Abstract
Opioids are the most effective option for severe pain. However, it is well documented that the side effects associated with prolonged opioid use significantly constrain dosage in the clinical setting. Recently, researchers have concentrated on the development of biased opioid receptor agonists that preferentially activate the G protein signaling pathway over β-arrestin signaling. This approach is based on the hypothesis that G protein signaling mediates analgesic effects, whereas β-arrestin signaling is implicated in adverse side effects. Although certain studies have demonstrated that the absence or inhibition of β-arrestin signaling can mitigate the incidence of side effects, recent research appears to challenge these earlier findings. In-depth investigations into biased signal transduction of opioid receptor agonists have been conducted, potentially offering novel insights for the development of biased opioid receptors. Consequently, this review elucidates the contradictory roles of β-arrestin signaling in the adverse reactions associated with opioid receptor activation. Furthermore, a comparative analysis was conducted to evaluate the efficacy of the classic G protein-biased agonists, TRV130 and PZM21, relative to the traditional non-biased agonist morphine. This review aims to inform the development of novel analgesic drugs that can optimize therapeutic efficacy and safety, while minimizing adverse reactions to the greatest extent possible.
1. Introduction
Opioids, such as morphine, fentanyl, and hydrocodone, are extensively utilized in clinical practice as the mainstay for managing various types of moderate to severe pain [1]. Despite potent analgesic properties, the extensive range of side effects greatly limits the use of opioid drugs, such as nausea and vomiting, constipation, respiratory depression, addiction, tolerance, opioid-induced hyperalgesia (OIH), etc. [2,3]. Even the Enhanced Recovery After Surgery (ERAS) protocols have increasingly advocated for the reduction or elimination of opioids, aiming for opioid-free analgesia [4]. However, opioids continue to be the most effective treatment for moderate to severe pain [4,5]. Therefore, the limitations inherent to conventional opioid drugs present significant challenges in clinical management. The addictive properties and euphoric effects of opioids threaten a significant market share in the illicit drug trade [6]. Furthermore, the mortality rate associated with opioids has remained consistently elevated in recent years, imposing considerable burdens on families and society [7,8,9]. These factors have made the development of analgesics targeting opioid receptors a challenging yet essential endeavor. Consequently, the pursuit of more potent analgesics with improved efficacy and minimal side effects has become a focused objective.
To mitigate the severe side effects caused by opioid receptor agonists as much as possible, researchers have proposed various strategies. Personalized administration is tailored to individual patients, using sustained-release formulations to reduce overall dosage and combined with low-dose antagonists to reduce adverse reactions [10,11]. Multiple opioid receptor agonists can also be used to counteract the side effects caused by one subtype, while preserving positive therapeutic efficacy through simultaneous activation of multiple opioid receptors [10]. Designing opioid receptor subtype-selective agonists and positive allosteric modulators has not effectively mitigated multiple side effects [12,13]. The advancement of selective agonists has consistently been a focal point for researchers. G protein-selective agonists, which are engineered to specifically interact with opioid receptors and specific second messenger systems, have demonstrated a substantial reduction in the incidence of adverse opioid reactions, including compounds such as TRV130 and PZM21 [14,15] (Table 1). Notably, TRV130 has received formal approval for clinical use.

Table 1.
Examples of biased ligands for opioid receptors.
In recent years, novel insights have emerged into the MOR-mediated biased agonistic signaling pathway, providing robust support for the prospective development of more precise and efficacious biased agonists. This review synthesizes the mechanisms underlying G protein-biased opioid receptor agonists and evaluates the therapeutic efficacy of classical biased opioid receptor agonists in comparison to the non-biased agonist morphine in both preclinical research and clinical practice. This comparative analysis establishes a foundational framework for the advancement of analgesics that are safer and have fewer side effects.
2. Mechanism of Opioid-Induced Analgesia and Adverse Reactions
Opioid receptors, which are G protein-coupled receptors (GPCRs), are primarily categorized into four subtypes: mu (μ)-opioid receptor (MOR), delta (δ)-opioid receptor (DOR), kappa (k)-opioid receptor (KOR), and nociceptive nocicepin/orphanin FQ peptide receptors (NOPR). All opioid receptor subtypes have analgesic implications. The analgesic mechanisms of opioids have been the subject of extensive investigation. Most clinically utilized opioids exert analgesic effects through interaction with MOR, an inhibitory G protein-coupled receptor [14,28] that is abundantly expressed in the central nervous system and gastrointestinal tract [29,30,31,32].
Upon binding to MOR, exogenous opioids induce conformational changes in the receptor, thereby modulating cellular activities through G protein and β-arrestin signaling pathways [33,34] (Figure 1). MOR is activated and coupled with G protein, which subsequently dissociates into alpha (Gα) and beta-gamma (Gβ/γ) subunits. Gα inhibits the activity of adenylate cyclase (AC), thereby preventing adenosine triphosphate (ATP) from producing cyclic adenosine monophosphate (cAMP) [35]. Gβ/γ leads to the inactivation of calcium channels (reducing Ca2+ influx) and activates potassium channels (increasing K+ efflux) [36,37]. Consequently, these effects inhibit the release of nociceptive neurotransmitters, thereby reducing the transmission of pain signals and exerting analgesic effects. In contrast, MOR is phosphorylated by G protein-coupled receptor kinase (GRK), which recruits β-arrestin. The binding of β-arrestin hinders the interaction between MOR and G protein, thereby blocking G protein-dependent signaling. Additionally, β-arrestin recruitment leads to endocytosis and desensitization of MOR and activation of the mitogen-activated protein kinase (MAPK) signaling pathway [34,38,39]. These processes are implicated in various adverse effects [40,41,42,43,44,45]. Empirical evidence has also confirmed these findings; for instance, in mice genetically deficient in β-arrestin, morphine administration has been associated with a reduction in constipation [46], respiratory suppression [47,48], and tolerance [14,47], among others.
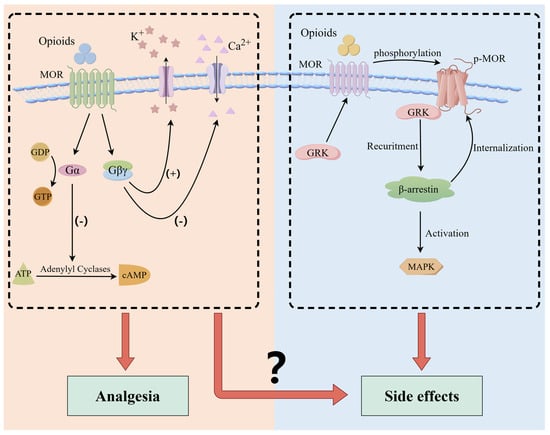
Figure 1.
Intracellular signaling mediated by opioid receptors. Activation of opioid receptors by exogenous agonists leads to dissociation of G protein heterotrimers into α and βγ subunits. Gα inhibits adenylate cyclase activity, thereby preventing adenosine ATP from producing cAMP. Gβ/γ leads to the inactivation of calcium channels and activation of potassium channels. Consequently, these effects inhibit the release of nociceptive neurotransmitters, thereby reducing the transmission of pain signals and exerting analgesic effects. The interaction of β-arrestin with phosphorylated MOR results in receptor internalization and desensitization, and activates the MAPK cascade pathways.
3. Conflicts in the β-Arrestin Signaling Pathway
The adverse effects of opioids can be reversed by MOR antagonists and are absent in animals with MOR gene knockout [29,30,49], suggesting that the incidence of side effects may not be associated with receptor selectivity. Previous studies have established that the analgesic effect of opioid receptor agonism is mediated by G protein signaling, whereas side effects such as respiratory depression, gastrointestinal dysfunction, and tolerance are mediated by β-arrestin signaling. However, a recent study demonstrated that both G protein signaling and β-arrestin contribute to side effects induced by morphine [50]. This study found that the β-arrestin pathway is involved in morphine tolerance, whereas the G protein signaling pathway is implicated in respiratory depression and constipation, achieved through the design of permeable peptides in combination with morphine. This challenges existing theories. Initially, β-arrestin was regarded as a negative regulator of the G protein signaling pathway. However, it was later discovered that β-arrestin can also function as a signal transducer of GPCR, mediating the transmission of downstream signals independently of G protein [51]. Indeed, the role of the β-arrestin signaling pathway in mediating the side effects of opioids remains unclear, and numerous studies investigating the adverse effects of opioids have focused on β-arrestin-2, although the findings remain contentious.
Early research in rodents revealed that the absence or reduction in the β-arrestin-2 gene amplifies the anti-nociceptive effects while simultaneously reducing tolerance to opioids [14,23,47,48,52,53,54,55], implying a role for β-arrestin-2 in the mechanisms underlying opioid-induced antinociceptive tolerance. Nonetheless, methadone, an opioid drug, exhibits a high affinity for β-arrestin-2, yet results in less tolerance and dependency than morphine [56]. This phenomenon blurs the role of β-arrestin-2 in analgesic tolerance. Subsequently, He and colleagues administered various G protein-biased and non-biased agonists to WT mice, revealing an inverse correlation between β-arrestin-2 recruitment and analgesic tolerance. Additionally, mice with the RMOR (Recycling MOR) genotype exhibit resistance to analgesic tolerance [57]. Contrary to previous findings, drugs that effectively recruit β-arrestin-2 reduce analgesic tolerance.
Opioid-induced analgesic tolerance is commonly regarded as centrally mediated; however, nociceptive dorsal root ganglia also play a significant role in tolerance development [58,59]. Research has demonstrated that desensitization of μ-Opioid receptors mediated by β-arrestin-2 is implicated in the acute analgesic tolerance of dorsal root ganglion nociceptive neurons to opioids [60]. Notably, the absence of β-arrestin-2 does not affect long-term tolerance to morphine in either male or female mice [60], indicating that non-β-arrestin-biased agonists may not alleviate tolerance induced by prolonged opioid use. The experimental data from various laboratories exhibit significant variability, which may be attributed to factors such as drug dosage, dosing frequency, physiological condition of animals, and laboratory settings. These divergent results could profoundly impact the clinical application of biased agonists, necessitating more rigorous experiments to obtain conclusive evidence.
Respiratory depression is another controversial side effect mediated by β-arrestin signaling. The risk of respiratory depression constrains the clinical dosage of opioids [61]. Numerous studies have demonstrated that the deletion of the β-arrestin-2 gene exhibits less morphine-induced respiratory depression [46,47,48,55,62], indicating a mediating role of β-arrestin-2 in respiratory dysfunction. However, previous perspectives have posited that opioid-induced respiratory depression is related to G protein-gated inwardly rectifying potassium channels rather than β-arrestin signaling [63,64,65]. Recent studies provide additional support for this perspective. Kliewer et al. proposed that morphine-induced respiratory depression is not associated with β-arrestin-2 signaling [66]. Afterward, He et al. investigated the drug response of three different genotypes—wild type (WT), β-arrestin-2 knockout (KO), and Recycling MOR (RMOR) C57BL/6 mice—to morphine. Their findings indicated that all three genotypes exhibited comparable levels of respiratory inhibition and analgesic effects in response to morphine [57]. Furthermore, when WT mice were administered different G protein-biased and non-biased agonists, no significant difference was observed in the maximum respiratory inhibition. These findings suggest that there may be no significant correlation between the recruitment level of β-arrestin-2 and the extent of respiratory depression.
Studies have shown that biased MOR agonists have the potential to mitigate abuse relative to standard opioids [67]. Notably, β-arrestin signaling may not be implicated in the addiction mechanism associated with opioid receptor agonists. Research has shown that in mice with β-arrestin-2 gene knockout, morphine-induced striatal dopamine release is significantly increased, and conditioned place preference (CPP) behavior is markedly enhanced [46,48,68]. This suggests that opioid receptor agonists that preferentially activate G protein over β-arrestin signaling may possess the potential to induce physical dependence [48].
4. What Is the Biased Ligand Agonist?
Previously, it was believed that ligands exert different pharmacological effects due to their interaction with distinct receptors [69]. However, it has gradually become evident that the effects of ligands are not entirely dependent on receptor selection; rather, they are closely associated with signaling molecules downstream of the receptors [70]. The activated conformation of GPCRs is dynamically variable, which allows for the recruitment of G proteins and β-arrestin to varying extents, thereby triggering different signaling pathways [71]. Recent studies have shown that ligands do not uniformly activate signaling pathways downstream of GPCRs; instead, they can selectively and preferentially activate certain pathways, a phenomenon known as “biased activation” [72]. Currently, there is no universally accepted standard for evaluating and assessing ligand biases. Some researchers have argued that ligand activation-specific signals are influenced by factors such as ligand bias, system bias, and dynamic bias [73,74,75]. The concept of the “bias factor” has been introduced as a measure of the selectivity of MOR agonists [73,76]. This involves systematically testing the degree of bias among different MOR agonists (i.e., the potency differences between G protein signals and other signaling pathways) to determine the therapeutic index [77].
Biased agonists stabilize receptor conformations and preferentially activate specific signaling pathways, allowing for more precise regulation of cellular functions. However, significant challenges persist in the development of biased agonists [78]. There is a pressing need for further technical advancements in the quantitative analysis of bias and the establishment of appropriate screening methods. Additionally, identifying which cell types express the drug targets, elucidating the signaling pathways that can elicit therapeutic effects, and understanding the regulatory mechanisms involved are all critical areas that require thorough investigation.
5. A New Approach to Biased Agonists
When non-biased agonists activate MOR, G protein and β-arrestin compete for the same binding site, which forms strong polar interactions with ICL2 (intracellular loop 2 (ICL2)) and ICL3 or the cytoplasmic region of TM6, and facilitates coupling with MOR [79]. Recent structural biology and pharmacological experiments have revealed that opioid receptor agonists attenuate or abolish the β-arrestin signaling pathway, resulting in a pattern of G protein-biased signal transduction. This bias reduces the interaction between opioids and the sixth and seventh transmembrane regions of the MOR [80]. Further verification was achieved through cellular-level functional analysis and molecular dynamics simulations. Specifically, studies have demonstrated that G protein-biased agonists (such as TRV130, PZM21, and SR-17018) preferentially bind to the TM2/3 region of the MOR binding site. This interaction induces the repositioning of TM6 in the cytoplasmic region of MOR, thereby preventing β-arrestin from forming a polar anchor with MOR, resulting in decreased affinity [79,80]. In contrast, balanced agonists such as fentanyl engage in more extensive and balanced interactions with the transmembrane region of the MOR [80]. Based on this, researchers meticulously modified fentanyl and synthesized two derivatives, FBD1 and FBD3, which exhibited a preference for the G protein pathway, thereby further substantiating the concept of biased binding sites. This discovery provides novel insights for further design of biased opioids. Nonetheless, the molecular binding model derived from molecular docking and dynamic simulation is intricate and may not fully replicate the actual drug binding mode. It is anticipated that this groundbreaking discovery will require further validation in animal models.
6. Comparison Between Biased Agonists and Morphine
MOR is the principal target of opioids, such as morphine and hydrocodone. Numerous pharmacological studies have focused on the ligand bias of this receptor. Some studies have measured the bias coefficient of MOR agonists at the molecular level as well as the analgesic and side effects at the animal level. These results indicate that when a biased ligand activates the G protein-dependent signaling pathway, the safety window is broad [21]. Consequently, biased ligand theory posits that biased agonists of the G protein-dependent signaling pathway may produce more selective pharmacological profiles and physiological responses. Based on this theory, extensive screening of compounds was conducted, resulting in the identification of a series of potentially biased ligands such as TRV-130 and PZM21.
6.1. TRV130
In 2013, TRV130 was designated as Oliceridine or OLINVO, demonstrating a selectivity for MOR receptors that exceeds 400-fold that of other opioid receptors [14]. Structurally distinct from other MOR agonists, TRV130 exhibits a degree of G protein coupling activation comparable to morphine; however, its efficiency in recruiting β-arrestin-2 is approximately 14% of that observed with morphine [14,81]. TRV130 possesses unique pharmacokinetic properties characterized by a rapid onset of action and prolonged duration. Clinical studies have indicated that its effects commence within 2-5 min and persist for up to 3 h [81]. The therapeutic window for dosage is broad, obviating the necessity for dosage adjustments across diverse patients, including variations in age, sex, race, body weight, and hepatic or renal function [82]. Consequently, TRV130 is regarded as a highly efficacious, safe, and selective drug characterized by its G protein-biased MOR agonist properties.
Similarly to morphine, TRV130 exhibits a high volume of distribution and significant monophasic clearance [14]. Its lipophilic properties facilitate rapid penetration into the brain, resulting in widespread tissue distribution and prompt access to the central nervous system where it binds to opioid receptors to exert its pharmacological effects [14]. Numerous animal studies have evaluated the efficacy of TRV130 in comparison with morphine, intending to assess the relative advantages and disadvantages of each, thereby improving clinical practices (Table 2). Empirical studies have demonstrated that TRV130 induces a more pronounced analgesic effect than respiratory depression and sedation in rat models. Furthermore, TRV130 has been shown to produce potent analgesic efficacy, with its effectiveness in acute thermal injury models in rats and mice ranging from 4.5 to 10 times greater than that of morphine [14,52,83,84]. However, at equivalent analgesic doses, TRV130 exhibits a less pronounced sedative effect than morphine, which aligns with its reduced inhibition of the central nervous system [14]. Additionally, the weak activation of β-arrestin-2 by TRV130 can lead to respiratory depression, constipation, antinociceptive tolerance, and behaviors associated with abuse [52,77,83,85,86], similar to other MOR agonists. However, multiple clinical trials have demonstrated that TRV130 elicits more rapid antinociceptive effects than morphine at equivalent analgesic doses in patients with moderate to severe pain. Furthermore, TRV130 is associated with a reduced incidence of adverse events, including nausea, vomiting, respiratory dysfunction, and gastrointestinal disturbances, thereby offering a broader therapeutic window [14,15,87,88,89,90,91]. Additionally, various rodent studies have indicated that TRV130 shows a lower propensity for tolerance development than conventional opioid medications [83,84,92,93]. Cellular experiments further substantiated that, compared to morphine, TRV130 exhibited reduced receptor phosphorylation and internalization, processes linked to opioid tolerance, when co-cultured with human MOR-expressing cells [14]. Interestingly, TRV130 has been observed to induce opioid-induced hyperalgesia (OIH), albeit to a lesser extent than morphine. In contrast, repeated or continuous administration of TRV-0109101, an analog of TRV130, does not result in OIH; however, it does lead to significant tolerance, with a rate of drug resistance comparable to that observed with conventional mu-opioid receptor (MOR) agonists [55,94].
Table 2.
Comparison of adverse reactions between TRV130 and morphine.
Conversely, empirical evidence suggests that TRV130 exhibits abuse potential comparable to that of hydrocodone [77]. Within the rat intracranial self-stimulation (ICSS) paradigm, TRV130 demonstrated reward-enhancing effects analogous to those of conventional opioids [83]. Notably, a single analgesic dose of TRV130 did not induce conditioned place preference (CPP) in mouse models [52]. Furthermore, at equianalgesic doses, TRV130 elicited a lower CPP response than morphine; however, this disparity was mitigated when higher doses of TRV130 were administered [84]. This phenomenon may be attributed to the regulation of brain exposure to opioid drugs by the P-glycoprotein (P-gp) efflux transporter (MDR1) [96,97,98,99,100,101]. The function of P-gp is a critical determinant of the pharmacokinetic properties of opioids. Both TRV130 and morphine are substrates of P-gp; however, administering morphine at doses at least ten times higher than TRV130 may saturate efflux transporters, thereby reducing morphine clearance and resulting in increased brain uptake. This suggests that the dosage required for TRV130 to achieve analgesic effects may not pose a significant risk of abuse. Moreover, TRV130 has demonstrated opioid withdrawal mitigation effects comparable to those of methadone [102], indicating its potential as a viable treatment option for patients with opioid use disorder (OUD). Empirical studies have revealed that TRV130 exerts a more pronounced impact on drug addiction relapse in male rats than in female rats [85], suggesting that maintenance therapy with TRV130 may generate greater efficacy in male patients. Furthermore, prolonged administration of TRV130 has been shown to prevent brain hypoxia induced by moderate doses of hydrocodone in both male and female rats [103], with this neuroprotective effect sustained across both sexes.
Morphine-6-glucuronide, the active metabolite of morphine, causes long-term respiratory inhibition [104,105]. In contrast, TRV130 lacks bioactive metabolites [106]. The high potency and rapid pharmacokinetics of TRV130 may contribute to a safer profile for pain management. Owing to its pharmacokinetic properties, TRV130 is limited to intravenous administration in humans [87]. In 2020, the U.S. Food and Drug Administration (FDA) approved TRV130 for short-term intravenous use in hospital settings, with a daily cumulative dose not exceeding 27 mg. The limitation associated with the unsuitability for daily maintenance can be mitigated through oral administration of the active analog TRV734 [17,107] or by formulating depot preparations of TRV130/TRV734 for periodic application. Additionally, a method for chronic pain management via transdermal patch administration, leveraging its high skin permeability, is currently under development.
The biased agonism of TRV130 has been shown to offer significant advantages in mitigating side effects relative to other MOR agonists. An economic model indicated that the administration of TRV130 in postoperative care for high-risk patients, including elderly and obese individuals who are susceptible to opioid-related adverse events (ORAEs), yields significant health and economic benefits [108]. Furthermore, preclinical studies have shown that TRV130 is well tolerated in animal models and exhibits superior therapeutic efficacy relative to morphine. However, numerous clinical studies conducted since 2014 have indicated the persistence of opioid-related side effects. Therefore, there is a pressing need for a more comprehensive investigation of biased agonists in the clinical setting.
6.2. PZM21
Unlike TRV130, the design strategy of PZM21 is based on analysis of the solved MOR structure and is further developed through high-throughput screening [52]. It exhibits high selectivity for the MOR and is associated with weak KOR activation [52]. Similarly to TRV130, PZM21 activates the G protein and reduces the recruitment of β-arrestin [52,109]. The respiratory effects of PZM21 differ significantly from those of morphine. Initial studies indicated that mice administered PZM21 do not experience respiratory depression, whereas equivalent doses of morphine result in respiratory inhibition [52]. However, recent research has indicated that PZM21 exhibits respiratory inhibitory effects comparable to morphine and induces antinociceptive tolerance [110]. These findings may be attributed to variations in experimental outcomes across different laboratories. Furthermore, studies in mice have demonstrated that PZM21 selectively inhibits the affective components of pain. Notably, PZM21 produced analgesic effects in the hot plate test, which assesses the advanced central nervous system and spinal cord injury circuits, but did not provide pain relief in the tail-flick test, which evaluates spinal reflexes [52]. This presents a novel distinction between PZM21 and other opioids. Through simulation, it was determined that PZM21 did not exhibit significant advantages in terms of analgesia or reduction in side effects, such as TRV130 [16,110]. Specifically, PZM21 was identified as a low-efficacy analgesic and a low-efficacy biased agonist. These findings are consistent with the experimental results reported by Hill et al., which demonstrated PZM21’s inefficacy in both the G protein and β-arrestin pathways [110]. Unlike TRV130, this study assessed the abuse potential of PZM21 using intracranial self-stimulation and found minimal enhancement of its use. These results suggest the need for further investigation of the potential applications and limitations of PZM21.
7. Other Biased Agonists
SR-17018 has a half-life of approximately 6 h, demonstrates brain permeability, and is associated with low respiratory depression [21,22]. Long-term oral administration of SR-17018 in mice produces analgesic effects comparable to those of morphine. Upon cessation of treatment, withdrawal symptoms were observed, albeit for a shorter duration than those associated with morphine. When administered to male mice at a dose of 24 mg/kg/day, SR-17018 did not induce drug resistance; however, a slight reduction in efficacy was noted at a dose of 48 mg/kg/day. These findings suggest that the tolerance to SR-17018 may be dose-dependent. The absence of tolerance in female mice administered SR-17018 at a dose of 48 mg/kg/day indicates the potential sex-specific effects of SR-17018 [23]. However, SR-17018 is more effective in various pain models and does not induce tolerance, which distinguishes it from other commonly used opioids [111]. Prolonged administration of SR-17018 preserves sensitivity to morphine in mice and mitigates morphine withdrawal symptoms, likely due to SR-17018’s role in stabilizing the G protein-coupled state. Thus, SR-17018 may be a viable strategy for restoring MOR responsiveness and sustaining opioid analgesic efficacy.
SR-14968 is a complete agonist with less respiratory depression and a broader therapeutic window [21]. SR-14968 exhibits a G protein bias approximately ten times greater than TRV130 [14,21]. This compound demonstrated dose-dependent analgesic and differential stimulatory effects. However, compared to morphine and methadone, SR-14968 showed a higher propensity to produce discriminative stimuli. The extended duration of action of SR-14968, relative to TRV130, morphine, and methadone in rodent models, raises questions about how pharmacokinetic properties influence enhancing effects [21]. Furthermore, no sex differences were observed in the efficacy of SR-14968 [95]. In addition, SR-17018 and SR-14968 are both non-competitive agonists that can stabilize the receptor active. Studies have demonstrated that SR-17018 and SR-14968 bind to MOR in an almost irreversible manner, resulting in sustained G protein signaling. However, MOR antagonists can completely reverse this stimulation, indicating that SR series compounds bind to different sites on the receptor with high affinity, promote G protein signaling, and maintain sensitivity to orthosteric antagonists [112]. Furthermore, SR-17018 can inhibit the effects of SR-14968, suggesting that there may be competition between them at the allosteric site [112].
8. Conclusions
With the progressive rise in demand for opioids, the impact of side effects is becoming increasingly apparent. The development of targeted, safer, and more potent analgesics with a broader therapeutic window aligns with contemporary trends in pharmaceutical research and clinical practice. Although the existing designs of biased opioid receptor agonists have not yet fully met the rigorous standard of “zero side effects”, the ongoing advancement of biased ligand technology offers promise for the creation of innovative opioid therapeutics.
Author Contributions
J.J.: Writing—Original Draft Preparation; Z.L.: Writing—Review and Editing; J.L.: Validation; X.P.: Validation; F.G.: Funding Acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 81974168.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Abdel-Zaher, A.O.; Mostafa, M.G.; Farghaly, H.S.; Hamdy, M.M.; Abdel-Hady, R.H. Role of oxidative stress and inducible nitric oxide synthase in morphine-induced tolerance and dependence in mice. Effect of alpha-lipoic acid. Behav. Brain Res. 2013, 247, 17–26. [Google Scholar] [CrossRef]
- Chan, H.C.S.; McCarthy, D.; Li, J.; Palczewski, K.; Yuan, S. Designing Safer Analgesics via μ-Opioid Receptor Pathways. Trends Pharmacol. Sci. 2017, 38, 1016–1037. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.D.; Henderson, G.; McKnight, A.T.; Paterson, S.J. 75 years of opioid research: The exciting but vain quest for the Holy Grail. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S153–S162. [Google Scholar] [CrossRef]
- Echeverria-Villalobos, M.; Stoicea, N.; Todeschini, A.B.; Fiorda-Diaz, J.; Uribe, A.A.; Weaver, T.; Bergese, S.D. Enhanced Recovery After Surgery (ERAS): A Perspective Review of Postoperative Pain Management Under ERAS Pathways and Its Role on Opioid Crisis in the United States. Clin. J. Pain 2020, 36, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kharasch, E.D.; Clark, J.D. Opioid-free Anesthesia: Time to Regain Our Balance. Anesthesiology 2021, 134, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, A.; Courtwright, D.T.; Hwang, C.S.; Kreiner, P.; Eadie, J.L.; Clark, T.W.; Alexander, G.C. The prescription opioid and heroin crisis: A public health approach to an epidemic of addiction. Annu. Rev. Public Health 2015, 36, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Vashishtha, D.; Mittal, M.L.; Werb, D. The North American opioid epidemic: Current challenges and a call for treatment as prevention. Harm Reduct. J. 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Rudd, R.A.; Noonan, R.K.; Haegerich, T.M. Quantifying the Epidemic of Prescription Opioid Overdose Deaths. Am. J. Public Health 2018, 108, 500–502. [Google Scholar] [CrossRef]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, G.W.; Shin, S.H.; Bruera, E. Opioid withdrawal syndrome after treatment with low-dose extended-release oxycodone and naloxone in a gastric cancer patient with portal vein thrombosis. J. Pain Symptom Manag. 2013, 46, e15–e17. [Google Scholar] [CrossRef]
- Gilbert, P.E.; Martin, W.R. The effects of morphine and nalorphine-like drugs in the nondependent, morphine-dependent and cyclazocine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 198, 66–82. [Google Scholar] [CrossRef]
- Burford, N.T.; Traynor, J.R.; Alt, A. Positive allosteric modulators of the μ-opioid receptor: A novel approach for future pain medications. Br. J. Pharmacol. 2015, 172, 277–286. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Viscusi, E.R.; Webster, L.; Kuss, M.; Daniels, S.; Bolognese, J.A.; Zuckerman, S.; Soergel, D.G.; Subach, R.A.; Cook, E.; Skobieranda, F. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain 2016, 157, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Huang, T.; Li, J. Molecular Dynamics Simulations to Investigate How PZM21 Affects the Conformational State of the μ-Opioid Receptor Upon Activation. Int. J. Mol. Sci. 2020, 21, 4699. [Google Scholar] [CrossRef] [PubMed]
- James, I.E.; Skobieranda, F.; Soergel, D.G.; Ramos, K.A.; Ruff, D.; Fossler, M.J. A First-in-Human Clinical Study With TRV734, an Orally Bioavailable G-Protein-Biased Ligand at the μ-Opioid Receptor. Clin. Pharmacol. Drug Dev. 2020, 9, 256–266. [Google Scholar] [CrossRef]
- Li, X.; He, W.; Chen, Y.; Yang, G.; Wan, H.; Zhang, L.; Hu, Q.; Feng, J.; Zhang, Z.; He, F.; et al. Discovery of SHR9352: A Highly Potent G Protein-Biased μ-Opioid Receptor Agonist. ACS Omega 2017, 2, 9261–9267. [Google Scholar] [CrossRef] [PubMed]
- Gach-Janczak, K.; Piekielna-Ciesielska, J.; Adamska-Bartłomiejczyk, A.; Wtorek, K.; Ferrari, F.; Calo, G.; Szymaszkiewicz, A.; Piasecka-Zelga, J.; Janecka, A. In vitro and in vivo activity of cyclopeptide Dmt-c[d-Lys-Phe-Asp]NH(2), a mu opioid receptor agonist biased toward β-arrestin. Peptides 2018, 105, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Váradi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabó, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e1113. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yan, W.; McCorvy, J.D.; Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61, 9841–9878. [Google Scholar] [CrossRef]
- Grim, T.W.; Schmid, C.L.; Stahl, E.L.; Pantouli, F.; Ho, J.H.; Acevedo-Canabal, A.; Kennedy, N.M.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 2020, 45, 416–425. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, L.; Wang, Y.; Zhou, J.; Wang, D.; Zhang, Y.; Zhang, X.; Wang, Z.; Yang, D.; Mou, L.; et al. MEL-N16: A Series of Novel Endomorphin Analogs with Good Analgesic Activity and a Favorable Side Effect Profile. ACS Chem. Neurosci. 2017, 8, 2180–2193. [Google Scholar] [CrossRef]
- Spahn, V.; Del Vecchio, G.; Labuz, D.; Rodriguez-Gaztelumendi, A.; Massaly, N.; Temp, J.; Durmaz, V.; Sabri, P.; Reidelbach, M.; Machelska, H.; et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gaztelumendi, A.; Spahn, V.; Labuz, D.; Machelska, H.; Stein, C. Analgesic effects of a novel pH-dependent μ-opioid receptor agonist in models of neuropathic and abdominal pain. Pain 2018, 159, 2277–2284. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Zuo, A.; Li, C.; Wang, W.; Jiang, W.; Zhang, X.; Che, X.; Zhang, Y.; Wu, W.; et al. Synthesis, biological, and structural explorations of a series of μ-opioid receptor (MOR) agonists with high G protein signaling bias. Eur. J. Med. Chem. 2022, 228, 113986. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef]
- Kieffer, B.L. Opioids: First lessons from knockout mice. Trends Pharmacol. Sci. 1999, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef]
- Mello, N.K.; Negus, S.S. Preclinical evaluation of pharmacotherapies for treatment of cocaine and opioid abuse using drug self-administration procedures. Neuropsychopharmacology 1996, 14, 375–424. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Sarton, E.; Teppema, L.; Olievier, C.; Nieuwenhuijs, D.; Matthes, H.W.; Kieffer, B.L. Anesthetic potency and influence of morphine and sevoflurane on respiration in mu-opioid receptor knockout mice. Anesthesiology 2001, 94, 824–832. [Google Scholar] [CrossRef]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Caron, M.G. G protein-coupled receptor kinase/beta-arrestin systems and drugs of abuse: Psychostimulant and opiate studies in knockout mice. Neuromolecular Med. 2004, 5, 41–50. [Google Scholar] [CrossRef]
- Hepler, J.R.; Gilman, A.G. G proteins. Trends Biochem. Sci. 1992, 17, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef]
- Che, T.; Roth, B.L. Molecular basis of opioid receptor signaling. Cell 2023, 186, 5203–5219. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Violin, J.D. Biased ligands for better cardiovascular drugs: Dissecting G-protein-coupled receptor pharmacology. Circ. Res. 2011, 109, 205–216. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef]
- Alvarez, V.A.; Arttamangkul, S.; Dang, V.; Salem, A.; Whistler, J.L.; Von Zastrow, M.; Grandy, D.K.; Williams, J.T. mu-Opioid receptors: Ligand-dependent activation of potassium conductance, desensitization, and internalization. J. Neurosci. 2002, 22, 5769–5776. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Dykstra, L.A.; Lefkowitz, R.J.; Caron, M.G.; Barak, L.S. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol. Pharmacol. 2004, 66, 106–112. [Google Scholar] [CrossRef]
- Connor, M.; Osborne, P.B.; Christie, M.J. Mu-opioid receptor desensitization: Is morphine different? Br. J. Pharmacol. 2004, 143, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Premont, R.T.; Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004, 27, 107–144. [Google Scholar] [CrossRef]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef]
- Bao, F.; Li, C.L.; Chen, X.Q.; Lu, Y.J.; Bao, L.; Zhang, X. Clinical opioids differentially induce co-internalization of μ- and δ-opioid receptors. Mol. Pain 2018, 14, 1744806918769492. [Google Scholar] [CrossRef]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the μ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Sora, I.; Takahashi, N.; Funada, M.; Ujike, H.; Revay, R.S.; Donovan, D.M.; Miner, L.L.; Uhl, G.R. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. USA 1997, 94, 1544–1549. [Google Scholar] [CrossRef]
- Xia, J.; Li, X.; Zhu, H.; Zhou, X.; Chen, J.; Li, Q.; Li, S.; Chu, H.; Dong, M. The μ-opioid receptor-mediated G(i/o) protein and β-arrestin2 signaling pathways both contribute to morphine-induced side effects. Eur. J. Pharmacol. 2024, 966, 176333. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, W.; Zhong, T.; Song, Z.; Zou, Y.; Ding, Z.; Guo, Q.; Dong, X.; Zou, W. miR-365 targets β-arrestin 2 to reverse morphine tolerance in rats. Sci. Rep. 2016, 6, 38285. [Google Scholar] [CrossRef]
- Yang, C.H.; Huang, H.W.; Chen, K.H.; Chen, Y.S.; Sheen-Chen, S.M.; Lin, C.R. Antinociceptive potentiation and attenuation of tolerance by intrathecal β-arrestin 2 small interfering RNA in rats. Br. J. Anaesth. 2011, 107, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Enquist, J.; Ferwerda, M.; Milan-Lobo, L.; Whistler, J.L. Chronic methadone treatment shows a better cost/benefit ratio than chronic morphine in mice. J. Pharmacol. Exp. Ther. 2012, 340, 386–392. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Gooding, S.W.; Lewis, E.; Felth, L.C.; Gaur, A.; Whistler, J.L. Pharmacological and genetic manipulations at the µ-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice. Neuropsychopharmacology 2021, 46, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Tawfik, V.L.; Wang, D.; Sypek, E.I.; Low, S.A.; Dickinson, J.R.; Sotoudeh, C.; Clark, J.D.; Barres, B.A.; Bohlen, C.J.; et al. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 2017, 23, 164–173. [Google Scholar] [CrossRef]
- Chen, S.R.; Prunean, A.; Pan, H.M.; Welker, K.L.; Pan, H.L. Resistance to morphine analgesic tolerance in rats with deleted transient receptor potential vanilloid type 1-expressing sensory neurons. Neuroscience 2007, 145, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Muchhala, K.H.; Jacob, J.C.; Dewey, W.L.; Akbarali, H.I. Role of β-arrestin-2 in short- and long-term opioid tolerance in the dorsal root ganglia. Eur. J. Pharmacol. 2021, 899, 174007. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A. Respiratory depression with opioids. J. Pain Palliat. Care Pharmacother. 2007, 21, 63–66. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. 2002, 22, 10494–10500. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef]
- Levitt, E.S.; Abdala, A.P.; Paton, J.F.; Bissonnette, J.M.; Williams, J.T. μ opioid receptor activation hyperpolarizes respiratory-controlling Kölliker-Fuse neurons and suppresses post-inspiratory drive. J. Physiol. 2015, 593, 4453–4469. [Google Scholar] [CrossRef] [PubMed]
- Montandon, G.; Ren, J.; Victoria, N.C.; Liu, H.; Wickman, K.; Greer, J.J.; Horner, R.L. G-protein-gated Inwardly Rectifying Potassium Channels Modulate Respiratory Depression by Opioids. Anesthesiology 2016, 124, 641–650. [Google Scholar] [CrossRef]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmiedel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020, 177, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Collins, F.S. The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med. 2017, 377, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Agonist-receptor efficacy II: Agonist trafficking of receptor signals. Trends Pharmacol. Sci. 1995, 16, 232–238. [Google Scholar] [CrossRef]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. The Effective Application of Biased Signaling to New Drug Discovery. Mol. Pharmacol. 2015, 88, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Gaddum Memorial Lecture 2014: Receptors as an evolving concept: From switches to biased microprocessors. Br. J. Pharmacol. 2015, 172, 4238–4253. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Seifert, R.; Bond, R.A. Dynamic bias and its implications for GPCR drug discovery. Nat. Rev. Drug Discov. 2014, 13, 869. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.L.; Zhou, L.; Ehlert, F.J.; Bohn, L.M. A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol. Pharmacol. 2015, 87, 866–877. [Google Scholar] [CrossRef]
- Zamarripa, C.A.; Edwards, S.R.; Qureshi, H.N.; Yi, J.N.; Blough, B.E.; Freeman, K.B. The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol Depend. 2018, 192, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Charlton, S.J. Biased Agonism in Drug Discovery-Is It Too Soon to Choose a Path? Mol. Pharmacol. 2018, 93, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Mafi, A.; Kim, S.K.; Goddard, W.A., 3rd. Mechanism of β-arrestin recruitment by the μ-opioid G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 16346–16355. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 2022, 185, 4361–4375.e4319. [Google Scholar] [CrossRef]
- Pedersen, M.F.; Wróbel, T.M.; Märcher-Rørsted, E.; Pedersen, D.S.; Møller, T.C.; Gabriele, F.; Pedersen, H.; Matosiuk, D.; Foster, S.R.; Bouvier, M.; et al. Biased agonism of clinically approved μ-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology 2020, 166, 107718. [Google Scholar] [CrossRef]
- Nafziger, A.N.; Arscott, K.A.; Cochrane, K.; Skobieranda, F.; Burt, D.A.; Fossler, M.J. The Influence of Renal or Hepatic Impairment on the Pharmacokinetics, Safety, and Tolerability of Oliceridine. Clin. Pharmacol. Drug Dev. 2020, 9, 639–650. [Google Scholar] [CrossRef]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Liang, D.Y.; Li, W.W.; Nwaneshiudu, C.; Irvine, K.A.; Clark, J.D. Pharmacological Characters of Oliceridine, a μ-Opioid Receptor G-Protein-Biased Ligand in Mice. Anesth. Analg. 2019, 129, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Bossert, J.M.; Kiyatkin, E.A.; Korah, H.; Hoots, J.K.; Afzal, A.; Perekopskiy, D.; Thomas, S.; Fredriksson, I.; Blough, B.E.; Negus, S.S.; et al. In a Rat Model of Opioid Maintenance, the G Protein-Biased Mu Opioid Receptor Agonist TRV130 Decreases Relapse to Oxycodone Seeking and Taking and Prevents Oxycodone-Induced Brain Hypoxia. Biol. Psychiatry 2020, 88, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.G.; Subach, R.A.; Sadler, B.; Connell, J.; Marion, A.S.; Cowan, C.L.; Violin, J.D.; Lark, M.W. First clinical experience with TRV130: Pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Minkowitz, H.S.; Soergel, D.G.; Burt, D.A.; Subach, R.A.; Salamea, M.Y.; Fossler, M.J.; Skobieranda, F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel µ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017, 10, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Viscusi, E.R.; Skobieranda, F.; Soergel, D.G.; Cook, E.; Burt, D.A.; Singla, N. APOLLO-1: A randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the µ-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J. Pain Res. 2019, 12, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.K.; Skobieranda, F.; Soergel, D.G.; Salamea, M.; Burt, D.A.; Demitrack, M.A.; Viscusi, E.R. APOLLO-2: A Randomized, Placebo and Active-Controlled Phase III Study Investigating Oliceridine (TRV130), a G Protein-Biased Ligand at the μ-Opioid Receptor, for Management of Moderate to Severe Acute Pain Following Abdominoplasty. Pain Pract. 2019, 19, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kuzumaki, N.; Arima, T.; Narita, M.; Tateishi, R.; Kondo, T.; Hamada, Y.; Kuwata, H.; Kawata, M.; Yamazaki, M.; et al. Usefulness for the combination of G-protein- and β-arrestin-biased ligands of μ-opioid receptors: Prevention of antinociceptive tolerance. Mol. Pain 2017, 13, 1744806917740030. [Google Scholar] [CrossRef]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Koblish, M.; Carr, R., 3rd; Siuda, E.R.; Rominger, D.H.; Gowen-MacDonald, W.; Cowan, C.L.; Crombie, A.L.; Violin, J.D.; Lark, M.W. TRV0109101, a G Protein-Biased Agonist of the µ-Opioid Receptor, Does Not Promote Opioid-Induced Mechanical Allodynia following Chronic Administration. J. Pharmacol. Exp. Ther. 2017, 362, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Schwienteck, K.L.; Faunce, K.E.; Rice, K.C.; Obeng, S.; Zhang, Y.; Blough, B.E.; Grim, T.W.; Negus, S.S.; Banks, M.L. Effectiveness comparisons of G-protein biased and unbiased mu opioid receptor ligands in warm water tail-withdrawal and drug discrimination in male and female rats. Neuropharmacology 2019, 150, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.J.; Koszdin, K.; Bernards, C.M. Opiate-induced analgesia is increased and prolonged in mice lacking P-glycoprotein. Anesthesiology 2000, 92, 1392–1399. [Google Scholar] [CrossRef]
- Letrent, S.P.; Pollack, G.M.; Brouwer, K.R.; Brouwer, K.L. Effects of a potent and specific P-glycoprotein inhibitor on the blood-brain barrier distribution and antinociceptive effect of morphine in the rat. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 827–834. [Google Scholar] [CrossRef]
- Zong, J.; Pollack, G.M. Morphine antinociception is enhanced in mdr1a gene-deficient mice. Pharm. Res. 2000, 17, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Kishioka, S.; Maeda, T.; Fukazawa, Y.; Yamamoto, C.; Ozaki, M.; Yamamoto, H. Role of pharmacokinetic effects in the potentiation of morphine analgesia by L-type calcium channel blockers in mice. J. Pharmacol. Sci. 2004, 94, 240–245. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hamabe, W.; Maeda, T.; Fukazawa, Y.; Kumamoto, K.; Shang, L.Q.; Yamamoto, A.; Yamamoto, C.; Tokuyama, S.; Kishioka, S. P-glycoprotein ATPase activating effect of opioid analgesics and their P-glycoprotein-dependent antinociception in mice. Pharmacol. Biochem. Behav. 2006, 85, 629–636. [Google Scholar] [CrossRef]
- King, M.; Su, W.; Chang, A.; Zuckerman, A.; Pasternak, G.W. Transport of opioids from the brain to the periphery by P-glycoprotein: Peripheral actions of central drugs. Nat. Neurosci. 2001, 4, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.A.; Blough, B.E.; Epstein, D.H.; Negus, S.S.; Shaham, Y.; Banks, M.L. Effect of TRV130 and methadone on fentanyl-vs.-food choice and somatic withdrawal signs in opioid-dependent and post-opioid-dependent rats. Neuropsychopharmacology 2022, 47, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Kiyatkin, E.A. Respiratory depression and brain hypoxia induced by opioid drugs: Morphine, oxycodone, heroin, and fentanyl. Neuropharmacology 2019, 151, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.L.; Hedner, T.; Hedner, J.; Björkman, R.; Nordberg, G. Antinociceptive and ventilatory effects of the morphine metabolites: Morphine-6-glucuronide and morphine-3-glucuronide. Eur. J. Pharmacol. 1991, 193, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Romberg, R.; Sarton, E.; Teppema, L.; Matthes, H.W.; Kieffer, B.L.; Dahan, A. Comparison of morphine-6-glucuronide and morphine on respiratory depressant and antinociceptive responses in wild type and mu-opioid receptor deficient mice. Br. J. Anaesth. 2003, 91, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Ventriglia, E.; Rizzo, A.; Gomez, J.L.; Friedman, J.; Lam, S.; Solís, O.; Rais, R.; Bonaventura, J.; Michaelides, M. Essential role of P-glycoprotein in the mechanism of action of oliceridine. Neuropsychopharmacology 2023, 48, 831–842. [Google Scholar] [CrossRef]
- Ramos, K.A.; James, I.E.; Skobieranda, F.; Soergel, D.G.; Ruff, D.; Fossler, M.J. Two-Part Phase 1 Multiple-Ascending-Dose Study to Evaluate the Safety, Tolerability, Pharmacodynamics, and Pharmacokinetics of TRV734 in Healthy Adults. Clin. Pharmacol. Drug Dev. 2022, 11, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Simpson, K.N.; Fossler, M.J.; Wase, L.; Demitrack, M.A.; Wandstrat, T.L. Budget impact and pharmacy costs with targeted use of oliceridine for postsurgical pain in patients at high risk of opioid-related adverse events. Expert Rev. Pharmacoeconomics Outcomes Res. 2022, 22, 671–681. [Google Scholar] [CrossRef]
- Kieffer, B.L. Drug discovery: Designing the ideal opioid. Nature 2016, 537, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef]
- Pantouli, F.; Grim, T.W.; Schmid, C.L.; Acevedo-Canabal, A.; Kennedy, N.M.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Comparison of morphine, oxycodone and the biased MOR agonist SR-17018 for tolerance and efficacy in mouse models of pain. Neuropharmacology 2021, 185, 108439. [Google Scholar] [CrossRef]
- Stahl, E.L.; Schmid, C.L.; Acevedo-Canabal, A.; Read, C.; Grim, T.W.; Kennedy, N.M.; Bannister, T.D.; Bohn, L.M. G protein signaling-biased mu opioid receptor agonists that produce sustained G protein activation are noncompetitive agonists. Proc. Natl. Acad. Sci. USA 2021, 118, e2102178118. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).