

Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis

Abstract
1. Introduction
2. Pathogenesis of MASH
2.1. Diet and Lifestyle Factors of MASH
2.2. Metabolic Factors of MASH
2.3. Genetic Factors of MASH
2.4. Epigenetic Factors
2.5. Microbiome in MASH
3. Therapeutic Targets in MASH
3.1. Targeting Lipid Metabolism and Reducing Lipotoxicity
3.1.1. Thyroid Hormone Receptor Beta Agonist
3.1.2. Acetyl-CoA Carboxylase Inhibitors
3.1.3. Diacylglycerol Acyltransferase 2 Inhibitor
3.1.4. Fibroblast Growth Factor Analogs
3.1.5. Farnesoid X Receptor Agonist
3.1.6. Peroxisome Proliferator-Activated Receptors
3.1.7. Stearoyl-CoA Desaturase 1 Modulators
3.1.8. Antisense Oligonucleotides Targeting Lipid Metabolism
3.2. Reducing Oxidative Stress and Inflammation
3.2.1. Apoptosis Signal-Regulating Kinase 1
3.2.2. C-C Chemokine Receptor Antagonists
3.2.3. Galectin-3 Inhibitors
3.2.4. RXFP1 Agonists
3.2.5. L-Ornithine L-Aspartate
3.3. Enhancing Insulin Sensitivity and Glucose Homeostasis
3.3.1. GLP-1 Receptor Agonists
3.3.2. Sodium–Glucose Cotransporter 2 Inhibitors
3.4. Targeting NASH Microbiome
3.4.1. Probiotics
3.4.2. Dietary Intervention
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ALT | Alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| APOC3 | Apolipoprotein C3 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| AST | Aspartate aminotransferase |
| ASOs | Antisense oligonucleotides |
| CCR | C-C chemokine receptor |
| CDCA | Chenodeoxycholic acid |
| CD98hc | CD98 heavy chain |
| CRD | Carbohydrate recognition domain |
| DGAT2 | Diacylglycerol acyltransferase 2 |
| DEX | Dexmedetomidine |
| DNL | De novo lipogenesis |
| ER | Endoplasmic reticulum |
| FATP5 | Fatty acid transport protein 5 |
| FGF4 | Fibroblast growth factor 4 |
| FGF19 | Fibroblast growth factor 19 |
| FMT | Fecal Microbiota Transplantation |
| FOS | Fructo-oligosaccharides |
| FXR | Farnesoid X receptor |
| G6Pase | Glucose-6-phophatase |
| GLP-1 | Glucagon-like peptide-1 |
| GSH | Glutathione |
| HDL | High-density lipoprotein |
| HFD | High-fat diet |
| HSCs | Hepatic stellate cells |
| HSD17B13 | Hydroxysteroid 17 beta dehydrogenase 13 |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| IL-6 | Interleukin 6 |
| LOLA | L-ornithine L-aspartate |
| IR | Insulin resistance |
| IRGM | Immunity-related GTPase family M |
| LPS | Lipopolysaccharides |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MBOAT7 | Membrane-bound O-acyltransferase domain-containing 7 |
| NAFLD | Non-alcoholic fatty liver disease |
| NAS | NAFLD activity score |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| NO | Nitric oxide |
| OCA | Obeticholic acid |
| PEPCK | Phosphoenolpyruvate carbpxykinase |
| PGC1a | Peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PNPLA3 | Patatin-like phospholipase domain-containing protein 3 |
| PPARs | Peroxisome proliferator-activated receptors |
| PRRs | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| RXFP1 | Relaxin/insulin-like family peptide receptor 1 |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SGLT2 | Sodium-glucose cotransporter 2 |
| SOD2 | Superoxide dismutase 2 |
| SHP | Small heterodimer partner |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| T2DM | Type 2 diabetes mellitus |
| TGF-β1 | Transforming growth factor beta 1 |
| THR-β | Thyroid hormone receptor beta |
| TNF-α | Tumor necrosis factor alpha |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| TMAO | Trimethylamine N-oxide |
| UPR | Unfolded protein response |
| VLDL | Very-low-density lipoprotein |
References
- Gofton, C.; Upendran, Y.; Zheng, M.H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef] [PubMed]
- Boccatonda, A.; Andreetto, L.; D’Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; Schiavone, C.; Cipollone, F.; Guagnano, M.T. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef]
- Huang, J.; Ou, W.; Wang, M.; Singh, M.; Liu, Y.; Liu, S.; Wu, Y.; Zhu, Y.; Kumar, R.; Lin, S. MAFLD Criteria Guide the Subtyping of Patients with Fatty Liver Disease. Risk Manag. Healthc. Policy 2021, 14, 491–501. [Google Scholar] [CrossRef]
- Alghamdi, W.; Mosli, M.; Alqahtani, S.A. Gut microbiota in MAFLD: Therapeutic and diagnostic implications. Ther. Adv. Endocrinol. Metab. 2024, 15, 20420188241242937. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Gabriel-Medina, P.; Ferrer-Costa, R.; Rodriguez-Frias, F.; Ciudin, A.; Augustin, S.; Rivera-Esteban, J.; Pericàs, J.M.; Selva, D.M. Influence of Type 2 Diabetes in the Association of PNPLA3 rs738409 and TM6SF2 rs58542926 Polymorphisms in NASH Advanced Liver Fibrosis. Biomedicines 2022, 10, 1015. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Tamura, A.; Kanai, S.; Tanaka, M.; Kanamori, Y.; Shirakawa, I.; Ito, A.; Oka, Y.; Hidaka, I.; Takami, T.; et al. Lysosomal cholesterol overload in macrophages promotes liver fibrosis in a mouse model of NASH. J. Exp. Med. 2023, 220, e20220681. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Jackson, K.L.; O’Sullivan, K.; Singh, J.; Giddy, T.; Deo, M.; Parry, L.J.; Ritchie, R.H.; Woodman, O.L.; Head, G.A.; et al. Endothelium-dependent relaxation is impaired in Schlager hypertensive (BPH/2J) mice by region-specific mechanisms in conductance and resistance arteries. Life Sci. 2023, 320, 121542. [Google Scholar] [CrossRef]
- Li, J.C.; Velagic, A.; Qin, C.X.; Li, M.; Leo, C.H.; Kemp-Harper, B.K.; Ritchie, R.H.; Woodman, O.L. Diabetes Attenuates the Contribution of Endogenous Nitric Oxide but Not Nitroxyl to Endothelium Dependent Relaxation of Rat Carotid Arteries. Front. Pharmacol. 2020, 11, 585740. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; Anthonisz, J.; Leo, C.H.; Kahlberg, N.; Velagic, A.; Li, M.; Jap, E.; Woodman, O.L.; Parry, L.J.; Horowitz, J.D.; et al. Nitric Oxide Resistance, Induced in the Myocardium by Diabetes, Is Circumvented by the Nitric Oxide Redox Sibling, Nitroxyl. Antioxid. Redox Signal. 2020, 32, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Kahlberg, N.; Leo, C.H.; Ng, H.H.; Rosli, S.; Deo, M.; Li, M.; Finlayson, S.; Walsh, J.; Parry, L.J.; et al. Annexin-A1 deficiency exacerbates pathological remodelling of the mesenteric vasculature in insulin-resistant, but not insulin-deficient, mice. Br. J. Pharmacol. 2020, 177, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Foo, S.Y.; Tan, J.C.W.; Tan, U.X.; Chua, C.K.; Ong, E.S. Green Extraction of Orange Peel Waste Reduces TNFα-Induced Vascular Inflammation and Endothelial Dysfunction. Antioxidants 2022, 11, 1768. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.S.; Low, J.; Tan, J.C.W.; Foo, S.Y.; Leo, C.H. Valorization of avocado seeds with antioxidant capacity using pressurized hot water extraction. Sci. Rep. 2022, 12, 13036. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.S.; Oh, C.L.Y.; Tan, J.C.W.; Foo, S.Y.; Leo, C.H. Pressurized Hot Water Extraction of Okra Seeds Reveals Antioxidant, Antidiabetic and Vasoprotective Activities. Plants 2021, 10, 1645. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e17. [Google Scholar] [CrossRef]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Askling, J.; Hultcrantz, R.; Kechagias, S. Cardiovascular risk factors in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Hydes, T.; Alam, U.; Cuthbertson, D.J. The Impact of Macronutrient Intake on Non-alcoholic Fatty Liver Disease (NAFLD): Too Much Fat, Too Much Carbohydrate, or Just Too Many Calories? Front. Nutr. 2021, 8, 640557. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Wu, P.S.; Lin, H.C. Pathogenesis and treatment of non-alcoholic steatohepatitis and its fibrosis. Clin. Mol. Hepatol. 2023, 29, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Harb, A.A.; Shechter, A.; Koch, P.A.; St-Onge, M.P. Ultra-processed foods and the development of obesity in adults. Eur. J. Clin. Nutr. 2023, 77, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Berná, G.; Romero-Gomez, M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int. 2020, 40 (Suppl. S1), 102–108. [Google Scholar] [CrossRef]
- Detopoulou, P.; Dedes, V.; Syka, D.; Tzirogiannis, K.; Panoutsopoulos, G.I. Relation of Minimally Processed Foods and Ultra-Processed Foods with the Mediterranean Diet Score, Time-Related Meal Patterns and Waist Circumference: Results from a Cross-Sectional Study in University Students. Int. J. Environ. Res. Public Health 2023, 20, 2806. [Google Scholar] [CrossRef]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Chen, L.; Deng, M.G.; Liang, Y.; Liu, F.; Nie, J.Q. Dietary choline intake and non-alcoholic fatty liver disease (NAFLD) in U.S. adults: National Health and Nutrition Examination Survey (NHANES) 2017–2018. Eur. J. Clin. Nutr. 2023, 77, 1160–1166. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, W.; Ma, X.; Ren, L.; Feng, M.; Hu, S.; Xue, C.; Chen, R. Roles and Mechanisms of Choline Metabolism in Nonalcoholic Fatty Liver Disease and Cancers. Front. Biosci. (Landmark Ed.) 2024, 29, 182. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and nonalcoholic fatty liver disease: Current perspectives. Diabetes Metab. Syndr. Obes. 2018, 11, 533–542. [Google Scholar] [CrossRef]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Tsung, A.; Huang, H. The Effects of Physical Exercise on Fatty Liver Disease. Gene Expr. 2018, 18, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Sohn, W.; Cho, Y.K. The effects of moderate alcohol consumption on non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S261–S267. [Google Scholar] [CrossRef] [PubMed]
- Um, Y.J.; Chang, Y.; Jung, H.S.; Cho, I.Y.; Shin, J.H.; Shin, H.; Wild, S.H.; Byrne, C.D.; Ryu, S. Sleep Duration, Sleep Quality, and the Development of Nonalcoholic Fatty Liver Disease: A Cohort Study. Clin. Transl. Gastroenterol. 2021, 12, e00417. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Ungprasert, P. Insomnia and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. J. Postgrad. Med. 2017, 63, 226–231. [Google Scholar] [CrossRef]
- Byrne, C.D. Fatty liver: Role of inflammation and fatty acid nutrition. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Felipo, V.; Urios, A.; Montesinos, E.; Molina, I.; Garcia-Torres, M.L.; Civera, M.; Olmo, J.A.; Ortega, J.; Martinez-Valls, J.; Serra, M.A.; et al. Contribution of hyperammonemia and inflammatory factors to cognitive impairment in minimal hepatic encephalopathy. Metab. Brain Dis. 2012, 27, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Shawcross, D.L.; Wright, G.; Olde Damink, S.W.; Jalan, R. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 125–138. [Google Scholar] [CrossRef]
- Thomsen, K.L.; Eriksen, P.L.; Kerbert, A.J.; De Chiara, F.; Jalan, R.; Vilstrup, H. Role of ammonia in NAFLD: An unusual suspect. JHEP Rep. 2023, 5, 100780. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. NAFLD: A critical role for the NLRP3 inflammasome in NASH. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Luci, C.; Bourinet, M.; Leclère, P.S.; Anty, R.; Gual, P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front. Endocrinol. 2020, 11, 597648. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. 2020, 245, 96–108. [Google Scholar] [CrossRef]
- Detopoulou, P.; Nomikos, T.; Fragopoulou, E.; Antonopoulou, S. Association of PAF and its Metabolic Enzymes with GGT and the Fatty Liver Index in Healthy Volunteers. Curr. Vasc. Pharmacol. 2021, 19, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Shi, A.; Wu, J. Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2022, 15, 2003–2030. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.K.; Tarzemani, S.; Aghajanzadeh, T.; Kasravi, M.; Hatami, B.; Zali, M.R.; Baghaei, K. Exploring the role of genetic variations in NAFLD: Implications for disease pathogenesis and precision medicine approaches. Eur. J. Med. Res. 2024, 29, 190. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Wieland, A.; Cree-Green, M.; Nadeau, K.; Sullivan, S.; Lanaspa, M.A.; Johnson, R.J.; Jensen, T. Lean NAFLD: An underrecognized and challenging disorder in medicine. Rev. Endocr. Metab. Disord. 2021, 22, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.Y.; Li, H.; Cao, H.X.; Pan, Q.; Fan, J.G. APOC3 rs2070667 Associates with Serum Triglyceride Profile and Hepatic Inflammation in Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2020, 2020, 8869674. [Google Scholar] [CrossRef]
- Zhu, X.; Xia, M.; Gao, X. Update on genetics and epigenetics in metabolic associated fatty liver disease. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221132138. [Google Scholar] [CrossRef] [PubMed]
- Auinger, A.; Valenti, L.; Pfeuffer, M.; Helwig, U.; Herrmann, J.; Fracanzani, A.L.; Dongiovanni, P.; Fargion, S.; Schrezenmeir, J.; Rubin, D. A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis. Horm. Metab. Res. 2010, 42, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Deng, X.; Liu, C.T.; Chung, R.T.; Long, M.T. The immunity-related GTPase M rs13361189 variant does not increase the risk for prevalent or incident steatosis; results from the Framingham Heart Study. Liver Int. 2019, 39, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Bellini, G.; Miraglia Del Giudice, E.; Nobili, V.; Rossi, F. The IRGM rs10065172 variant increases the risk for steatosis but not for liver damage progression in Italian obese children. J. Hepatol. 2017, 67, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Chang, T.E.; Perng, C.L.; Huang, Y.H. Genetic variations of three important antioxidative enzymes SOD2, CAT, and GPX1 in nonalcoholic steatohepatitis. J. Chin. Med. Assoc. 2021, 84, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Moretti, V.; Romeo, S.; Valenti, L. The contribution of genetics and epigenetics to MAFLD susceptibility. Hepatol. Int. 2024, 18, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Zobeiry, M.; Abdolahi, S.; Hatami, B.; Zali, M.R.; Baghaei, K. A crosstalk between epigenetic modulations and non-alcoholic fatty liver disease progression. Pathol. Res. Pract. 2023, 251, 154809. [Google Scholar] [CrossRef] [PubMed]
- Zaiou, M.; Amrani, R.; Rihn, B.; Hajri, T. Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines 2021, 9, 1256. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.R.; Glastras, S.J. Maternal Weight Management to Prevent the Developmental Programming of MAFLD in Offspring of Obese Mothers. Nutrients 2023, 15, 2155. [Google Scholar] [CrossRef] [PubMed]
- Fouda, S.; Vennikandam, M.M.; Pappachan, J.M.; Fernandez, C.J. Pregnancy and Metabolic-associated Fatty Liver Disease: A Clinical Update. J. Clin. Transl. Hepatol. 2022, 10, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Breij, L.M.; Kerkhof, G.F.; Hokken-Koelega, A.C. Accelerated infant weight gain and risk for nonalcoholic fatty liver disease in early adulthood. J. Clin. Endocrinol. Metab. 2014, 99, 1189–1195. [Google Scholar] [CrossRef]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care 2007, 30, 2638–2640. [Google Scholar] [CrossRef] [PubMed]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood predictors of adult fatty liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Romeo, S. Destined to develop NAFLD? The predictors of fatty liver from birth to adulthood. J. Hepatol. 2016, 65, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011, 6, e21662. [Google Scholar] [CrossRef]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Kazeminasab, F.; Marandi, S.M.; Baharlooie, M.; Nasr-Esfahani, M.H.; Ghaedi, K. Modulation and bioinformatics screening of hepatic mRNA-lncRNAs (HML) network associated with insulin resistance in prediabetic and exercised mice. Nutr. Metab. 2021, 18, 75. [Google Scholar] [CrossRef]
- Macvanin, M.T.; Gluvic, Z.; Bajic, V.; Isenovic, E.R. Novel insights regarding the role of noncoding RNAs in diabetes. World J. Diabetes 2023, 14, 958–976. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.; Urrata, V.; Gilardini, L.; Bertoli, S.; Calcaterra, V.; Zuccotti, G.V.; Cancello, R.; Carelli, S. Role of long non-coding RNAs in adipogenesis: State of the art and implications in obesity and obesity-associated diseases. Obes. Rev. 2021, 22, e13203. [Google Scholar] [CrossRef] [PubMed]
- Hochreuter, M.Y.; Dall, M.; Treebak, J.T.; Barrès, R. MicroRNAs in non-alcoholic fatty liver disease: Progress and perspectives. Mol. Metab. 2022, 65, 101581. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory Non-coding RNAs Network in Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Cai, Z.R.; Liu, J.; Wang, D.S.; Ju, H.Q.; Xu, R.H. Circular RNA: Metabolism, functions and interactions with proteins. Mol. Cancer 2020, 19, 172. [Google Scholar] [CrossRef]
- Xiao, J.; Joseph, S.; Xia, M.; Teng, F.; Chen, X.; Huang, R.; Zhai, L.; Deng, W. Circular RNAs Acting as miRNAs’ Sponges and Their Roles in Stem Cells. J. Clin. Med. 2022, 11, 2909. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z.; Loomba, R. The Intestinal Microbiome, Plasma Metabolome, and Liver Transcriptome: A Conspiracy Driving Hepatic Steatosis. Hepatology 2019, 70, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Duan, Y.; Yang, L.; Schnabl, B. Small metabolites, possible big changes: A microbiota-centered view of non-alcoholic fatty liver disease. Gut 2019, 68, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Salatino, A.; Castaño, G.O.; Landa, M.S.; Fijalkowky, C.; Garaycoechea, M.; Pirola, C.J. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut 2020, 69, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Lam-Galvez, B.R.; Kirsop, J.; Wang, Z.; Levison, B.S.; Gu, X.; Copeland, M.F.; Bartlett, D.; Cody, D.B.; Dai, H.J.; et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J. Clin. Investig. 2019, 129, 373–387. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900257. [Google Scholar] [CrossRef]
- Shanmugham, M.; Bellanger, S.; Leo, C.H. Gut-Derived Metabolite, Trimethylamine-N-oxide (TMAO) in Cardio-Metabolic Diseases: Detection, Mechanism, and Potential Therapeutics. Pharmaceuticals 2023, 16, 504. [Google Scholar] [CrossRef]
- Shanmugham, M.; Devasia, A.G.; Chin, Y.L.; Cheong, K.H.; Ong, E.S.; Bellanger, S.; Ramasamy, A.; Leo, C.H. Time-dependent specific molecular signatures of inflammation and remodelling are associated with trimethylamine-N-oxide (TMAO)-induced endothelial cell dysfunction. Sci. Rep. 2023, 13, 20303. [Google Scholar] [CrossRef]
- Tan, S.S.Y.; Shanmugham, M.; Chin, Y.L.; An, J.; Chua, C.K.; Ong, E.S.; Leo, C.H. Pressurized Hot Water Extraction of Mangosteen Pericarp and Its Associated Molecular Signatures in Endothelial Cells. Antioxidants 2023, 12, 1932. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Mesmin, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Chassaing, B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7 (Suppl. S2), 771–781. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Food, immunity, and the microbiome. Gastroenterology 2015, 148, 1107–1119. [Google Scholar] [CrossRef]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021, 27, 1836–1848. [Google Scholar] [CrossRef] [PubMed]
- Pfizer. A Phase 2a, Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging, Parallel Group Study to Evaluate Safety, Tolerability, and Pharmacodynamics of PF-05221304 Administered Daily for 16-weeks to Adult Subjects with Nonalcoholic Fatty Liver Disease. Available online: https://clinicaltrials.gov/study/NCT03248882 (accessed on 12 September 2024).
- Pfizer. A Study to Assess Pharmacodynamics, Safety and Tolerability of PF-05221304 and PF-06865571 Co-Administered for 6 Weeks In Adults with Non-Alcoholic Fatty Liver Disease. Available online: https://clinicaltrials.gov/study/NCT03776175 (accessed on 12 September 2024).
- Zhang, X.J.; Ji, Y.X.; Cheng, X.; Cheng, Y.; Yang, H.; Wang, J.; Zhao, L.P.; Huang, Y.P.; Sun, D.; Xiang, H.; et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci. Transl. Med. 2021, 13, eabg8116. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef] [PubMed]
- Puengel, T.; Tacke, F. Efruxifermin, an investigational treatment for fibrotic or cirrhotic nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2023, 32, 451–461. [Google Scholar] [CrossRef]
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jackson, J.P.; St Claire, R.L., 3rd; Freeman, K.; Brouwer, K.R.; Edwards, J.E. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacol. Res. Perspect. 2017, 5, e00329. [Google Scholar] [CrossRef]
- Roy, P.P.; Mahtab, M.A.; Rahim, M.A.; Yesmin, S.S.; Islam, S.B.; Akbar, S.M.F. Treatment of Nonalcoholic Steatohepatitis by Obeticholic Acid: Current Status. Euroasian J. Hepatogastroenterol. 2022, 12 (Suppl. S1), S46–S50. [Google Scholar]
- NIDDK. The Farnesoid X Receptor (FXR) Ligand Obeticholic Acid in NASH Treatment Trial (FLINT). Available online: https://clinicaltrials.gov/study/NCT01265498 (accessed on 12 September 2024).
- Markham, A.; Keam, S.J. Obeticholic Acid: First Global Approval. Drugs 2016, 76, 1221–1226. [Google Scholar] [CrossRef]
- Ratziu, V. Obeticholic Acid for the Treatment of Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2021, 17, 398–400. [Google Scholar] [CrossRef]
- Eslam, M.; Alvani, R.; Shiha, G. Obeticholic acid: Towards first approval for NASH. Lancet 2019, 394, 2131–2133. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated with the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non-Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. Study of Safety and Efficacy of Tropifexor (LJN452) in Patients with Non-alcoholic Steatohepatitis (NASH) (FLIGHT-FXR). Available online: https://clinicaltrials.gov/study/NCT02855164 (accessed on 12 September 2024).
- Sanyal, A.J.; Lopez, P.; Lawitz, E.J.; Lucas, K.J.; Loeffler, J.; Kim, W.; Goh, G.B.B.; Huang, J.F.; Serra, C.; Andreone, P.; et al. Tropifexor for nonalcoholic steatohepatitis: An adaptive, randomized, placebo-controlled phase 2a/b trial. Nat. Med. 2023, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients with Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Königshofer, P.; Peck-Radosavljevic, M.; et al. The Non-Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Radreau, P.; Joly, S.; Dubos, C.; Vonderscher, J.; Scalfaro, P.; Meldrum, E.; Darteil, R. In vitro and in vivo characterization of EYP001, a novel, potent and selective FXR agonist now in a Phase 2 clinical trial in NASH. Hepatology 2019, 70, 1267A. [Google Scholar]
- Ratziu, V.; Harrison, S.A.; Loustaud-Ratti, V.; Bureau, C.; Lawitz, E.; Abdelmalek, M.; Alkhouri, N.; Francque, S.; Girma, H.; Darteil, R.; et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J. Hepatol. 2023, 78, 479–492. [Google Scholar] [CrossRef]
- Enyo Pharma. Safety, Tolerability, Pharmacokinetics and Efficacy of EYP001a in Patients with Nonalcoholic Steatohepatitis (NASH). Available online: https://clinicaltrials.gov/study/NCT03812029 (accessed on 12 September 2024).
- Karim, G.; Bansal, M.B. Resmetirom: An Orally Administered, Smallmolecule, Liver-directed, β-selective THR Agonist for the Treatment of Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. touchREV. Endocrinol. 2023, 19, 60–70. [Google Scholar] [CrossRef]
- Petta, S.; Targher, G.; Romeo, S.; Pajvani, U.B.; Zheng, M.H.; Aghemo, A.; Valenti, L.V.C. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024, 44, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Madrigal Pharmaceuticals, Inc. Phase 2 Study of MGL-3196 in Patients with Non-Alcoholic Steatohepatitis (NASH). Available online: https://clinicaltrials.gov/study/NCT02912260 (accessed on 11 September 2024).
- Madrigal Pharmaceuticals, Inc. A Phase 3 Study to Evaluate Safety and Biomarkers of Resmetirom (MGL-3196) in Non Alcoholic Fatty Liver Disease Patients (MAESTRO-NAFLD1). Available online: https://clinicaltrials.gov/study/NCT04197479 (accessed on 11 September 2024).
- Harrison, S.A.; Ratziu, V.; Anstee, Q.M.; Noureddin, M.; Sanyal, A.J.; Schattenberg, J.M.; Bedossa, P.; Bashir, M.R.; Schneider, D.; Taub, R.; et al. Design of the phase 3 MAESTRO clinical program to evaluate resmetirom for the treatment of nonalcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2024, 59, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Taub, R.; Neff, G.W.; Lucas, K.J.; Labriola, D.; Moussa, S.E.; Alkhouri, N.; Bashir, M.R. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2023, 29, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Madrigal Pharmaceuticals, Inc. A Phase 3 Study to Evaluate the Efficacy and Safety of MGL-3196 (Resmetirom) in Patients with NASH and Fibrosis (MAESTRO-NASH). Available online: https://clinicaltrials.gov/study/NCT03900429 (accessed on 12 September 2024).
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef]
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Inventiva Pharma. A Phase 3 Study Evaluating Efficacy and Safety of Lanifibranor Followed by an Active Treatment Extension in Adult Patients with (NASH) and Fibrosis Stages F2 and F3 (NATiV3) (NATiV3). Available online: https://clinicaltrials.gov/study/NCT04849728 (accessed on 13 September 2024).
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- van den Hoek, A.M.; Verschuren, L.; Caspers, M.P.M.; Worms, N.; Menke, A.L.; Princen, H.M.G. Beneficial effects of elafibranor on NASH in E3L.CETP mice and differences between mice and men. Sci. Rep. 2021, 11, 5050. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Padole, P.; Arora, A.; Sharma, P.; Chand, P.; Verma, N.; Kumar, A. Saroglitazar for Nonalcoholic Fatty Liver Disease: A Single Centre Experience in 91 Patients. J. Clin. Exp. Hepatol. 2022, 12, 435–439. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.C.; et al. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Guo, X.; Xu, M.; Wang, Y.; Xie, W.; Chen, H.; Ma, M.; Li, X. Dexmedetomidine ameliorates high-fat diet-induced nonalcoholic fatty liver disease by targeting SCD1 in obesity mice. Pharmacol. Res. Perspect. 2021, 9, e00700. [Google Scholar] [CrossRef] [PubMed]
- Ionis Pharmaceuticals. ION839. Available online: https://www.ionis.com/medicines/ionis-az6-2-5-lrxazd2693/ (accessed on 13 September 2024).
- AstraZeneca. A Study to Evaluate AZD2693 in Participants Who Are Carriers of the PNPLA3 148M Risk Allele with Non-cirrhotic Non-alcoholic Steatohepatitis with Fibrosis (FORTUNA). Available online: https://clinicaltrials.gov/study/NCT05809934 (accessed on 13 September 2024).
- Ionis Pharmaceuticals. ION224. Available online: https://www.ionis.com/medicines/ion224/ (accessed on 13 September 2024).
- Choi, C.S.; Savage, D.B.; Kulkarni, A.; Yu, X.X.; Liu, Z.X.; Morino, K.; Kim, S.; Distefano, A.; Samuel, V.T.; Neschen, S.; et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem. 2007, 282, 22678–22688. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Murray, S.F.; Pandey, S.K.; Booten, S.L.; Bao, D.; Song, X.Z.; Kelly, S.; Chen, S.; McKay, R.; Monia, B.P.; et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42, 362–371. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Ionis Pharmaceuticals. Ionis Announces Positive Results from Phase 2 Study of ION224, an Investigational Medicine Demonstrating Clinical Efficacy in the Treatment of NASH/MASH. Available online: https://ir.ionis.com/news-releases/news-release-details/ionis-announces-positive-results-phase-2-study-ion224 (accessed on 13 September 2024).
- Ionis Pharmaceuticals. A Study to Assess the Safety, Efficacy, and Pharmacokinetics of Multiple Doses of ION224. Available online: https://clinicaltrials.gov/study/NCT04932512 (accessed on 13 September 2024).
- Nerstedt, A.; Cansby, E.; Andersson, C.X.; Laakso, M.; Stančáková, A.; Blüher, M.; Smith, U.; Mahlapuu, M. Serine/threonine protein kinase 25 (STK25): A novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia 2012, 55, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Cansby, E.; Nuñez-Durán, E.; Magnusson, E.; Amrutkar, M.; Booten, S.L.; Kulkarni, N.M.; Svensson, L.T.; Borén, J.; Marschall, H.U.; Aghajan, M.; et al. Targeted Delivery of Stk25 Antisense Oligonucleotides to Hepatocytes Protects Mice Against Nonalcoholic Fatty Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 597–618. [Google Scholar] [CrossRef]
- Sprint Bioscience. STK25. Available online: https://sprintbioscience.com/en/pipeline/internal-programs/#stk25 (accessed on 13 September 2024).
- Mak, L.Y.; Gane, E.; Schwabe, C.; Yoon, K.T.; Heo, J.; Scott, R.; Lee, J.H.; Lee, J.I.; Kweon, Y.O.; Weltman, M.; et al. A phase I/II study of ARO-HSD, an RNA interference therapeutic, for the treatment of non-alcoholic steatohepatitis. J. Hepatol. 2023, 78, 684–692. [Google Scholar] [CrossRef]
- Arrowhead Pharmaceuticals. Study of ARO-HSD in Healthy Volunteers and Patients with Non-Alcoholic Steatohepatitis (NASH) or Suspected NASH. Available online: https://clinicaltrials.gov/study/NCT04202354 (accessed on 13 September 2024).
- AstraZeneca. A Study to Assess the Safety, Tolerability and Pharmacokinetics of AZD7503 in Healthy Participants. Available online: https://clinicaltrials.gov/study/NCT05143905 (accessed on 13 September 2024).
- AstraZeneca. Knockdown of HSD17B13 mRNA, Pharmacokinetics, Safety, and Tolerability, of AZD7503 in Non-Alcoholic Fatty Liver Disease. Available online: https://clinicaltrials.gov/study/NCT05560607 (accessed on 13 September 2024).
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Gilead Sciences. Safety and Efficacy of Selonsertib in Adults with Nonalcoholic Steatohepatitis (NASH) and Bridging (F3) Fibrosis (STELLAR-3). Available online: https://clinicaltrials.gov/study/NCT03053050 (accessed on 14 September 2024).
- Gilead Sciences. Safety and Efficacy of Selonsertib in Adults with Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (NASH) (STELLAR-4). Available online: https://clinicaltrials.gov/study/NCT03053063 (accessed on 14 September 2024).
- Tamura, Y.; Sugimoto, M.; Murayama, T.; Minami, M.; Nishikaze, Y.; Ariyasu, H.; Akamizu, T.; Kita, T.; Yokode, M.; Arai, H. C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice. J. Atheroscler. Thromb. 2010, 17, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients with Nonalcoholic Steatohepatitis with Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef] [PubMed]
- Galectin Therapeutics Inc. Study Evaluating the Efficacy and Safety of Belapectin for the Prevention of Esophageal Varices in NASH Cirrhosis (NAVIGATE). Available online: https://clinicaltrials.gov/study/NCT04365868 (accessed on 14 September 2024).
- Mariño, K.V.; Cagnoni, A.J.; Croci, D.O.; Rabinovich, G.A. Targeting galectin-driven regulatory circuits in cancer and fibrosis. Nat. Rev. Drug Discov. 2023, 22, 295–316. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, F.R.; MacKinnon, A.; Brimert, T.; Gravelle, L.; Johnsson, R.E.; Kahl-Knutson, B.; Leffler, H.; Nilsson, U.J.; Pedersen, A.; Peterson, K.; et al. Discovery and Optimization of the First Highly Effective and Orally Available Galectin-3 Inhibitors for Treatment of Fibrotic Disease. J. Med. Chem. 2022, 65, 12626–12638. [Google Scholar] [CrossRef] [PubMed]
- Galecto Biotech AB. A Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Orally Administered GB1211 in Participants with Suspected or Confirmed Non-alcoholic Steatohepatitis (NASH) and Liver Fibrosis. Available online: https://clinicaltrials.gov/study/NCT04607655 (accessed on 14 September 2024).
- Jelinic, M.; Marshall, S.A.; Leo, C.H.; Parry, L.J.; Tare, M. From pregnancy to cardiovascular disease: Lessons from relaxin-deficient animals to understand relaxin actions in the vascular system. Microcirculation 2019, 26, e12464. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Marshall, S.A.; Stewart, D.; Unemori, E.; Parry, L.J.; Leo, C.H. Peptide hormone relaxin: From bench to bedside. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R753–R760. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Fernando, D.T.; Tran, L.; Ng, H.H.; Marshall, S.A.; Parry, L.J. Serelaxin Treatment Reduces Oxidative Stress and Increases Aldehyde Dehydrogenase-2 to Attenuate Nitrate Tolerance. Front. Pharmacol. 2017, 8, 141. [Google Scholar] [CrossRef]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Marshall, S.A.; Novak, J.; Tare, M.; Conrad, K.P.; Parry, L.J. Vascular actions of relaxin: Nitric oxide and beyond. Br. J. Pharmacol. 2017, 174, 1002–1014. [Google Scholar] [CrossRef]
- Leo, C.H.; Ng, H.H.; Marshall, S.A.; Jelinic, M.; Rupasinghe, T.; Qin, C.; Roessner, U.; Ritchie, R.H.; Tare, M.; Parry, L.J. Relaxin reduces endothelium-derived vasoconstriction in hypertension: Revealing new therapeutic insights. Br. J. Pharmacol. 2020, 177, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Ou, J.L.M.; Ong, E.S.; Qin, C.X.; Ritchie, R.H.; Parry, L.J.; Ng, H.H. Relaxin elicits renoprotective actions accompanied by increasing bile acid levels in streptozotocin-induced diabetic mice. Biomed. Pharmacother. 2023, 162, 114578. [Google Scholar] [CrossRef] [PubMed]
- Devasia, A.G.; Shanmugham, M.; Ramasamy, A.; Bellanger, S.; Parry, L.J.; Leo, C.H. Therapeutic potential of relaxin or relaxin mimetics in managing cardiovascular complications of diabetes. Biochem. Pharmacol. 2024, 229, 116507. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Parry, L.J.; Tare, M. Recent developments in relaxin mimetics as therapeutics for cardiovascular diseases. Curr. Opin. Pharmacol. 2019, 45, 42–48. [Google Scholar] [CrossRef]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Tare, M.; Parry, L.J. Serelaxin: A Novel Therapeutic for Vascular Diseases. Trends Pharmacol. Sci. 2016, 37, 498–507. [Google Scholar] [CrossRef]
- Ng, H.H.; Leo, C.H.; Parry, L.J.; Ritchie, R.H. Relaxin as a Therapeutic Target for the Cardiovascular Complications of Diabetes. Front. Pharmacol. 2018, 9, 501. [Google Scholar] [CrossRef]
- Bennett, R.G.; Heimann, D.G.; Singh, S.; Simpson, R.L.; Tuma, D.J. Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. 2014, 34, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, J.A.; Hayden, A.L.; Snowdon, V.K.; Aucott, R.L.; Stutchfield, B.M.; Mole, D.J.; Pellicoro, A.; Gordon-Walker, T.T.; Henke, A.; Schrader, J.; et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 2014, 59, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Hsieh, Y.C.; Chan, C.C.; Sun, H.J.; Huang, Y.H.; Hou, M.C.; Lin, H.C. Human relaxin-2 attenuates hepatic steatosis and fibrosis in mice with non-alcoholic fatty liver disease. Lab. Investig. 2019, 99, 1203–1216. [Google Scholar] [CrossRef]
- McBride, A.; Hoy, A.M.; Bamford, M.J.; Mossakowska, D.E.; Ruediger, M.P.; Griggs, J.; Desai, S.; Simpson, K.; Caballero-Hernandez, I.; Iredale, J.P.; et al. In search of a small molecule agonist of the relaxin receptor RXFP1 for the treatment of liver fibrosis. Sci. Rep. 2017, 7, 10806. [Google Scholar] [CrossRef] [PubMed]
- Kaftanovskaya, E.M.; Ng, H.H.; Soula, M.; Rivas, B.; Myhr, C.; Ho, B.A.; Cervantes, B.A.; Shupe, T.D.; Devarasetty, M.; Hu, X.; et al. Therapeutic effects of a small molecule agonist of the relaxin receptor ML290 in liver fibrosis. FASEB J. 2019, 33, 12435–12446. [Google Scholar] [CrossRef]
- Hu, M.; Wang, Y.; Liu, Z.; Yu, Z.; Guan, K.; Liu, M.; Wang, M.; Tan, J.; Huang, L. Hepatic macrophages act as a central hub for relaxin-mediated alleviation of liver fibrosis. Nat. Nanotechnol. 2021, 16, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, V.K.; Lachlan, N.J.; Hoy, A.M.; Hadoke, P.W.; Semple, S.I.; Patel, D.; Mungall, W.; Kendall, T.J.; Thomson, A.; Lennen, R.J.; et al. Serelaxin as a potential treatment for renal dysfunction in cirrhosis: Preclinical evaluation and results of a randomized phase 2 trial. PLoS Med. 2017, 14, e1002248. [Google Scholar] [CrossRef] [PubMed]
- Kobalava, Z.; Villevalde, S.; Kotovskaya, Y.; Hinrichsen, H.; Petersen-Sylla, M.; Zaehringer, A.; Pang, Y.; Rajman, I.; Canadi, J.; Dahlke, M.; et al. Pharmacokinetics of serelaxin in patients with hepatic impairment: A single-dose, open-label, parallel group study. Br. J. Clin. Pharmacol. 2015, 79, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Leo, C.H.; Girling, J.E.; Tare, M.; Beard, S.; Hannan, N.J.; Parry, L.J. Relaxin treatment reduces angiotensin II-induced vasoconstriction in pregnancy and protects against endothelial dysfunction†. Biol. Reprod. 2017, 96, 895–906. [Google Scholar] [CrossRef]
- Marshall, S.A.; O’Sullivan, K.; Ng, H.H.; Bathgate, R.A.D.; Parry, L.J.; Hossain, M.A.; Leo, C.H. B7-33 replicates the vasoprotective functions of human relaxin-2 (serelaxin). Eur. J. Pharmacol. 2017, 807, 190–197. [Google Scholar] [CrossRef]
- Ng, H.H.; Leo, C.H.; Parry, L.J. Serelaxin (recombinant human relaxin-2) prevents high glucose-induced endothelial dysfunction by ameliorating prostacyclin production in the mouse aorta. Pharmacol. Res. 2016, 107, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Leo, C.H.; Prakoso, D.; Qin, C.; Ritchie, R.H.; Parry, L.J. Serelaxin treatment reverses vascular dysfunction and left ventricular hypertrophy in a mouse model of Type 1 diabetes. Sci. Rep. 2017, 7, 39604. [Google Scholar] [CrossRef]
- Canbay, A.; Sowa, J.P. L-Ornithine L-Aspartate (LOLA) as a Novel Approach for Therapy of Non-alcoholic Fatty Liver Disease. Drugs 2019, 79 (Suppl. S1), 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Davuluri, G.; Silva, R.N.E.; Engelen, M.; Ten Have, G.A.M.; Prayson, R.; Deutz, N.E.P.; Dasarathy, S. Ammonia lowering reverses sarcopenia of cirrhosis by restoring skeletal muscle proteostasis. Hepatology 2017, 65, 2045–2058. [Google Scholar] [CrossRef]
- Rose, C.; Michalak, A.; Pannunzio, P.; Therrien, G.; Quack, G.; Kircheis, G.; Butterworth, R.F. L-ornithine-L-aspartate in experimental portal-systemic encephalopathy: Therapeutic efficacy and mechanism of action. Metab. Brain Dis. 1998, 13, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Cai, F.; Lin, N.; Ye, J.; Zheng, Q.; Ding, G. Effects of glutamine on oxidative stress and nuclear factor-κB expression in the livers of rats with nonalcoholic fatty liver disease. Exp. Ther. Med. 2014, 7, 365–370. [Google Scholar] [CrossRef]
- Sellmann, C.; Jin, C.J.; Degen, C.; De Bandt, J.P.; Bergheim, I. Oral Glutamine Supplementation Protects Female Mice from Nonalcoholic Steatohepatitis. J. Nutr. 2015, 145, 2280–2286. [Google Scholar] [CrossRef]
- Grungreiff, K.; Lambert-Baumann, J. Efficacy of L-ornithin-L-aspartate-granules in chronic liver diseases. Med. Welt Stuttg. 2001, 52, 219–226. [Google Scholar]
- Tian, L.Y.; Lu, L.G.; Tang, C.W.; Xie, Y.; Luo, H.S.; Tan, S.Y.; Pang, Z.; Zhang, Y.L.; Gong, L.B.; Li, Y.M.; et al. Aspartate-ornithine granules in the treatment of nonalcoholic steatohepatitis: A multiple-dose parallel controlled clinical trial. Zhonghua Gan Zang Bing Za Zhi 2013, 21, 528–532. [Google Scholar]
- Ermolova, T.; Ermolov, S. Correction of intrahepatic microcirculation disorders by L-ornithine-L-aspartate at the chronic liver diseases patients. Гастрoэнтерoл. Санкт-Петербурга 2018, 108–109. [Google Scholar] [CrossRef]
- Jackson, S.H.; Martin, T.S.; Jones, J.D.; Seal, D.; Emanuel, F. Liraglutide (victoza): The first once-daily incretin mimetic injection for type-2 diabetes. Pharm. Ther. 2010, 35, 498–529. [Google Scholar]
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.; le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wharton, S.; Liu, A.; Pakseresht, A.; Nørtoft, E.; Haase, C.L.; Mancini, J.; Power, G.S.; Vanderlelie, S.; Christensen, R.A.G. Real-World Clinical Effectiveness of Liraglutide 3.0 mg for Weight Management in Canada. Obesity 2019, 27, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, J.; Shen, Y.; Li, W.; Liu, M.; Wang, J.; Zhu, S.; Wu, M. Comparative Study of Liraglutide and Insulin Glargine on Glycemic Control and Pancreatic β-Cell Function in db/db Mice. Med. Sci. Monit. 2018, 24, 3293–3300. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, C.; Yang, C.; Li, B.; Wei, J.; Lin, Y.; Ye, P.; Hu, G.; Li, J. Liraglutide reduces hepatic glucolipotoxicity-induced liver cell apoptosis through NRF2 signaling in Zucker diabetic fatty rats. Mol. Med. Rep. 2018, 17, 8316–8324. [Google Scholar] [CrossRef]
- Khalifa, O.; Al-Akl, N.S.; Errafii, K.; Arredouani, A. Exendin-4 alleviates steatosis in an in vitro cell model by lowering FABP1 and FOXA1 expression via the Wnt/-catenin signaling pathway. Sci. Rep. 2022, 12, 2226. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Soto-Catalán, M.; Opazo-Ríos, L.; Quiceno, H.; Lázaro, I.; Moreno, J.A.; Gómez-Guerrero, C.; Egido, J.; Mas-Fontao, S. Semaglutide Improves Liver Steatosis and De Novo Lipogenesis Markers in Obese and Type-2-Diabetic Mice with Metabolic-Dysfunction-Associated Steatotic Liver Disease. Int. J. Mol. Sci. 2024, 25, 2961. [Google Scholar] [CrossRef] [PubMed]
- Reis-Barbosa, P.H.; Marcondes-de-Castro, I.A.; Marinho, T.S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. The mTORC1/AMPK pathway plays a role in the beneficial effects of semaglutide (GLP-1 receptor agonist) on the liver of obese mice. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101922. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhan, Y.; Wang, Y. Semaglutide May Ameliorate Fibrosis and Inhibit Epithelial-Mesenchymal Transition in Intrauterine Adhesion Models. Int. J. Mol. Sci. 2024, 25, 6196. [Google Scholar] [CrossRef]
- Mao, T.; Zhang, C.; Yang, S.; Bi, Y.; Li, M.; Yu, J. Semaglutide alters gut microbiota and improves NAFLD in db/db mice. Biochem. Biophys. Res. Commun. 2024, 710, 149882. [Google Scholar] [CrossRef]
- Ruda, A.I.; Ciobanu, D.M.; Inceu, G.; Rusu, A.; Roman, G. The effect of Dulaglutide on glycemic and weight control in patients with type 2 diabetes. Med. Pharm. Rep. 2023, 96, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Wen, S.; Zhou, L. The Relationship Between the Blood-Brain-Barrier and the Central Effects of Glucagon-Like Peptide-1 Receptor Agonists and Sodium-Glucose Cotransporter-2 Inhibitors. Diabetes Metab. Syndr. Obes. 2022, 15, 2583–2597. [Google Scholar] [CrossRef]
- Cusi, K.; Sattar, N.; García-Pérez, L.E.; Pavo, I.; Yu, M.; Robertson, K.E.; Karanikas, C.A.; Haupt, A. Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: A post hoc analysis of the AWARD programme. Diabet. Med. 2018, 35, 1434–1439. [Google Scholar] [CrossRef]
- Nevola, R.; Epifani, R.; Imbriani, S.; Tortorella, G.; Aprea, C.; Galiero, R.; Rinaldi, L.; Marfella, R.; Sasso, F.C. GLP-1 Receptor Agonists in Non-Alcoholic Fatty Liver Disease: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 1703. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Raciti, G.A.; Nigro, C.; Mannino, G.C.; Procopio, T.; Davalli, A.M.; Beguinot, F.; Sesti, G.; Miele, C.; Folli, F. The GLP-1 receptor agonists exenatide and liraglutide activate Glucose transport by an AMPK-dependent mechanism. J. Transl. Med. 2016, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cao, H.; Chen, Z.; Gu, H.; Guo, W.; Lin, B.; Weng, J. Short-term GLP-1 receptor agonist exenatide ameliorates intramyocellular lipid deposition without weight loss in ob/ob mice. Int. J. Obes. 2020, 44, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Ionut, V.; Woolcott, O.O.; Mkrtchyan, H.J.; Stefanovski, D.; Kabir, M.; Iyer, M.S.; Liu, H.; Castro, A.V.; Wu, Q.; Broussard, J.L.; et al. Exenatide Treatment Alone Improves β-Cell Function in a Canine Model of Pre-Diabetes. PLoS ONE 2016, 11, e0158703. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- AstraZeneca. Exenatide Study of Cardiovascular Event Lowering Trial (EXSCEL): A Trial To Evaluate Cardiovascular Outcomes After Treatment with Exenatide Once Weekly in Patients with Type 2 Diabetes Mellitus. Available online: https://clinicaltrials.gov/study/NCT01144338 (accessed on 15 September 2024).
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, L.; Xiao, J.J.; Liu, Q.; Ni, L.; Hu, J.W.; Yu, H.; Wu, X.; Zhang, B.F. Empagliflozin attenuates the renal tubular ferroptosis in diabetic kidney disease through AMPK/NRF2 pathway. Free Radic. Biol. Med. 2023, 195, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, M.; Fang, Z.; Li, C.; Zou, F.; Hu, L.; Zhang, W. Efficacy and safety of empagliflozin at different doses in patients with type 2 diabetes mellitus: A network meta-analysis based on randomized controlled trials. J. Clin. Pharm. Ther. 2022, 47, 270–286. [Google Scholar] [CrossRef]
- Xu, J.; Hirai, T.; Koya, D.; Kitada, M. Effects of SGLT2 Inhibitors on Atherosclerosis: Lessons from Cardiovascular Clinical Outcomes in Type 2 Diabetic Patients and Basic Researches. J. Clin. Med. 2021, 11, 137. [Google Scholar] [CrossRef]
- Nomura, S.; Sakamaki, S.; Hongu, M.; Kawanishi, E.; Koga, Y.; Sakamoto, T.; Yamamoto, Y.; Ueta, K.; Kimata, H.; Nakayama, K.; et al. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J. Med. Chem. 2010, 53, 6355–6360. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, N.; Meininger, G.; Ways, K.; Polidori, D.; Desai, M.; Qiu, R.; Alba, M.; Vercruysse, F.; Balis, D.; Shaw, W.; et al. Canagliflozin: A sodium glucose co-transporter 2 inhibitor for the treatment of type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2015, 1358, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Zhang, G.; He, L.; Ma, S.; Ma, H.; Zhai, J.; Wang, Z.; Zhang, T.; Wang, Y.; Guo, Y. Canagliflozin Attenuates Hepatic Steatosis and Atherosclerosis Progression in Western Diet-Fed ApoE-Knockout Mice. Drug Des. Dev. Ther. 2022, 16, 4161–4177. [Google Scholar] [CrossRef] [PubMed]
- Rahadian, A.; Fukuda, D.; Salim, H.M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Sata, M. Canagliflozin Prevents Diabetes-Induced Vascular Dysfunction in ApoE-Deficient Mice. J. Atheroscler. Thromb. 2020, 27, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Komoroski, B.; Vachharajani, N.; Boulton, D.; Kornhauser, D.; Geraldes, M.; Li, L.; Pfister, M. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin. Pharmacol. Ther. 2009, 85, 520–526. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Li, L.; Li, Q.; Huang, W.; Han, Y.; Tan, H.; An, M.; Xiang, Q.; Zhou, R.; Yang, L.; Cheng, Y. Dapagliflozin Alleviates Hepatic Steatosis by Restoring Autophagy via the AMPK-mTOR Pathway. Front. Pharmacol. 2021, 12, 589273. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sun, P.; Wang, Y.; Chen, Y.; Niu, Y.; Ding, Y.; Xu, N.; Zhang, Y.; Xie, W. Dapagliflozin attenuates steatosis in livers of high-fat diet-induced mice and oleic acid-treated L02 cells via regulating AMPK/mTOR pathway. Eur. J. Pharmacol. 2021, 907, 174304. [Google Scholar] [CrossRef]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 Inhibitors in NAFLD: Expanding Their Role beyond Diabetes and Cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar] [CrossRef]
- Ohkura, T. Ipragliflozin: A novel sodium-glucose cotransporter 2 inhibitor developed in Japan. World J. Diabetes 2015, 6, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Salim, H.M.; Fukuda, D.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front. Cardiovasc. Med. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Sasaki, J.; Tanimura-Inagaki, K.; Sakuma, I.; Sugihara, H.; Oikawa, S. Ipragliflozin and sitagliptin differentially affect lipid and apolipoprotein profiles in type 2 diabetes: The SUCRE study. Cardiovasc. Diabetol. 2024, 23, 56. [Google Scholar] [CrossRef]
- Kaku, K.; Kadowaki, T.; Seino, Y.; Okamoto, T.; Shirakawa, M.; Sato, A.; O’Neill, E.A.; Engel, S.S.; Kaufman, K.D. Efficacy and safety of ipragliflozin in Japanese patients with type 2 diabetes and inadequate glycaemic control on sitagliptin. Diabetes Obes. Metab. 2021, 23, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Shin, M.J.; Youn, G.S.; Yoon, S.J.; Choi, Y.R.; Kim, H.S.; Gupta, H.; Han, S.H.; Kim, B.K.; Lee, D.Y.; et al. Lactobacillus attenuates progression of nonalcoholic fatty liver disease by lowering cholesterol and steatosis. Clin. Mol. Hepatol. 2021, 27, 110–124. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X.; Liu, W.; Wei, H.; Liang, W.; Zhou, Y.; Ding, Y.; Ji, F.; Ho-Kwan Cheung, A.; Wong, N.; et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 2023, 79, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Carpi, R.Z.; Barbalho, S.M.; Sloan, K.P.; Laurindo, L.F.; Gonzaga, H.F.; Grippa, P.C.; Zutin, T.L.M.; Girio, R.J.S.; Repetti, C.S.F.; Detregiachi, C.R.P.; et al. The Effects of Probiotics, Prebiotics and Synbiotics in Non-Alcoholic Fat Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH): A Systematic Review. Int. J. Mol. Sci. 2022, 23, 8805. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, M.; Yi, X.; Lu, X.; Zhu, M.; Xue, M.; Tang, Y.; Zhu, Y. Short-chain fatty acids in nonalcoholic fatty liver disease: New prospects for short-chain fatty acids as therapeutic targets. Heliyon 2024, 10, e26991. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.; Mousa, N.; Shabana, W.; Refaey, M.; Elzehery, R.; Elhelaly, R.; Zalata, K.; Abdelsalam, M.; Eldeeb, A.A.; Awad, M.; et al. Rifaximin in nonalcoholic fatty liver disease: Hit multiple targets with a single shot. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jee, J.J.; Lee, Y.S.; Kim, D.Y.; Bang, J.Y.; Lee, H.W.; Koh, H.; Bae, S.H. Fecal microbiota transplantation improves hepatic fibro-inflammation via regulating oxidative stress in experimental NASH. Dig. Liver Dis. 2023, 55, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Selective Agonists of Thyroid Hormone Receptor Beta for the Treatment of NASH. N. Engl. J. Med. 2024, 390, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Cevallos, P.; Murúa-Beltrán Gall, S.; Uribe, M.; Chávez-Tapia, N.C. Understanding the Relationship between Nonalcoholic Fatty Liver Disease and Thyroid Disease. Int. J. Mol. Sci. 2023, 24, 14605. [Google Scholar] [CrossRef]
- López, M.; Varela, L.; Vázquez, M.J.; Rodríguez-Cuenca, S.; González, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yamada, M.; Matsumoto, S.; Monden, T.; Satoh, T.; Mori, M. Mouse sterol response element binding protein-1c gene expression is negatively regulated by thyroid hormone. Endocrinology 2006, 147, 4292–4302. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Amin, N.B.; Darekar, A.; Anstee, Q.M.; Wong, V.W.; Tacke, F.; Vourvahis, M.; Lee, D.S.; Charlton, M.; Alkhouri, N.; Nakajima, A.; et al. Efficacy and safety of an orally administered DGAT2 inhibitor alone or coadministered with a liver-targeted ACC inhibitor in adults with non-alcoholic steatohepatitis (NASH): Rationale and design of the phase II, dose-ranging, dose-finding, randomised, placebo-controlled MIRNA (Metabolic Interventions to Resolve NASH with fibrosis) study. BMJ Open 2022, 12, e056159. [Google Scholar] [PubMed]
- Aaldijk, A.S.; Verzijl, C.R.C.; Jonker, J.W.; Struik, D. Biological and pharmacological functions of the FGF19- and FGF21-coreceptor beta klotho. Front. Endocrinol. 2023, 14, 1150222. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef]
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 From Mice to Human. Front. Endocrinol. 2020, 11, 601349. [Google Scholar] [CrossRef]
- Jung, D.; Inagaki, T.; Gerard, R.D.; Dawson, P.A.; Kliewer, S.A.; Mangelsdorf, D.J.; Moschetta, A. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J. Lipid Res. 2007, 48, 2693–2700. [Google Scholar] [CrossRef]
- Kerr, T.A.; Saeki, S.; Schneider, M.; Schaefer, K.; Berdy, S.; Redder, T.; Shan, B.; Russell, D.W.; Schwarz, M. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev. Cell 2002, 2, 713–720. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; Baldelli, F.; Zampella, A.; Distrutti, E.; Fiorucci, S. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J. 2012, 26, 3021–3031. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Zhu, L.; Liu, H.N.; Tseng, Y.J.; Weng, S.Q.; Liu, T.T.; Dong, L.; Shen, X.Z. The protective effect and mechanism of the FXR agonist obeticholic acid via targeting gut microbiota in non-alcoholic fatty liver disease. Drug Des. Dev. Ther. 2019, 13, 2249–2270. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. Study of Safety, Tolerability, and Efficacy of a Combination Treatment of LJN452 and CVC in Adult Patients with NASH and Liver Fibrosis (TANDEM). Available online: https://clinicaltrials.gov/study/NCT03517540 (accessed on 12 September 2024).
- Adorini, L.; Trauner, M. FXR agonists in NASH treatment. J. Hepatol. 2023, 79, 1317–1331. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Lamas Bervejillo, M.; Ferreira, A.M. Understanding Peroxisome Proliferator-Activated Receptors: From the Structure to the Regulatory Actions on Metabolism. Adv. Exp. Med. Biol. 2019, 1127, 39–57. [Google Scholar] [PubMed]
- Qiu, Y.Y.; Zhang, J.; Zeng, F.Y.; Zhu, Y.Z. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol. Res. 2023, 192, 106786. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Peeters, A.; Baes, M. Role of PPARα in Hepatic Carbohydrate Metabolism. PPAR Res. 2010, 2010, 572405. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef]
- Walczak, R.; Tontonoz, P. PPARadigms and PPARadoxes: Expanding roles for PPARgamma in the control of lipid metabolism. J. Lipid Res. 2002, 43, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mao, S.; Chen, S.; Zhang, W.; Liu, C. PPARs-Orchestrated Metabolic Homeostasis in the Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 8974. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current Clinical Trial Status and Future Prospects of PPAR-Targeted Drugs for Treating Nonalcoholic Fatty Liver Disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Butruille, L.; Francque, S. Treating NASH by targeting peroxisome proliferator-activated receptors. J. Hepatol. 2023, 79, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, S.M.; Vajreswari, A. Stearoyl-CoA desaturase 1: A potential target for non-alcoholic fatty liver disease?-perspective on emerging experimental evidence. World J. Hepatol. 2022, 14, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Cohen, P.; Asilmaz, E.; Hardie, D.G.; Friedman, J.M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 6409–6414. [Google Scholar] [CrossRef]
- Miyazaki, M.; Kim, Y.C.; Gray-Keller, M.P.; Attie, A.D.; Ntambi, J.M. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J. Biol. Chem. 2000, 275, 30132–30138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Y.; Zhang, J.; Liu, B.; Deng, X.; Xin, S.; Xu, K. N-glycosylation of CREBH improves lipid metabolism and attenuates lipotoxicity in NAFLD by modulating PPARα and SCD-1. FASEB J. 2020, 34, 15338–15363. [Google Scholar] [CrossRef]
- Flowers, J.B.; Rabaglia, M.E.; Schueler, K.L.; Flowers, M.T.; Lan, H.; Keller, M.P.; Ntambi, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes 2007, 56, 1228–1239. [Google Scholar] [CrossRef]
- Jiang, G.; Li, Z.; Liu, F.; Ellsworth, K.; Dallas-Yang, Q.; Wu, M.; Ronan, J.; Esau, C.; Murphy, C.; Szalkowski, D.; et al. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Investig. 2005, 115, 1030–1038. [Google Scholar] [CrossRef]
- Zhang, M.; Tang, Y.; Tang, E.; Lu, W. MicroRNA-103 represses hepatic de novo lipogenesis and alleviates NAFLD via targeting FASN and SCD1. Biochem. Biophys. Res. Commun. 2020, 524, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, J.; Wang, C.; Li, K.; Liu, B.; Du, Y.; Xiao, F.; Chen, S.; Guo, F. miR-212-5p suppresses lipid accumulation by targeting FAS and SCD1. J. Mol. Endocrinol. 2017, 59, 205–217. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, W.; Zhou, M.; Tang, Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep. 2017, 7, 14493. [Google Scholar] [CrossRef] [PubMed]
- Kurikawa, N.; Takagi, T.; Wakimoto, S.; Uto, Y.; Terashima, H.; Kono, K.; Ogata, T.; Ohsumi, J. A novel inhibitor of stearoyl-CoA desaturase-1 attenuates hepatic lipid accumulation, liver injury and inflammation in model of nonalcoholic steatohepatitis. Biol. Pharm. Bull. 2013, 36, 259–267. [Google Scholar] [CrossRef]
- Rahimi, Y.; Mehdizadeh, A.; Nozad Charoudeh, H.; Nouri, M.; Valaei, K.; Fayezi, S.; Darabi, M. Hepatocyte differentiation of human induced pluripotent stem cells is modulated by stearoyl-CoA desaturase 1 activity. Dev. Growth Differ. 2015, 57, 667–674. [Google Scholar] [CrossRef]
- Iida, T.; Ubukata, M.; Mitani, I.; Nakagawa, Y.; Maeda, K.; Imai, H.; Ogoshi, Y.; Hotta, T.; Sakata, S.; Sano, R.; et al. Discovery of potent liver-selective stearoyl-CoA desaturase-1 (SCD1) inhibitors, thiazole-4-acetic acid derivatives, for the treatment of diabetes, hepatic steatosis, and obesity. Eur. J. Med. Chem. 2018, 158, 832–852. [Google Scholar] [CrossRef] [PubMed]
- Issandou, M.; Bouillot, A.; Brusq, J.M.; Forest, M.C.; Grillot, D.; Guillard, R.; Martin, S.; Michiels, C.; Sulpice, T.; Daugan, A. Pharmacological inhibition of stearoyl-CoA desaturase 1 improves insulin sensitivity in insulin-resistant rat models. Eur. J. Pharmacol. 2009, 618, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiong, F.; Zhang, S.; Liu, J.; Gao, G.; Xie, J.; Wang, Y. Oligonucleotide therapies for nonalcoholic steatohepatitis. Mol. Ther. Nucleic Acids 2024, 35, 102184. [Google Scholar] [CrossRef]
- Kefala, G.; Tziomalos, K. Apoptosis signal-regulating kinase-1 as a therapeutic target in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 189–191. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, R.; Hayakawa, T.; Takeda, K.; Ichijo, H. Therapeutic targets in the ASK1-dependent stress signaling pathways. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 434–453. [Google Scholar] [CrossRef]
- Yamamoto, E.; Dong, Y.F.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension 2008, 52, 573–580. [Google Scholar] [CrossRef]
- Doulberis, M.; Papadimitriou, K.; Papaefthymiou, A.; Kountouras, J.; Polyzos, S.A. The therapeutic potential of C-C chemokine receptor antagonists in nonalcoholic steatohepatitis. Explor. Med. 2020, 1, 170–183. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310-21. [Google Scholar] [CrossRef] [PubMed]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Townsend, S.A.; Newsome, P.N. Review article: New treatments in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 46, 494–507. [Google Scholar] [CrossRef]
- Friedman, S.L.; Pinzani, M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 2022, 75, 473–488. [Google Scholar] [CrossRef]
- Traber, P.G.; Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 2013, 8, e83481. [Google Scholar] [CrossRef] [PubMed]
- Al Attar, A.; Antaramian, A.; Noureddin, M. Review of galectin-3 inhibitors in the treatment of nonalcoholic steatohepatitis. Expert Rev. Clin. Pharmacol. 2021, 14, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Dings, R.P.M.; Miller, M.C.; Griffin, R.J.; Mayo, K.H. Galectins as Molecular Targets for Therapeutic Intervention. Int. J. Mol. Sci. 2018, 19, 905. [Google Scholar] [CrossRef]
- Bai, L.; Lu, W.; Tang, S.; Tang, H.; Xu, M.; Liang, C.; Zheng, S.; Liu, S.; Kong, M.; Duan, Z.; et al. Galectin-3 critically mediates the hepatoprotection conferred by M2-like macrophages in ACLF by inhibiting pyroptosis but not necroptosis signalling. Cell Death Dis. 2022, 13, 775. [Google Scholar] [CrossRef] [PubMed]
- Kram, M. Galectin-3 inhibition as a potential therapeutic target in non-alcoholic steatohepatitis liver fibrosis. World J. Hepatol. 2023, 15, 201–207. [Google Scholar] [CrossRef]
- Fujita, K.; Iwama, H.; Sakamoto, T.; Okura, R.; Kobayashi, K.; Takano, J.; Katsura, A.; Tatsuta, M.; Maeda, E.; Mimura, S.; et al. Galectin-9 suppresses the growth of hepatocellular carcinoma via apoptosis in vitro and in vivo. Int. J. Oncol. 2015, 46, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Hsu, D.K.; Baghy, K.; Serizawa, N.; Scott, F.; Takada, Y.; Takada, Y.; Fukada, H.; Chen, J.; et al. Galectin-3 modulates phagocytosis-induced stellate cell activation and liver fibrosis in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G439–G446. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, F.L.; Panera, N.; De Stefanis, C.; Mosca, A.; D’Oria, V.; Crudele, A.; De Vito, R.; Nobili, V.; Alisi, A. The Number of Liver Galectin-3 Positive Cells Is Dually Correlated with NAFLD Severity in Children. Int. J. Mol. Sci. 2019, 20, 3460. [Google Scholar] [CrossRef] [PubMed]
- Coelho, I.; Duarte, N.; Barros, A.; Macedo, M.P.; Penha-Gonçalves, C. Trem-2 Promotes Emergence of Restorative Macrophages and Endothelial Cells During Recovery from Hepatic Tissue Damage. Front. Immunol. 2020, 11, 616044. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, J.; Berger, H.; Tacke, F. Single Cell RNA Sequencing in NASH. Methods Mol. Biol. 2022, 2455, 181–202. [Google Scholar]
- Salvatore, T.; Nevola, R.; Pafundi, P.C.; Monaco, L.; Ricozzi, C.; Imbriani, S.; Rinaldi, L.; Sasso, F.C. Incretin Hormones: The Link between Glycemic Index and Cardiometabolic Diseases. Nutrients 2019, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Henriksen, T.I.; Pedersen, B.K.; Solomon, T.P. Glucagon like peptide-1-induced glucose metabolism in differentiated human muscle satellite cells is attenuated by hyperglycemia. PLoS ONE 2012, 7, e44284. [Google Scholar] [CrossRef]
- Gao, H.; Wang, X.; Zhang, Z.; Yang, Y.; Yang, J.; Li, X.; Ning, G. GLP-1 amplifies insulin signaling by up-regulation of IRbeta, IRS-1 and Glut4 in 3T3-L1 adipocytes. Endocrine 2007, 32, 90–95. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Alobaid, S.M.; Alshahrani, R.M.; Alonazi, A.S.; Alrasheed, N.M.; Alamin, M.A.; Alshammari, T.K.; Bin Dayel, A.F.; Elnagar, D.M.; Alotaibi, R.R.; Almuthnabi, L.A.; et al. Liraglutide Attenuates Diabetic Cardiomyopathy via the ILK/PI3K/AKT/PTEN Signaling Pathway in Rats with Streptozotocin-Induced Type 2 Diabetes Mellitus. Pharmaceuticals 2024, 17, 374. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Sumida, Y.; Tanaka, S.; Mori, K.; Taketani, H.; Ishiba, H.; Hara, T.; Okajima, A.; Umemura, A.; Nishikawa, T.; et al. Effect of 12-week dulaglutide therapy in Japanese patients with biopsy-proven non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatol. Res. 2017, 47, 1206–1211. [Google Scholar] [CrossRef]
- Robinson, S.; Boye, K.S.; Mody, R.; Strizek, A.A.; Konig, M.; Malik, R.E.; Kennedy-Martin, T. Real-World Effectiveness of Dulaglutide in Patients with Type 2 Diabetes Mellitus: A Literature Review. Diabetes Ther. 2020, 11, 1437–1466. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Wu, W.; Fan, X.; Zhong, X.; Wang, N.; Wang, Y.; Pan, T.; Du, Y. Dulaglutide Protects Mice against Diabetic Sarcopenia-Mediated Muscle Injury by Inhibiting Inflammation and Regulating the Differentiation of Myoblasts. Int. J. Endocrinol. 2023, 2023, 9926462. [Google Scholar] [CrossRef]
- Padda, I.S.; Mahtani, A.U.; Parmar, M. Sodium-Glucose Transport Protein 2 (SGLT2) Inhibitors; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Ugalde-Nicalo, P.; Welsh, J.A.; Angeles, J.E.; Cordero, M.; Harlow, K.E.; Alazraki, A.; Durelle, J.; Knight-Scott, J.; Newton, K.P.; et al. Effect of a Low Free Sugar Diet vs Usual Diet on Nonalcoholic Fatty Liver Disease in Adolescent Boys: A Randomized Clinical Trial. JAMA 2019, 321, 256–265. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e5. [Google Scholar] [CrossRef]
- Meslier, V.; Laiola, M.; Roager, H.M.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
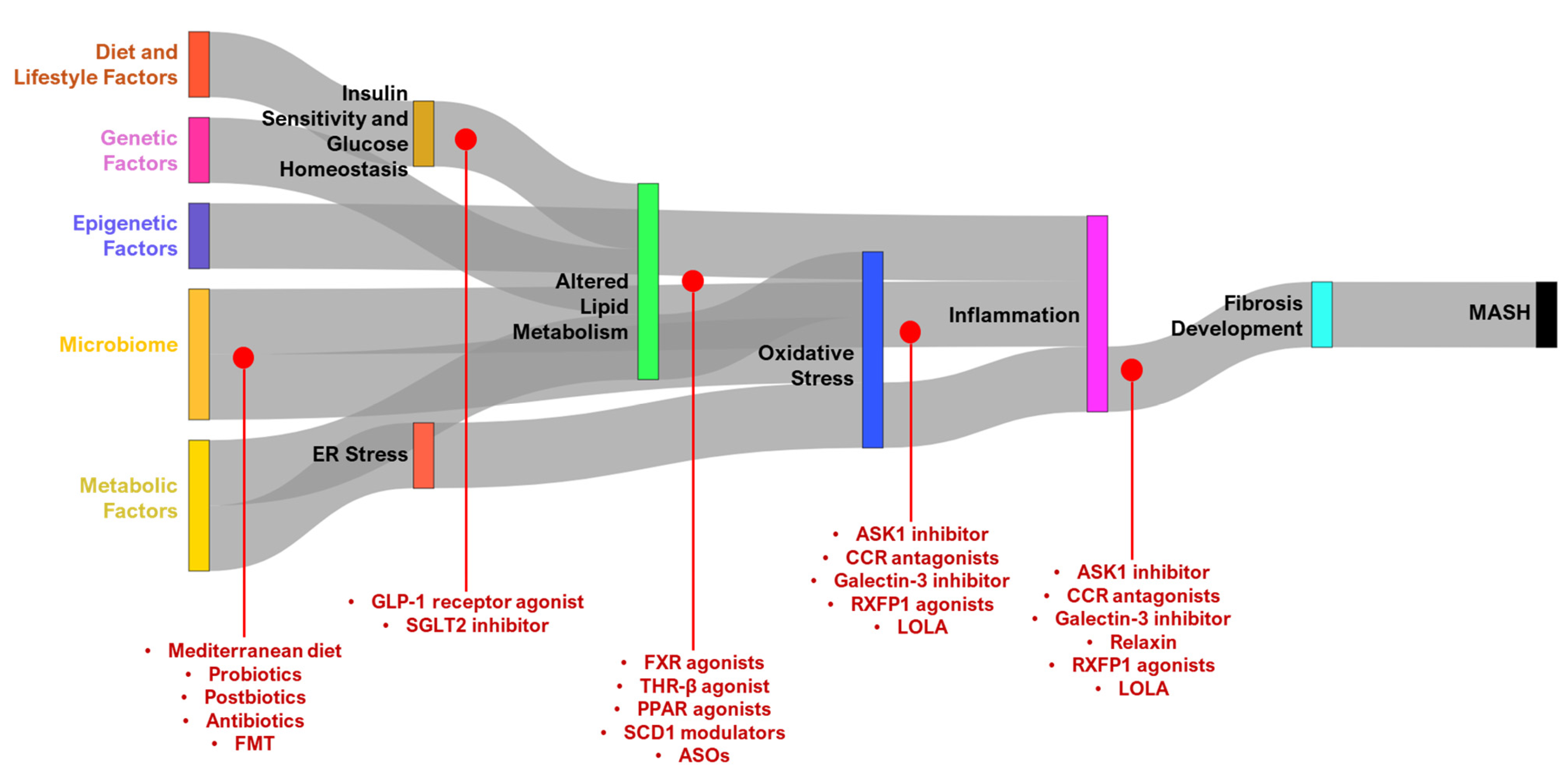

{kind=link}
| Category | Target | Drug Name | Target/Mechanism | Features/Benefits |
|---|---|---|---|---|
| Targeting Lipid Metabolism and Reducing Lipotoxicity | ACC1/2 inhibitor | PF-05221304 [105,106,107] | Inhibits acetyl-CoA carboxylase (ACC), targeting lipid synthesis pathways | Small molecule; dose-dependent declines in liver fat; minimal adverse events; phase 2a trials showed effectiveness in reducing liver fat percentage |
| IMA-1 [108] | Inhibits indoleamine 2,3-dioxygenase 1 (IDO1), modulating tryptophan metabolism and immune response | Small molecule; does not induce hyperlipidemia; halting diet induced NASH progression in male mice and macaque models; avoids side effects | ||
| DGAT2 inhibitor | PF-06865571 [105,106] | Inhibits diacylglycerol O-acyltransferase 2 (DGAT2), targeting triglyceride synthesis | Small molecule; combined therapy with PF-05221304 showed significant reduction in liver fat | |
| FGF analogs | Aldafermin (NGM282) [109] | Analog of FGF19, modulating bile acid synthesis, glucose metabolism, and lipid metabolism | Peptide; phase 2 study showed fibrosis improvement without worsening NASH | |
| Efruxifermin (EFX, also known as AKR-001 or AMG876) [110] | Fc-FGF21 fusion protein, targeting FGF21 receptor pathways involved in glucose and lipid metabolism | Peptide; improved pharmacokinetics and pharmacodynamics; showed potential in improving glycemic control; has good safety profile; demonstrated antifibrotic effects meeting FDA criteria for phase 3 trials | ||
| BFKB8488A [111] | Monoclonal antibody targeting FGFR1c/KLB, involved in metabolic regulation | Peptide; effective in reducing liver fat and enhancing metabolic parameters; favorable safety profile | ||
| FXR agonists | Obeticholic Acid (OCA) [112,113,114,115,116,117,118,119] | Semi-synthetic bile acid, potent FXR agonist | Regulates bile acid synthesis, improves insulin sensitivity; reduces liver fat, inflammation, and fibrosis.; demonstrated significant histological improvements in clinical trials but was associated with side effects like pruritus and altered lipid profiles; investigated in phase 2 and 3 trials | |
| Tropifexor (LJN452) [120,121,122,123,124] | Non-bile acid FXR agonist | Potent FXR agonist with significant reductions in hepatic triglycerides, liver fibrosis, and inflammation in preclinical studies; phase 2 trials showed efficacy in reducing liver enzymes and liver fat but with side effects like pruritus and changes in lipid profiles | ||
| Cilofexor (GS-9674) [125,126,127,128,129] | Non-bile acid FXR agonist | Shown to reduce hepatic steatosis and improve liver biochemistry in preclinical and phase 2 studies; demonstrated potential to reduce fibrosis and portal hypertension; side effects include pruritus at higher doses | ||
| Vonafexor (EYP001) [130,131,132] | Non-bile acid FXR agonist | Selective FXR agonist with promising results in preclinical studies, showing reductions in liver fat, fibrosis, and inflammation; phase 2 studies demonstrated reduced liver fat content and liver enzymes; mild pruritus observed at higher doses | ||
| THRβ agonist | Resmetirom [133,134,135,136,137,138,139,140,141] | Agonist of thyroid hormone receptor beta (THR-β), influencing cholesterol and lipid metabolism | Small molecule; enhances fatty acid oxidation and reduces lipogenesis in liver; promotes mitochondrial biogenesis and mitophagy and reduces lipid accumulation, inflammation, and fibrosis; decreases triglyceride synthesis, promotes LDL cholesterol uptake, and enhances bile acid synthesis, leading to improved lipid profiles by upregulating deiodinase type 1 (DIO1); improves intrahepatic thyroid hormone signaling and lipid metabolism, reducing hepatic lipotoxicity; first-ever FDA-approved treatment for NASH | |
| PPAR agonists | Lanifibranor [142,143,144] | Pan-PPAR agonist (PPARα, PPARδ, and PPARγ), targeting lipid and glucose metabolism | Small molecule; clinical studies in highly active NASH showed significant liver histology improvements without worsening fibrosis | |
| Elafibranor [145,146] | Dual PPARα/δ agonist, affecting lipid metabolism, inflammation, and glucose homeostasis | Small molecule; showed improving NASH-related histological parameters, failed to meet primary endpoint of NASH resolution without fibrosis progression, leading to its discontinuation | ||
| Saroglitazar [147,148] | Dual PPARα/γ agonist, modulating lipid and glucose metabolism | Small molecule; improved liver enzyme levels, hepatic fat content, and metabolic parameters; approved in India for NASH, with pending broader international approval | ||
| Pemafibrate [149,150,151] | Selective PPARα modulator, targeting lipid metabolism | Small molecule; showed benefits in hepatic inflammation, fibrosis, and biochemical scores | ||
| SCD1 modulators | Dexmedetomidine (DEX) [152] | Selective α2-adrenergic agonist, affecting central nervous system and sedation pathways | Small molecule; improved insulin sensitivity and reduced hepatic steatosis and inflammation in NAFLD mice | |
| Antisense oligonucleotides (ASOs) | AZD2693 (ION839) [153,154] | Targets PNPLA3 gene, specifically I148M variant linked to increased liver triglyceride accumulation | Reduces expression of mutated PNPLA3 protein, lowering hepatic lipid storage; shown to reduce liver triglyceride levels and alleviate steatosis in animal models; targeted NASH patients with PNPLA3 I148M variant, which is genetic risk factor in phase 2 trials | |
| DGAT2 ASO (ION224) [155,156,157,158,159,160] | Targets DGAT2 enzyme, involved in final step of triglyceride synthesis | Reduces hepatic lipid accumulation by inhibiting triglyceride synthesis; shown to improve liver histology in preclinical studies, reducing inflammation and fibrosis; phase 2 trials in NASH patients showed improvements in liver histology without worsening fibrosis | ||
| STK25 ASO [161,162,163] | Targets STK25, regulator of lipid metabolism and insulin sensitivity | Reduces liver fat accumulation and improves β-oxidation by decreasing STK25 activity; preclinical studies demonstrated significant reductions in liver steatosis and improved insulin sensitivity; currently in preclinical development | ||
| HSD17B13 ASO (ARO-HSD) [164,165,166,167] | Targets HSD17B13 gene, leveraging protective effects of loss-of-function variants associated with reduced risk of liver diseases | Reduces inflammation and fibrosis by silencing HSD17B13; early clinical trials showed reduced hepatic HSD17B13 expression and protein levels, with promising results in mitigating liver damage in MASLD and MASH patients | ||
| Reducing Oxidative Stress and Inflammation | ASK1 inhibitor | Selonsertib [168,169,170] | Inhibits apoptosis signal-regulating kinase 1 (ASK1), involved in inflammation and apoptosis pathways | Small molecule; reduces hepatic macrophage and stellate cell activation, thereby decreasing hepatic inflammation and fibrosis |
| CCR antagonists | Cenicriviroc (CVC) [171,172,173] | Dual CCR2/CCR5 antagonist, targeting inflammatory and fibrotic pathways | Small molecule; reduces influx and activation of these immune cells, thereby mitigating hepatic inflammation | |
| Galectin-3 inhibitor | Belapectin [174,175] | Inhibits galectin-3, targeting fibrosis and inflammation pathways | Large molecule galactoarabino-rhamnogalacturonan polysaccharide inhibitor derived from natural sources; prevents formation of oligomeric structures and lattice-like assemblies that are essential for activating pro-fibrotic signaling pathways, thereby halting fibrosis and reducing liver inflammation | |
| GB1107 [176,177] | Inhibits galectin-3, targeting fibrosis and inflammation pathways | Small molecule thiogalactoside inhibitor targeting carbohydrate recognition domain (CRD) of galectin-3 | ||
| GB1211 [178] | Inhibits galectin-3, targeting fibrosis and inflammation pathways | Small molecule; analog of GB1107 | ||
| RXFP1 agonists | Relaxin and its mimetics [179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200] | Activates RXFP1 | Induces multiple effects including anti-fibrosis, anti-inflammatory, vasoprotective actions in various organs | |
| L-ornithine L-aspartate (LOLA) [201,202,203,204,205,206,207,208] | Metabolic modulator, targeting ammonia detoxification pathways in liver and skeletal muscle | Small molecule; reduces hyperammonemia, enhances urea cycle function; lowers oxidative stress; improves microcirculation | ||
| Enhancing Insulin Sensitivity and Glucose Homeostasis | GLP-1 receptor agonist | Liraglutide [209,210,211,212,213,214,215] | GLP-1 receptor agonist, enhancing insulin secretion and glucose homeostasis | Peptide; promotes glucose uptake in muscle and adipose tissues; enhances glucose-dependent insulin secretion and inhibits glucagon secretion, slowing gastric emptying, and reducing appetite, collectively leading to improved glycemic control and weight loss |
| Semaglutide [216,217,218,219,220] | GLP-1 receptor agonist, enhancing insulin secretion and glucose homeostasis | Peptide; reduces liver fat and inflammation, improves hepatic insulin sensitivity, and slows fibrosis progression through central mechanisms in brain | ||
| Dulaglutide [221,222,223] | GLP-1 receptor agonist fused to Fc fragment, enhancing insulin secretion and glucose homeostasis | Peptide; enhances insulin sensitivity by stimulating insulin secretion and suppressing glucagon release; improves glycemic control and reducing hepatic glucose production; promotes weight loss by decreasing appetite | ||
| Exenatide [146,224,225,226,227,228,229] | Synthetic version of exendin-4, GLP-1 receptor agonist, enhancing insulin secretion and glucose homeostasis | Peptide; targets GLP-1 receptor, insulin signaling, AMPK, and inflammatory pathways, reducing hepatic inflammation and oxidative stress | ||
| SGLT2 inhibitor | Empagliflozin [230,231,232,233] | SGLT2 inhibitor specifically targets insulin signaling pathway | Small molecule; pre-clinical studies demonstrated reduction in hepatic fat accumulation, improved insulin resistance in various rodent models of NAFLD, including high-fat-diet-induced obese mice and humans | |
| Canagliflozin [234,235,236,237] | SGLT2 inhibitor targets pathways involved in de novo lipogenesis and fatty acid oxidation | Small molecule; reduced hepatic lipid content and inflammation by modulating key metabolic pathways in pre-clinical rodent models | ||
| Dapagliflozin [238,239,240,241] | SGLT2 inhibitor targets insulin signaling pathway involved in fatty acid oxidation, inflammation reduction, and fibrosis mitigation | Small molecule; reduce hepatic steatosis, inflammation, and liver fat content in high-fat-induced obese and diabetic mice models | ||
| Ipragliflozin [242,243,244,245,246] | SGLT2 inhibitor targeting AMPK pathway and reducing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling for reducing hepatic inflammation and oxidative stress | Small molecule; significantly reduced hepatic steatosis and inflammation in animal models of NAFLD and patients with type 2 diabetes in pre-clinical studies | ||
| Targeting NASH Microbiome | Probiotics | Lactobacillus [247] and Bifidobacterium species [248]; probiotics and prebiotics such as inulin and FOS [249] | Modulates gut microbiota to improve liver health | Reduce liver fat content and improve liver enzyme levels; shown to improve liver health outcomes in clinical studies |
| Postbiotics | Short-chain fatty acids (SCFAs) [250] | Anti-inflammatory compounds produced during bacterial fermentation of dietary fibers | Enhance gut barrier function and exhibit anti-inflammatory properties, contributing to better liver health | |
| Dietary intervention | Mediterranean diet, rich in fiber, polyphenols, and healthy fats | Promotes liver health through gut-friendly dietary patterns | Improves liver function by promoting healthy gut microbiota; linked to reduced liver fat and improved liver health outcomes in patients with NASH | |
| Antibiotics | Rifaximin [251] | Reduces endotoxin-producing bacteria in gut | Improves liver function by decreasing harmful bacteria; long-term use is controversial due to potential antibiotic resistance | |
| Fecal Microbiota Transplantation (FMT) [252,253] | Restores balanced gut microbiome through transfer of fecal matter from healthy donors | Improves insulin sensitivity and reduces liver fat, with early clinical trials showing positive outcomes in NASH patients. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devasia, A.G.; Ramasamy, A.; Leo, C.H. Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis. Int. J. Mol. Sci. 2025, 26, 1778. https://doi.org/10.3390/ijms26041778
Devasia AG, Ramasamy A, Leo CH. Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis. International Journal of Molecular Sciences. 2025; 26(4):1778. https://doi.org/10.3390/ijms26041778
Chicago/Turabian StyleDevasia, Arun George, Adaikalavan Ramasamy, and Chen Huei Leo. 2025. "Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis" International Journal of Molecular Sciences 26, no. 4: 1778. https://doi.org/10.3390/ijms26041778
APA StyleDevasia, A. G., Ramasamy, A., & Leo, C. H. (2025). Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis. International Journal of Molecular Sciences, 26(4), 1778. https://doi.org/10.3390/ijms26041778