-processing protein EBP2 |
| Chr 1 | 13_74 | MCH002388.1 | - | - | - |
| Chr 1 | 13_74 | MCH002389.1 | - | - | - |
| Chr 1 | 2754_69 | MCH002393.1 | - | - | - |
| Chr 3 | 4790_45 | MCH018322.1 | - | - | - |
| Chr 3 | 4790_45 | MCH018323.1 | - | - | - |
| Chr 3 | 4790_45 | MCH018324.1 | - | - | - |
| Chr 3 | 9239_55; 6678_42 | MCH018404.1 | eggNOG | 8010.XP_010901236.1 | acid-sensing proton-gated ion channel |
| Chr 3 | 9239_55; 6678_42 | MCH018405.1 | eggNOG | 10224.XP_006826007.1 | zinc ion binding |
| Chr 4 | 836_12 | MCH019553.1 | eggNOG | 7739.XP_002593026.1 | centromere complex assembly |
| Chr 4 | 616_22 | MCH019761.1 | - | - | - |
| Chr 4 | 7299_61 | MCH019944.1 | BLAST NR | XP_022330633.1 | ninein-like protein |
| Chr 4 | 8684_32 | MCH019979.1 | BLAST NR | OWF51276.1 | neuronal acetylcholine receptor subunit a-3 |
| Chr 4 | 8684_32 | MCH019980.1 | - | - | - |
| Chr 4 | 4041_7 | MCH020207.1 | BLAST NR | XP_021356125.1 | transcription factor Sox-14-like |
| Chr 6 | 7434_34 | MCH025417.1 | Pfam | PF05721.12 | phytanoyl-CoA dioxygenase |
| Chr 6 | 3097_32 | MCH026223.1 | eggNOG | 28377.ENSACAP01319 | sphingosine N-acyltransferase activity |
| Chr 6 | 1203_57 | MCH026423.1 | BLAST NR | AGU13048.1 | myostatin |
| Chr 6 | 1870_40 | MCH026543.1 | Swissprot | K1QMC6 | uncharacterized protein |
| Chr 6 | 1870_40 | MCH026544.1 | Swissprot | K1QGI1 | uncharacterized protein |
| Chr 7 | 9179_59 | MCH028892.1 | - | - | - |
| Chr 7 | 9179_59 | MCH028893.1 | BLAST NR | XP_005093289.1 | histidine triad nucleotide-binding protein |
| Chr 8 | 7678_48 | MCH031237.1 | eggNOG | 10224.XP_002734847.1 | B-cell translocation gene |
| Chr 8 | 6819_55 | MCH031669.1 | eggNOG | 7739.XP_002597116.1 | Maelstrom spermatogenic transposon silencer |
| Chr 8 | 6819_55 | MCH031670.1 | BLAST NR | XP_022327614.1 | histone H4 transcription factor-like |
| Chr 8 | 6819_55 | MCH031671.1 | Swissprot | K1PE14 | Pogo transposable element with KRAB domain |
| Chr 9 | 1292_19 | MCH033323.1 | eggNOG | 6500.XP_005092544.1 | homeobox protein unc-4 homolog |
| Chr 9 | 7598_24 | MCH034354.1 | Pfam | PF03732.16 | retrotransposon gag protein |
| Chr 10 | 160_49 | MCH003892.1 | Pfam | PF00023.29 | ankyrin repeat |
| Chr 10 | 2547_39 | MCH004021.1 | Swissprot | A0A210PPH0 | protein mab-21-like 3 |
| Chr 10 | 2547_39 | MCH004022.1 | BLAST NR | XP_021378478.1 | protein mab-21-like 3 |
| Chr 10 | 6715_15 | MCH004774.1 | Pfam | PF15433.5 | mitochondrial 28S ribosomal protein S31 |
| Chr 10 | 6715_15 | MCH004775.1 | BLAST NR | XP_021362947.1 | osteopetrosis-associated transmembrane |
| Chr 11 | 3041_66 | MCH006832.1 | Pfam | PF03281.13 | mab-21 protein |
| Chr 11 | 3041_66 | MCH006833.1 | Pfam | PF00643.23 | B-box zinc finger |
| Chr 11 | 3041_66 | MCH006834.1 | eggNOG | 10224.XP_002733914.1 | RAB28, member RAS oncogene family |
| Chr 13 | 2134_59 | MCH011299.1 | Pfam | PF00022.18 | actin |
| Chr 13 | 2134_59 | MCH011300.1 | BLAST NR | XP_021366265.1 | dnaJ homolog subfamily C member 13-like |
| Chr 14 | 3073_57 | MCH012310.1 | Pfam | PF00654.19 | voltage-gated chloride channel |
| Chr 14 | 3422_30 | MCH012728.1 | BLAST NR | EKC34371.1 | Protein jagged-2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yévenes, M.; Gajardo, G.; Gallardo-Escárate, C. Decoding Local Adaptation in the Exploited Native Marine Mussel Mytilus chilensis: Genomic Evidence from a Reciprocal Transplant Experiment. Int. J. Mol. Sci. 2025, 26, 931. https://doi.org/10.3390/ijms26030931
Yévenes M, Gajardo G, Gallardo-Escárate C. Decoding Local Adaptation in the Exploited Native Marine Mussel Mytilus chilensis: Genomic Evidence from a Reciprocal Transplant Experiment. International Journal of Molecular Sciences. 2025; 26(3):931. https://doi.org/10.3390/ijms26030931
Chicago/Turabian StyleYévenes, Marco, Gonzalo Gajardo, and Cristian Gallardo-Escárate. 2025. "Decoding Local Adaptation in the Exploited Native Marine Mussel Mytilus chilensis: Genomic Evidence from a Reciprocal Transplant Experiment" International Journal of Molecular Sciences 26, no. 3: 931. https://doi.org/10.3390/ijms26030931
APA StyleYévenes, M., Gajardo, G., & Gallardo-Escárate, C. (2025). Decoding Local Adaptation in the Exploited Native Marine Mussel Mytilus chilensis: Genomic Evidence from a Reciprocal Transplant Experiment. International Journal of Molecular Sciences, 26(3), 931. https://doi.org/10.3390/ijms26030931