
Bulge-Derived Epithelial Cells Isolated from Human Hair Follicles Using Enzymatic Digestion or Explants Result in Comparable Tissue-Engineered Skin

 , ,
, , 
Abstract
:1. Introduction
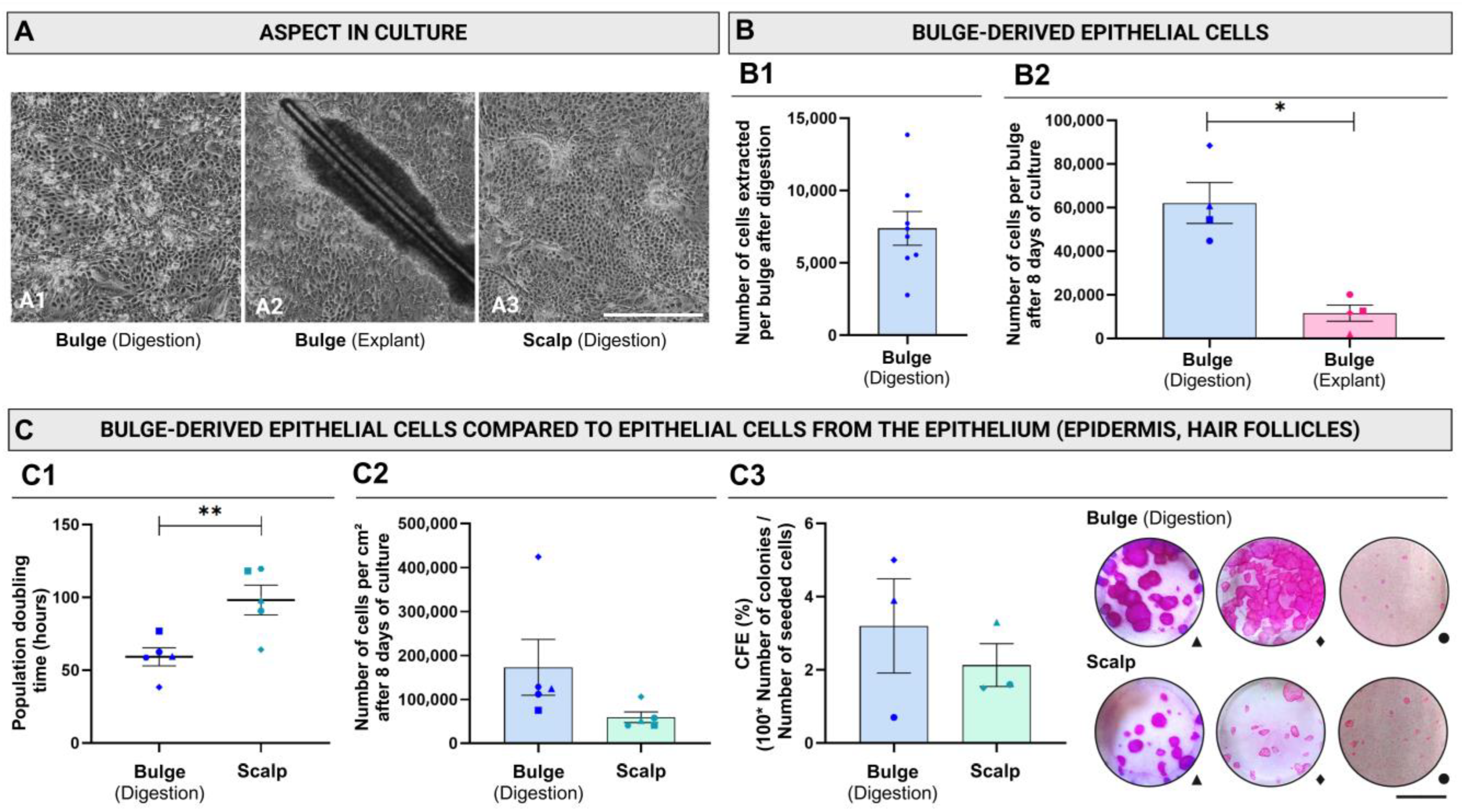
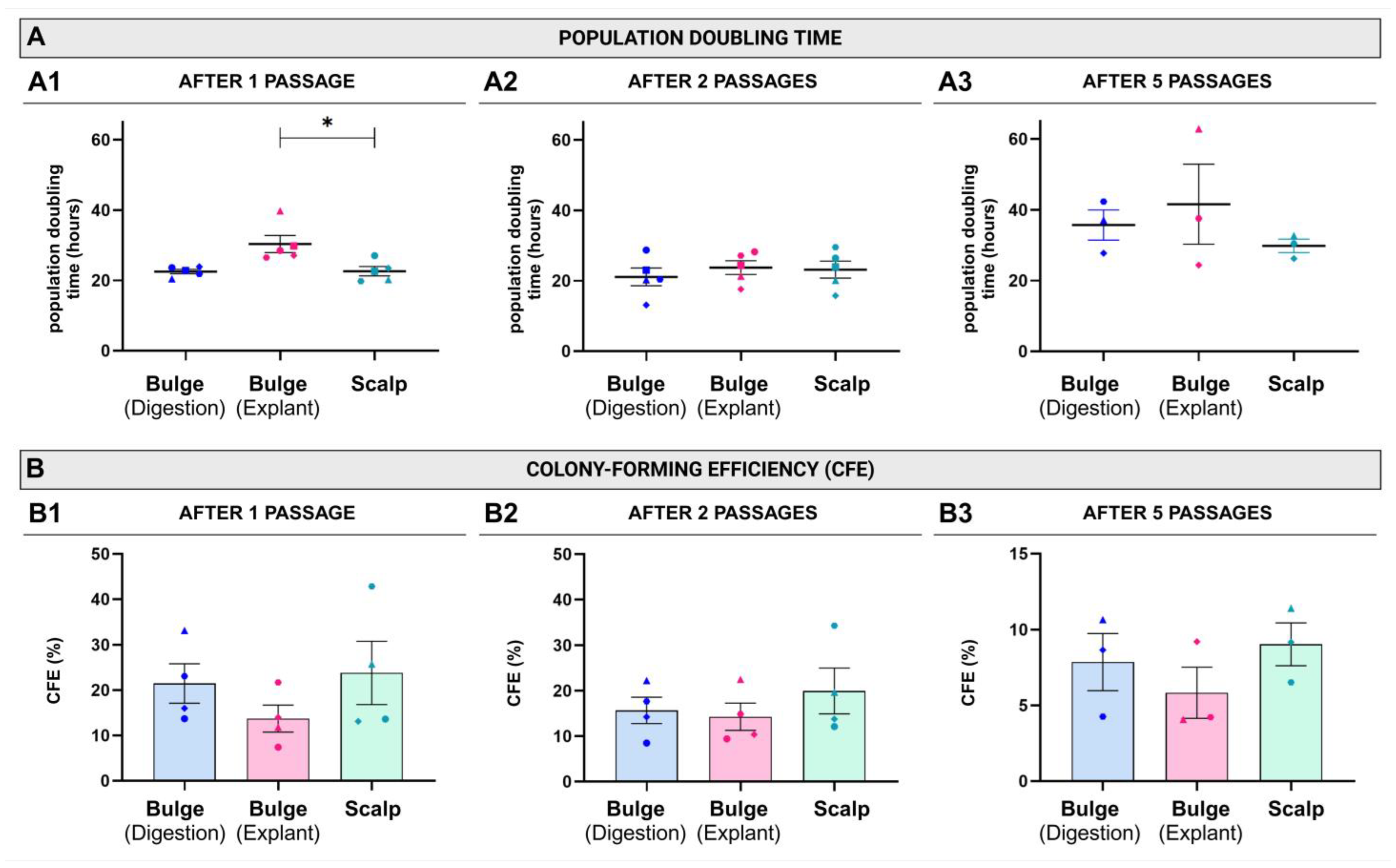
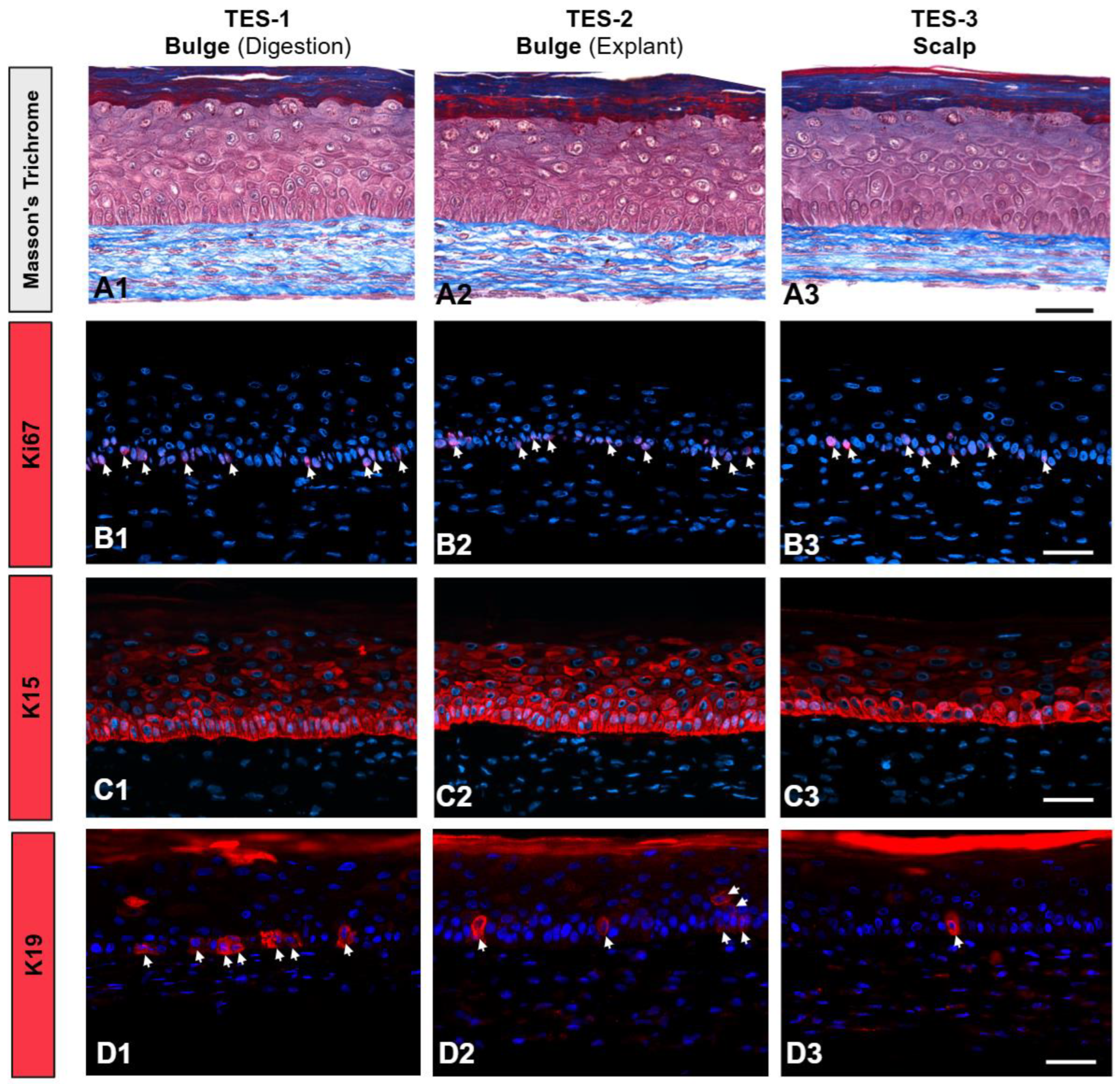
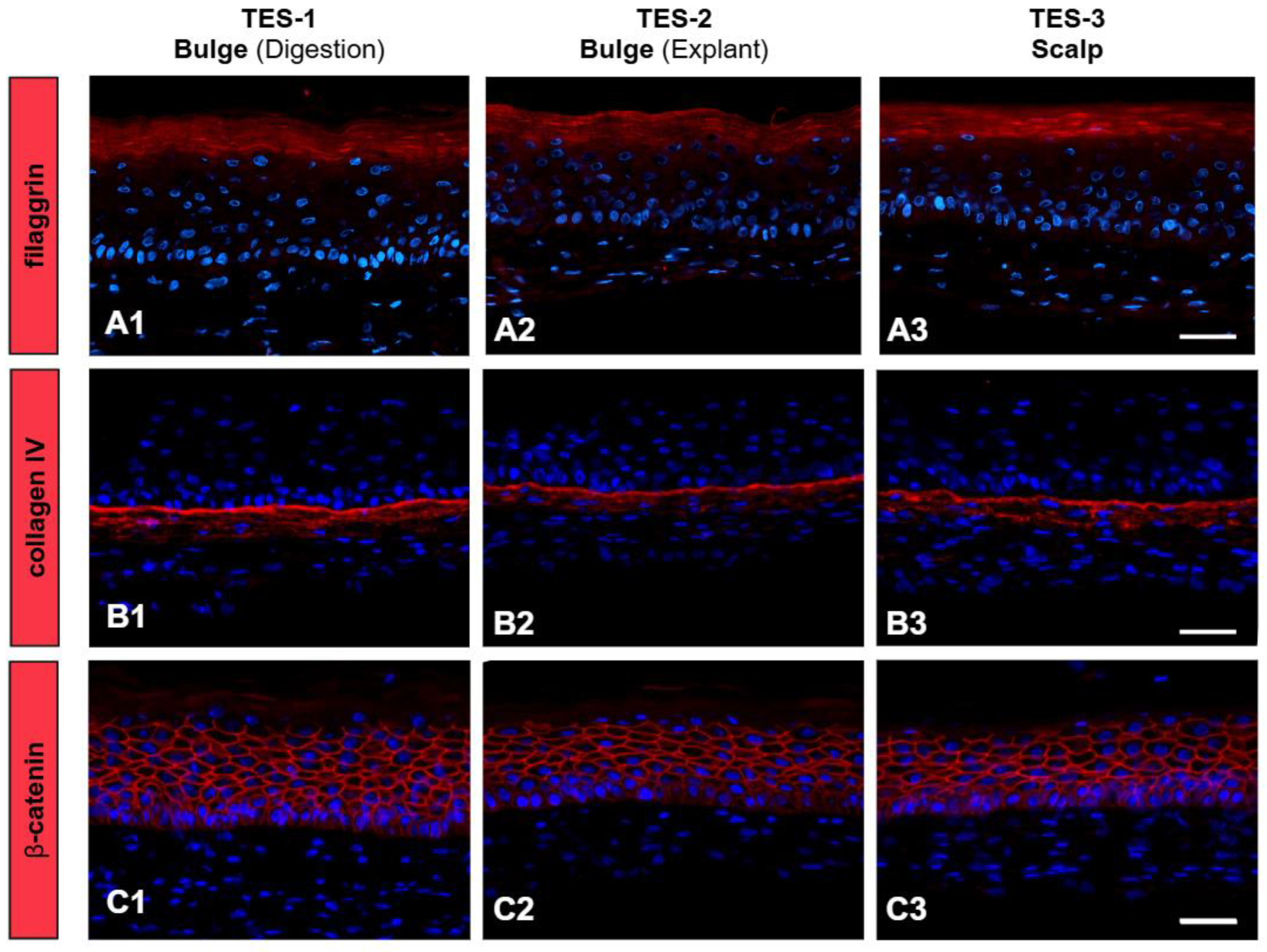
2. Results
3. Discussion
4. Materials and Methods
4.1. Tissue Collection
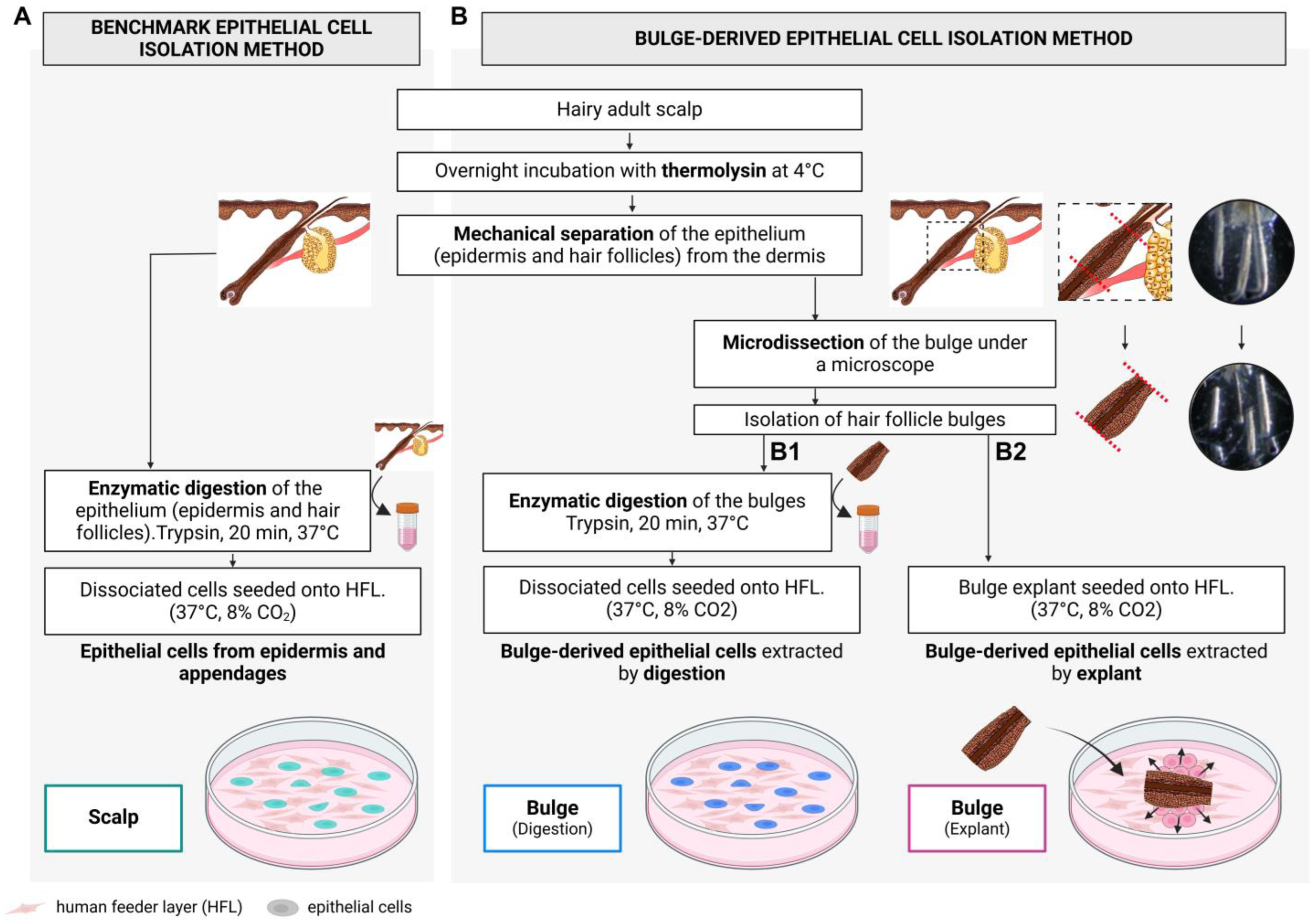
4.2. Skin Cell Isolation
4.2.1. Total Epithelium: Interfollicular Epidermis, Hair Follicles, and Sebaceous Glands
4.2.2. Bulge-Derived Cell Isolation: Microdissection
4.2.3. Bulge-Derived Cells Isolation: Enzymatic Digestion
4.2.4. Bulge-Derived Cells Isolation: Explant Approach
4.3. Subcultures
4.4. Skin Substitute Production
4.5. Histology and Immunofluorescence
4.6. Population Doubling Time and Cell-Yield Evaluation After the First Passage (P0)
4.7. Colony-Forming Efficiency (CFE)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kretzschmar, K.; Watt, F.M. Markers of Epidermal Stem Cell Subpopulations in Adult Mammalian Skin. Cold Spring Harb. Perspect. Med. 2014, 4, a013631. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-Renewal, Multipotency, and the Existence of Two Cell Populations within an Epithelial Stem Cell Niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The Hair Follicle as a Dynamic Miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef] [PubMed]
- Stenn, K.S.; Paus, R. Controls of Hair Follicle Cycling. Physiol. Rev. 2001, 81, 449–494. [Google Scholar] [CrossRef]
- Ohyama, M. Hair Follicle Bulge: A Fascinating Reservoir of Epithelial Stem Cells. J. Dermatol. Sci. 2007, 46, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Cotsarelis, G.; Sun, T.-T.; Lavker, R.M. Label-Retaining Cells Reside in the Bulge Area of Pilosebaceous Unit: Implications for Follicular Stem Cells, Hair Cycle, and Skin Carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Kloepper, J.E.; Tiede, S.; Brinckmann, J.; Reinhardt, D.P.; Meyer, W.; Faessler, R.; Paus, R. Immunophenotyping of the Human Bulge Region: The Quest to Define Useful in Situ Markers for Human Epithelial Hair Follicle Stem Cells and Their Niche. Exp. Dermatol. 2008, 17, 592–609. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Green, H. Serial Cultivation of Strains of Human Epidermal Keratinocytes: The Formation of Keratinizing Colonies from Single Cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Wüstner, L.S.; Klingenstein, M.; Frey, K.G.; Nikbin, M.R.; Milazzo, A.; Kleger, A.; Liebau, S.; Klingenstein, S. Generating IPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming. Cells 2022, 11, 1955. [Google Scholar] [CrossRef] [PubMed]
- Germain, L.; Larouche, D.; Nedelec, B.; Perreault, I.; Duranceau, L.; Bortoluzzi, P.; Cloutier, C.B.; Genest, H.; Caouette-Laberge, L.; Dumas, A.; et al. Autologous Bilayered Self-Assembled Skin Substitutes (Sasss) as Permanent Grafts: A Case Series of 14 Severely Burned Patients Indicating Clinical Effectiveness. Eur. Cell Mater. 2018, 36, 128–141. [Google Scholar] [CrossRef]
- Guiraud, B.; Hernandez-Pigeon, H.; Ceruti, I.; Mas, S.; Palvadeau, Y.; Saint-Martory, C.; Castex-Rizzi, N.; Duplan, H.; Bessou-Touya, S. Characterization of a Human Epidermis Model Reconstructed from Hair Follicle Keratinocytes and Comparison with Two Commercially Models and Native Skin. Int. J. Cosmet. Sci. 2014, 36, 485–493. [Google Scholar] [CrossRef]
- O’Connor, N.E.; Mulliken, J.B.; Banks-Schlegel, S.; Kehinde, O.; Green, H. Grafting of Burns with Cultured Epithelium Prepared from Autologous Epidermal Cell. Lancet 1981, 317, 75–78. [Google Scholar] [CrossRef]
- Cortez Ghio, S.; Larouche, D.; Doucet, E.J.; Germain, L. The Role of Cultured Autologous Bilayered Skin Substitutes as Epithelial Stem Cell Niches after Grafting: A Systematic Review of Clinical Studies. Burn. Open 2021, 5, 56–66. [Google Scholar] [CrossRef]
- Peterson, A.; Nair, L.S. Hair Follicle Stem Cells for Tissue Regeneration. Tissue Eng. Part B Rev. 2021, 28, 695–706. [Google Scholar] [CrossRef]
- Ji, S.; Zhu, Z.; Sun, X.; Fu, X. Functional Hair Follicle Regeneration: An Updated Review. Signal Transduct. Target. Ther. 2021, 6, 66. [Google Scholar] [CrossRef]
- Cheng, X.; Yu, Z.; Song, Y.; Zhang, Y.; Du, J.; Su, Y.; Ma, X. Hair Follicle Bulge-Derived Stem Cells Promote Tissue Regeneration during Skin Expansion. Biomed. Pharmacother. 2020, 132, 110805. [Google Scholar] [CrossRef] [PubMed]
- Goedkoop, R.J.; Hunziker, T. EPIDEX, A NOVEL TREATMENT FOR RECALCITRANT CHRONIC LEG ULCERS. ASAIO J. 2002, 48, 187. [Google Scholar] [CrossRef]
- Mignone, J.L.; Roig-Lopez, J.L.; Fedtsova, N.; Schones, D.E.; Manganas, L.N.; Maletic-Savatic, M.; Keyes, W.M.; Mills, A.A.; Gleiberman, A.; Zhang, M.Q.; et al. Neural Potential of a Stem Cell Population in the Hair Follicle. Cell Cycle 2007, 6, 2161–2170. [Google Scholar] [CrossRef]
- Yashiro, M.; Mii, S.; Aki, R.; Hamada, Y.; Arakawa, N.; Kawahara, K.; Hoffman, R.M.; Amoh, Y. From Hair to Heart: Nestin-Expressing Hairfollicle- Associated Pluripotent (HAP) Stem Cells Differentiate to Beating Cardiac Muscle Cells. Cell Cycle 2015, 14, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
- Urano-Morisawa, E.; Takami, M.; Suzawa, T.; Matsumoto, A.; Osumi, N.; Baba, K.; Kamijo, R. Induction of Osteoblastic Differentiation of Neural Crest-Derived Stem Cells from Hair Follicles. PLoS ONE 2017, 12, e0174940. [Google Scholar] [CrossRef] [PubMed]
- Amoh, Y.; Li, L.; Katsuoka, K.; Penman, S.; Hoffman, R.M. Multipotent Nestin-Positive, Keratin-Negative Hair-Follicle Bulge Stem Cells Can Form Neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 5530–5534. [Google Scholar] [CrossRef] [PubMed]
- Amoh, Y.; Li, L.; Campillo, R.; Kawahara, K.; Katsuoka, K.; Penman, S.; Hoffman, R.M. Implanted Hair Follicle Stem Cells Form Schwann Cells That Support Repair of Severed Peripheral Nerves. Proc. Natl. Acad. Sci. USA 2005, 102, 17734–17738. [Google Scholar] [CrossRef] [PubMed]
- Fatehi, A.; Sadat, M.; Fayyad, M.; Tang, J.; Han, D.; Rogers, I.M.; Taylor, D. Efficient Generation of Pancreatic Progenitor Cells from Induced Pluripotent Stem Cells Derived from a Non-Invasive and Accessible Tissue Source—The Plucked Hair Follicle. Cells 2024, 13, 1010. [Google Scholar] [CrossRef]
- Limat, A.; Noser, F.K. Serial Cultivation of Single Keratinocytes from the Outer Root Sheath of Human Scalp Hair Follicles. J. Investig. Dermatol. 1986, 87, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Gho, C.G.; Braun, J.E.F.; Tilli, C.M.L.J.; Neumann, H.A.M.; Ramaekers, F.C.S. Human Follicular Stem Cells: Their Presence in Plucked Hair and Follicular Cell Culture. Br. J. Dermatol. 2004, 150, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, M.; Terunuma, A.; Tock, C.L.; Radonovich, M.F.; Pise-Masison, C.A.; Hopping, S.B.; Brady, J.N.; Udey, M.C.; Vogel, J.C. Characterization and Isolation of Stem Cell-Enriched Human Hair Follicle Bulge Cells. J. Clin. Investig. 2006, 116, 249–260. [Google Scholar] [CrossRef]
- Oh, J.H.; Mohebi, P.; Farkas, D.L.; Tajbakhsh, J. Towards Expansion of Human Hair Follicle Stem Cells in Vitro. Cell Prolif. 2011, 44, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Draheim, K.; Lyle, S. Isolation and Culture of Adult Epithelial Stem Cells from Human Skin. J. Vis. Exp. 2011, 49, 2561. [Google Scholar] [CrossRef]
- Molina, B.J.; Finol, H.J. Isolation, Cultivation, and Morphological Characteristics of Hair Follicle Adult Stem Cells in the Bulge Region in Mouse and Human. Microsc. Res. 2020, 8, 9–30. [Google Scholar] [CrossRef]
- Li, H.; Masieri, F.F.; Schneider, M.; Bartella, A.; Gaus, S.; Hahnel, S.; Zimmerer, R.; Sack, U.; Maksimovic-Ivanic, D.; Mijatovic, S.; et al. The Middle Part of the Plucked Hair Follicle Outer Root Sheath Is Identified as an Area Rich in Lineage-Specific Stem Cell Markers. Biomolecules 2021, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gupta, S.; Mohanty, S.; Bhargava, B.; Airan, B. Stem Cell Niche Is Partially Lost during Follicular Plucking: A Preliminary Pilot Study. Int. J. Trichology 2013, 5, 97–100. [Google Scholar] [CrossRef]
- Walzer, C.; Benathan, M.; Frenk, E. Thermolysin Treatment: A New Method for Dermo-Epidermal Separation. J. Investig. Dermatol. 1989, 92, 78–81. [Google Scholar] [CrossRef]
- Germain, L.; Rouabhia, M.; Guignard, R.; Carrier, L.; Bouvard, V.; Auger, F.A. Improvement of Human Keratinocyte Isolation and Culture Using Thermolysin. Burns 1993, 19, 99–104. [Google Scholar] [CrossRef]
- Rochat, A.; Kobayashi, K.; Barrandon, Y. Location of Stem Cells of Human Hair Follicles by Clonal Analysis. Cell 1994, 76, 1063–1073. [Google Scholar] [CrossRef]
- Lavoie, A.; Fugère, C.; Fradette, J.; Larouche, D.; Paquet, C.; Beauparlant, A.; Gauvin, R.; Têtu, F.A.; Roy, A.; Bouchard, M.; et al. Considerations in the Choice of a Skin Donor Site for Harvesting Keratinocytes Containing a High Proportion of Stem Cells for Culture in Vitro. Burns 2011, 37, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; L’heureux, N.; Pouliot, R.; Xu, W.; Auger, F.A.; Germain, L. Characterization of a New Tissue-Engineered Human Skin Equivalent with Hair. In Vitro Cell. Dev. Biol.-Anim. 1999, 35, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Barrandon, Y.; Green, H. Three Clonal Types of Keratinocyte with Different Capacities for Multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar] [CrossRef]
- Larouche, D.; Cantin-Warren, L.; Desgagné, M.; Guignard, R.; Martel, I.; Ayoub, A.; Lavoie, A.; Gauvin, R.; Auger, F.A.; Moulin, V.J.; et al. Improved Methods to Produce Tissue-Engineered Skin Substitutes Suitable for the Permanent Closure of Full-Thickness Skin Injuries. Biores. Open Access 2016, 5, 320–329. [Google Scholar] [CrossRef]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a Mouse Monoclonal Antibody Reactive with a Human Nuclear Antigen Associated with Cell Proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef]
- Liu, Y.; Lyle, S.; Yang, Z.; Cotsarelis, G. Keratin 15 Promoter Targets Putative Epithelial Stem Cells in the Hair Follicle Bulge. J. Investig. Dermatol. 2003, 121, 963–968. [Google Scholar] [CrossRef]
- Lyle, S.; Christofidou-Solomidou, M.; Liu, Y.; Elder, D.E.; Albelda, S.; Cotsarelis, G. The C8/144B Monoclonal Antibody Recognizes Cytokeratin 15 and Defines the Location of Human Hair Follicle Stem Cells. J. Cell Sci. 1998, 111 Pt 21, 3179–3188. [Google Scholar] [CrossRef]
- Michel, M.; Török, N.; Godbout, M.J.; Lussier, M.; Gaudreau, P.; Royal, A.; Germain, L. Keratin 19 as a Biochemical Marker of Skin Stem Cells in Vivo and in Vitro: Keratin 19 Expressing Cells Are Differentially Localized in Function of Anatomic Sites, and Their Number Varies with Donor Age and Culture Stage. J. Cell Sci. 1996, 109 Pt 5, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, A.E.; Kajava, A.V.; Steinert, P.M. Epithelial Barrier Function: Assembly and Structural Features of the Cornified Cell Envelope. Bioessays 2002, 24, 789–800. [Google Scholar] [CrossRef]
- Steinert, P.M.; Cantieri, J.S.; Teller, D.C.; Lonsdale-Eccles, J.D.; Dale, B.A. Characterization of a Class of Cationic Proteins That Specifically Interact with Intermediate Filaments. Proc. Natl. Acad. Sci. USA 1981, 78, 4097–4101. [Google Scholar] [CrossRef]
- Jamora, C.; Fuchs, E. Intercellular Adhesion, Signalling and the Cytoskeleton. Nat. Cell Biol. 2002, 4, E101–E108. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Lavker, R.M.; Sun, T.T. Upper Human Hair Follicle Contains a Subpopulation of Keratinocytes with Superior in Vitro Proliferative Potential. J. Investig. Dermatol. 1993, 101, 652–659. [Google Scholar] [CrossRef]
- Cotsarelis, G. Epithelial Stem Cells: A Folliculocentric View. J. Investig. Dermatol. 2006, 126, 1459–1468. [Google Scholar] [CrossRef]
- Kobayashi, K.; Rochat, A.; Barrandon, Y. Segregation of Keratinocyte Colony-Forming Cells in the Bulge of the Rat Vibrissa. Proc. Natl. Acad. Sci. USA 1993, 90, 7391–7395. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, W.; Lin, K.; Wang, X.; Gao, Y. β-Catenin Activation in Gli-1+ Stem Cells Leads to Reprograming of the Hair Follicle. Eur. J. Dermatol. 2023, 33, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lin, K.; Choo, H.Q.; Chen, Y.; Zhang, X.; Xu, R.H.; Wang, X.; Wu, Y. Distinct Bulge Stem Cell Populations Maintain the Pilosebaceous Unit in a β-Catenin-Dependent Manner. iScience 2022, 26, 105805. [Google Scholar] [CrossRef]
- Gao, Y.; Fan, Z.; Xiao, X.; Kong, D.; Han, J.; Chu, W. Epidermal ET-1 Signal Induces Activation of Resting Hair Follicles by Upregulating the PI3K/AKT Pathway in the Dermis. FASEB J. 2024, 38, e23476. [Google Scholar] [CrossRef]
- Rowden, G. Ultrastructural Studies of the Keratinized Epithelia of the Mouse III. Determination of the Volumes of Nuclei and Cytoplasm of Cells in Murine Epidermis. J. Investig. Dermatol. 1975, 64, 1. [Google Scholar] [CrossRef]
- Inoue, K.; Yoshimura, K. Isolation and Characterization of Human Hair Follicle Epithelial Cells. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2013; Volume 946, pp. 411–421. ISBN 9781627031271. [Google Scholar]
- Raxworthy, M.; Cunliffe, W.; Wood, E. The Influence of Proteases on the Colony-Forming Efficiency of Human Keratinocytes in Culture. Biochem. Soc. Trans. 1987, 15, 519–520. [Google Scholar] [CrossRef]
- Myung, P.; Ito, M. Dissecting the Bulge in Hair Regeneration. J. Clin. Investig. 2012, 122, 448–454. [Google Scholar] [CrossRef]
- Jiang, S.; Zhao, L.; Purandare, B.; Hantash, B.M. Differential Expression of Stem Cell Markers in Human Follicular Bulge and Interfollicular Epidermal Compartments. Histochem. Cell Biol. 2010, 133, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Orazizadeh, M.; Hashemitabar, M.; Bahramzadeh, S.; Dehbashi, F.N.; Saremy, S. Comparison of the Enzymatic and Explant Methods for the Culture of Keratinocytes Isolated from Human Foreskin. Biomed. Rep. 2015, 3, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Veltri, A.; Lang, C.; Lien, W.H. Concise Review: Wnt Signaling Pathways in Skin Development and Epidermal Stem Cells. Stem Cells 2018, 36, 22–35. [Google Scholar] [CrossRef]
- Amoh, Y.; Hoffman, R.M. Hair Follicle-Associated-Pluripotent (HAP) Stem Cells. Cell Cycle 2017, 16, 2169–2175. [Google Scholar] [CrossRef]
- Hamida, O.B.; Kim, M.K.; Sung, Y.K.; Kim, M.K.; Kwack, M.H. Hair Regeneration Methods Using Cells Derived from Human Hair Follicles and Challenges to Overcome. Cells 2024, 14, 7. [Google Scholar] [CrossRef]
- Abaci, H.E.; Coffman, A.; Doucet, Y.; Chen, J.; Jacków, J.; Wang, E.; Guo, Z.; Shin, J.U.; Jahoda, C.A.; Christiano, A.M. Tissue Engineering of Human Hair Follicles Using a Biomimetic Developmental Approach. Nat. Commun. 2018, 9, 5301. [Google Scholar] [CrossRef]
- Jahoda, C.A.B.A.B.; Horne, K.A.A.; Oliver, R.F.F. Induction of Hair Growth by Implantation of Cultured Dermal Papilla Cells. Nature 1984, 311, 560–562. [Google Scholar] [CrossRef]
- Higgins, C.A.; Chen, J.C.; Cerise, J.E.; Jahoda, C.A.B.B.; Christiano, A.M. Microenvironmental Reprogramming by Threedimensional Culture Enables Dermal Papilla Cells to Induce de Novo Human Hair-Follicle Growth. Proc. Natl. Acad. Sci. USA 2013, 110, 19679–19688. [Google Scholar] [CrossRef]
- Weinberg, W.C.; Goodman, L.V.; George, C.; Morgan, D.L.; Ledbetter, S.; Yuspa, S.H.; Lichti, U. Reconstitution of Hair Follicle Development in Vivo: Determination of Follicle Formation, Hair Growth, and Hair Quality by Dermal Cells. J. Investig. Dermatol. 1993, 100, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Lichti, U.; Weinberg, W.C.; Goodman, L.; Ledbetter, S.; Dooley, T.; Morgan, D.; Yuspa, S.H. In Vivo Regulation of Murine Hair Growth: Insights from Grafting Defined Cell Populations onto Nude Mice. J. Investig. Dermatol. 1993, 101, 124S–129S. [Google Scholar] [CrossRef] [PubMed]
- Larouche, D.; Cuffley, K.; Paquet, C.; Germain, L. Tissue-Engineered Skin Preserving the Potential of Epithelial Cells to Differentiate into Hair after Grafting. Tissue Eng. Part A 2011, 17, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Cortez Ghio, S.; Le-Bel, G.; Lavoie, A.; Larouche, D.; Germain, L. Isolation and Culture of Human Keratinocytes. In Skin Tissue Engineering: Methods and Protocols; Böttcher-Haberzeth, S., Biedermann, T., Eds.; Springer: New York, NY, USA, 2019; pp. 3–13. ISBN 978-1-4939-9473-1. [Google Scholar]
- Bisson, F.; Rochefort, É.; Lavoie, A.; Larouche, D.; Zaniolo, K.; Simard-Bisson, C.; Damour, O.; Auger, F.A.; Guérin, S.L.; Germain, L. Irradiated Human Dermal Fibroblasts Are as Efficient as Mouse Fibroblasts as a Feeder Layer to Improve Human Epidermal Cell Culture Lifespan. Int. J. Mol. Sci. 2013, 14, 4684. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Age (y.o. 1) | Color Code Digested or Explanted Bulge, Scalp 2 |
|---|---|---|
| Male | 64 | ▲ ▲ ▲ |
| Female | 61 | ● ● ● |
| Female | 55 | ⬣ ⬣ ⬣ |
| Female | 68 | ♦ ♦ ♦ |
| Female | 61 | ■ ■ ■ |
| Female | 60 | ● Digestion |
| Female | 63 | |
| Female | 71 | |
| Mean = 63 (S.D. 3 = 5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattier, B.; Guignard, R.; Martel, I.; Martel, C.; Simard-Bisson, C.; Larouche, D.; Guiraud, B.; Bessou-Touya, S.; Germain, L. Bulge-Derived Epithelial Cells Isolated from Human Hair Follicles Using Enzymatic Digestion or Explants Result in Comparable Tissue-Engineered Skin. Int. J. Mol. Sci. 2025, 26, 1852. https://doi.org/10.3390/ijms26051852
Cattier B, Guignard R, Martel I, Martel C, Simard-Bisson C, Larouche D, Guiraud B, Bessou-Touya S, Germain L. Bulge-Derived Epithelial Cells Isolated from Human Hair Follicles Using Enzymatic Digestion or Explants Result in Comparable Tissue-Engineered Skin. International Journal of Molecular Sciences. 2025; 26(5):1852. https://doi.org/10.3390/ijms26051852
Chicago/Turabian StyleCattier, Bettina, Rina Guignard, Israël Martel, Christian Martel, Carolyne Simard-Bisson, Danielle Larouche, Béatrice Guiraud, Sandrine Bessou-Touya, and Lucie Germain. 2025. "Bulge-Derived Epithelial Cells Isolated from Human Hair Follicles Using Enzymatic Digestion or Explants Result in Comparable Tissue-Engineered Skin" International Journal of Molecular Sciences 26, no. 5: 1852. https://doi.org/10.3390/ijms26051852
APA StyleCattier, B., Guignard, R., Martel, I., Martel, C., Simard-Bisson, C., Larouche, D., Guiraud, B., Bessou-Touya, S., & Germain, L. (2025). Bulge-Derived Epithelial Cells Isolated from Human Hair Follicles Using Enzymatic Digestion or Explants Result in Comparable Tissue-Engineered Skin. International Journal of Molecular Sciences, 26(5), 1852. https://doi.org/10.3390/ijms26051852