
MD Simulation Reveals a Trimerization-Enhanced Interaction of CD137L with CD137


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
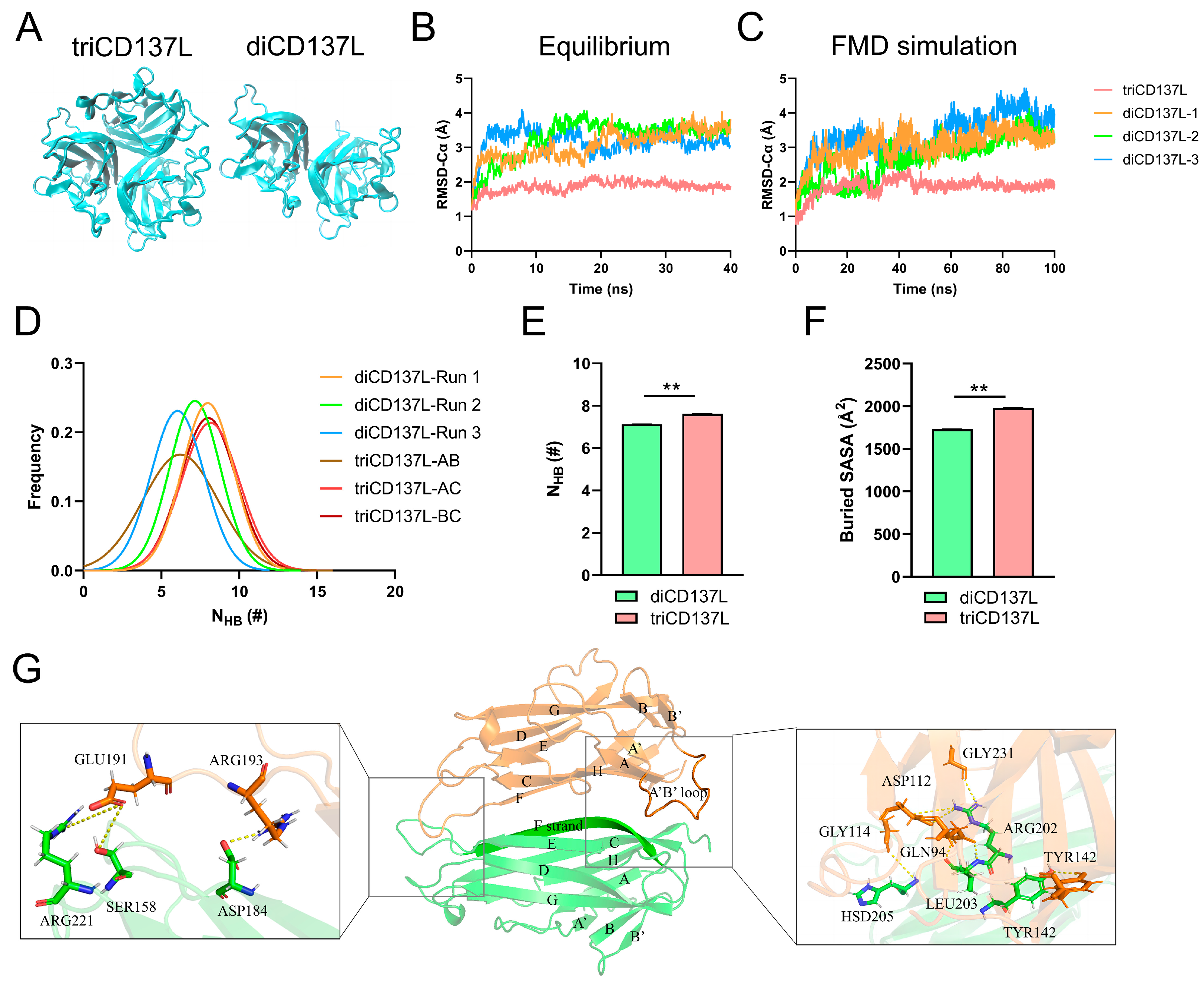
2.1. CD137L Trimer Has Better Thermostability than the Dimer Complex
2.2. Key Residues and Structural Variations Are Responsible for Improved triCD137L Thermostability
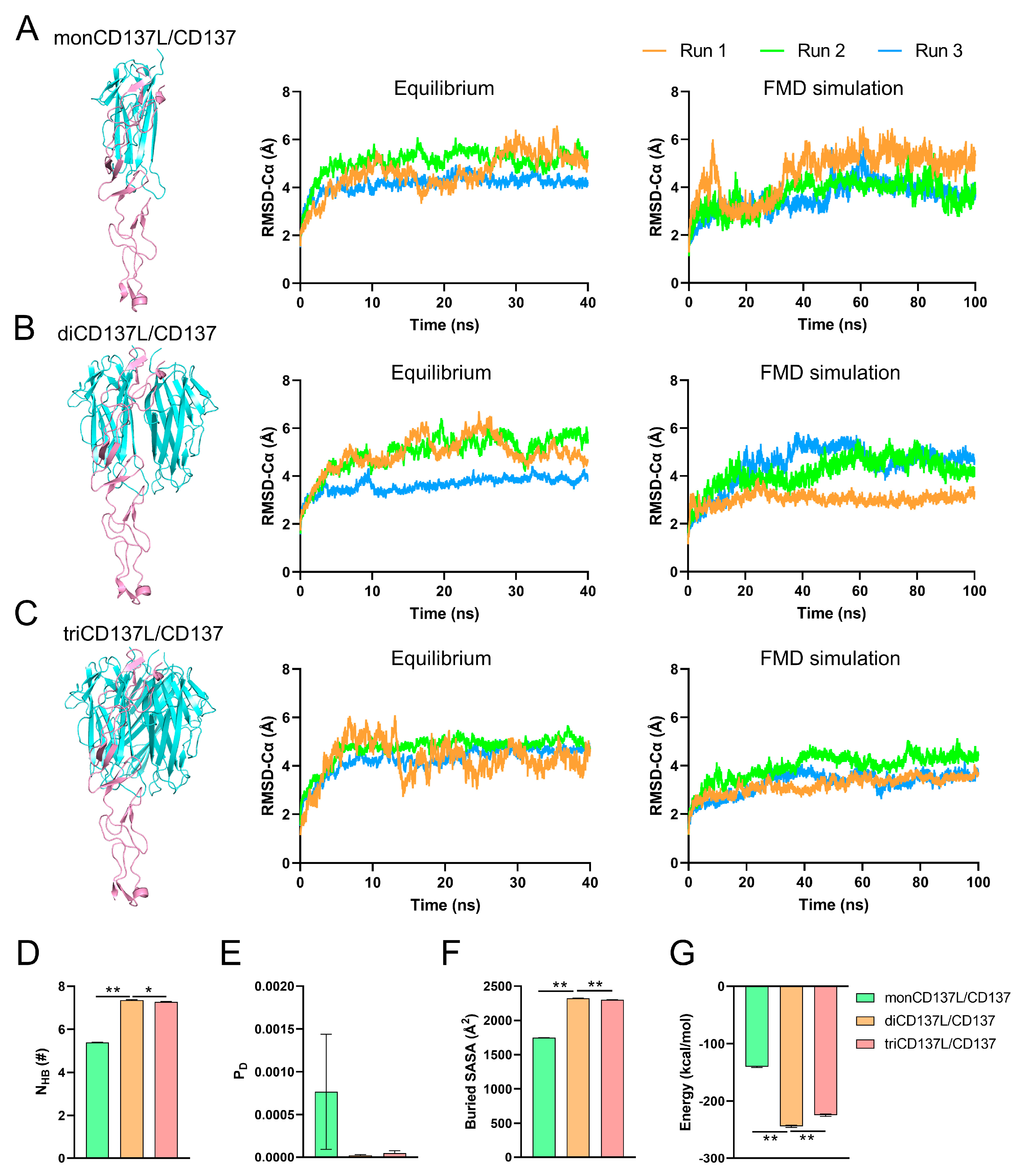
2.3. CD137L Dimerization and Trimerization Enhances Its Binding Affinity to CD137
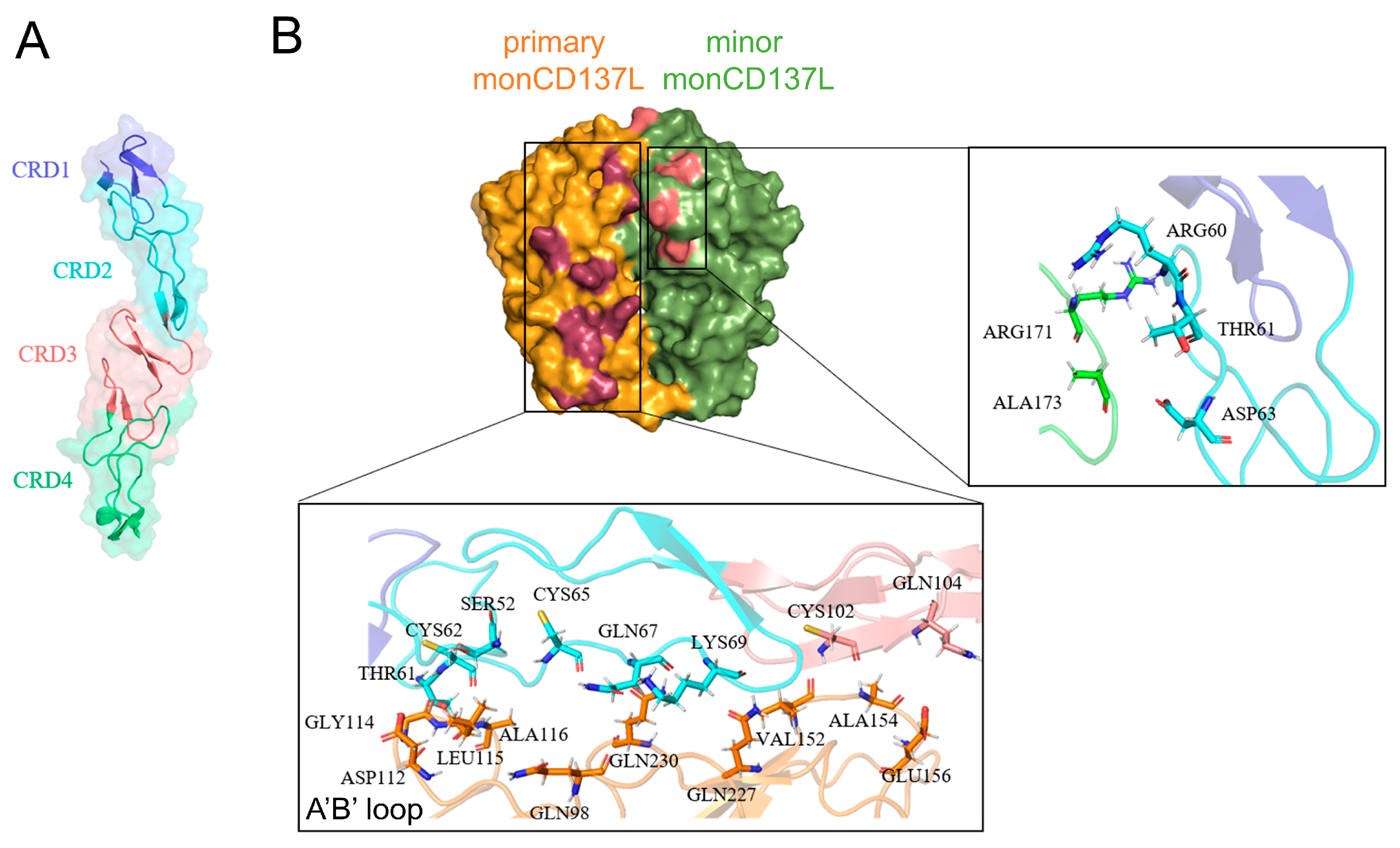
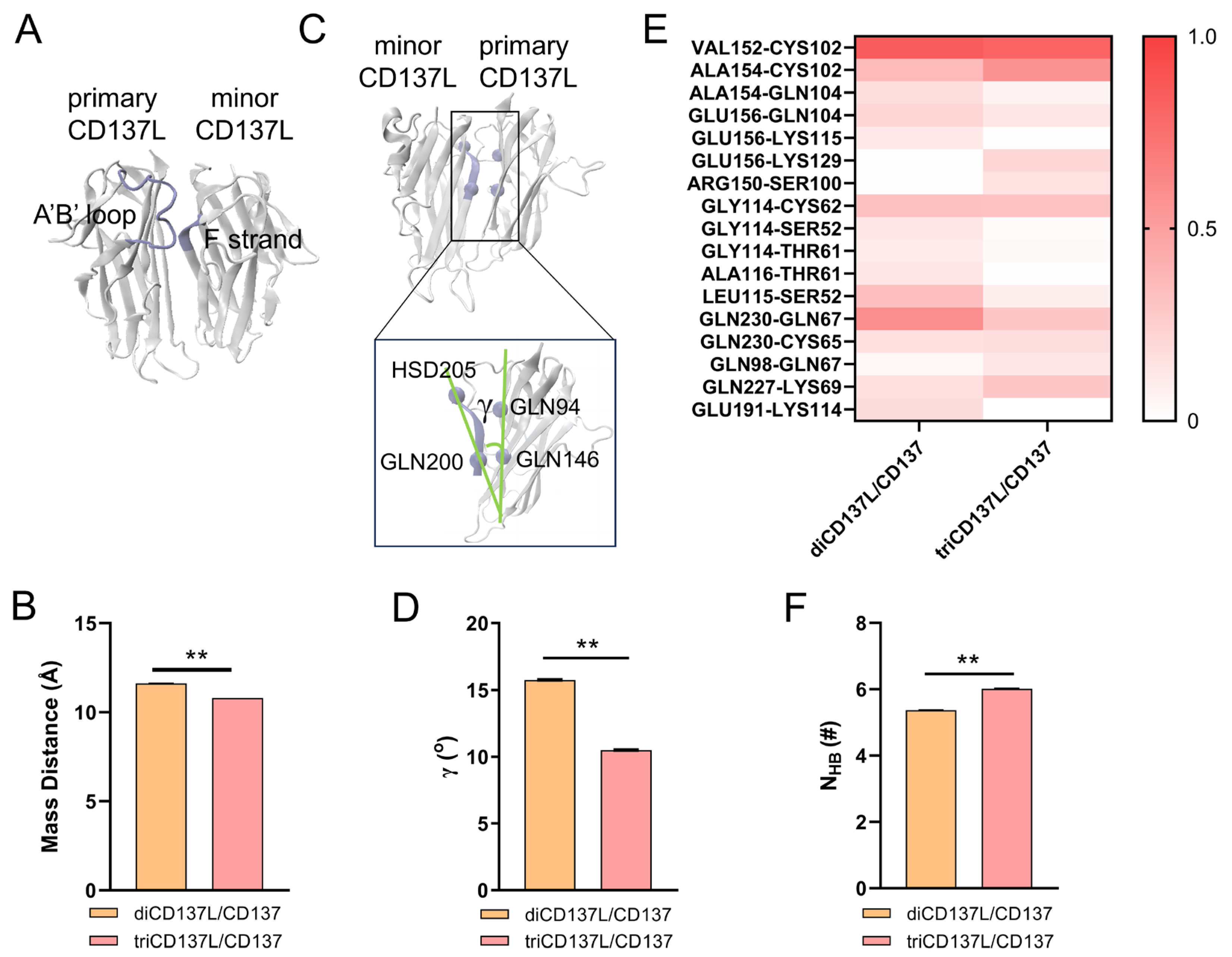
2.4. Allosteric Modulation of CD137 Binding Affinity Through Conformational Dynamics and Hydrogen Bonding in CD137L Complexes
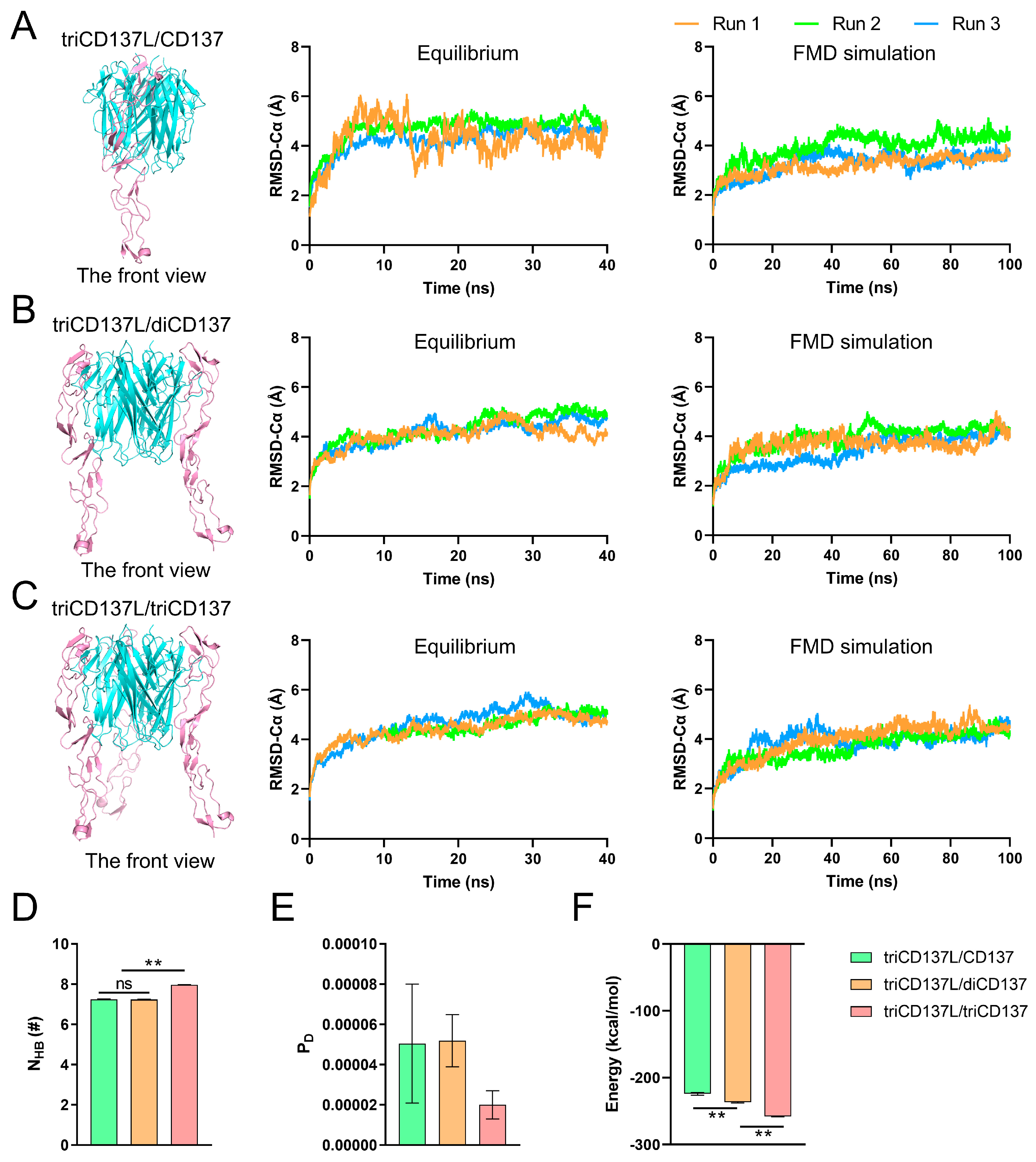
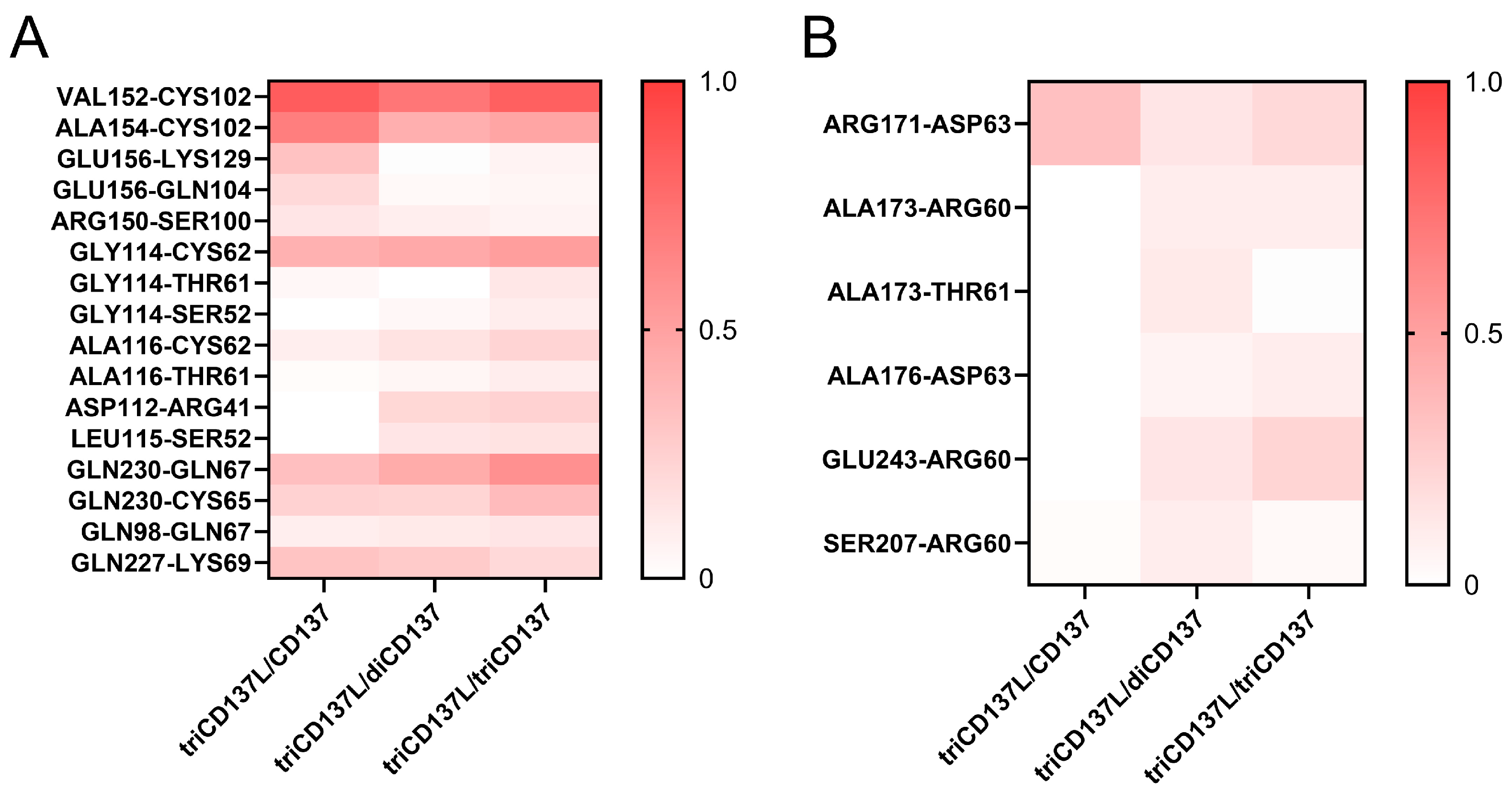
2.5. Trimerization of monoCD137 Induced by triCD137L Enhances the Binding Affinity Between CD137L and CD137 and CD137L Stability
3. Discussion
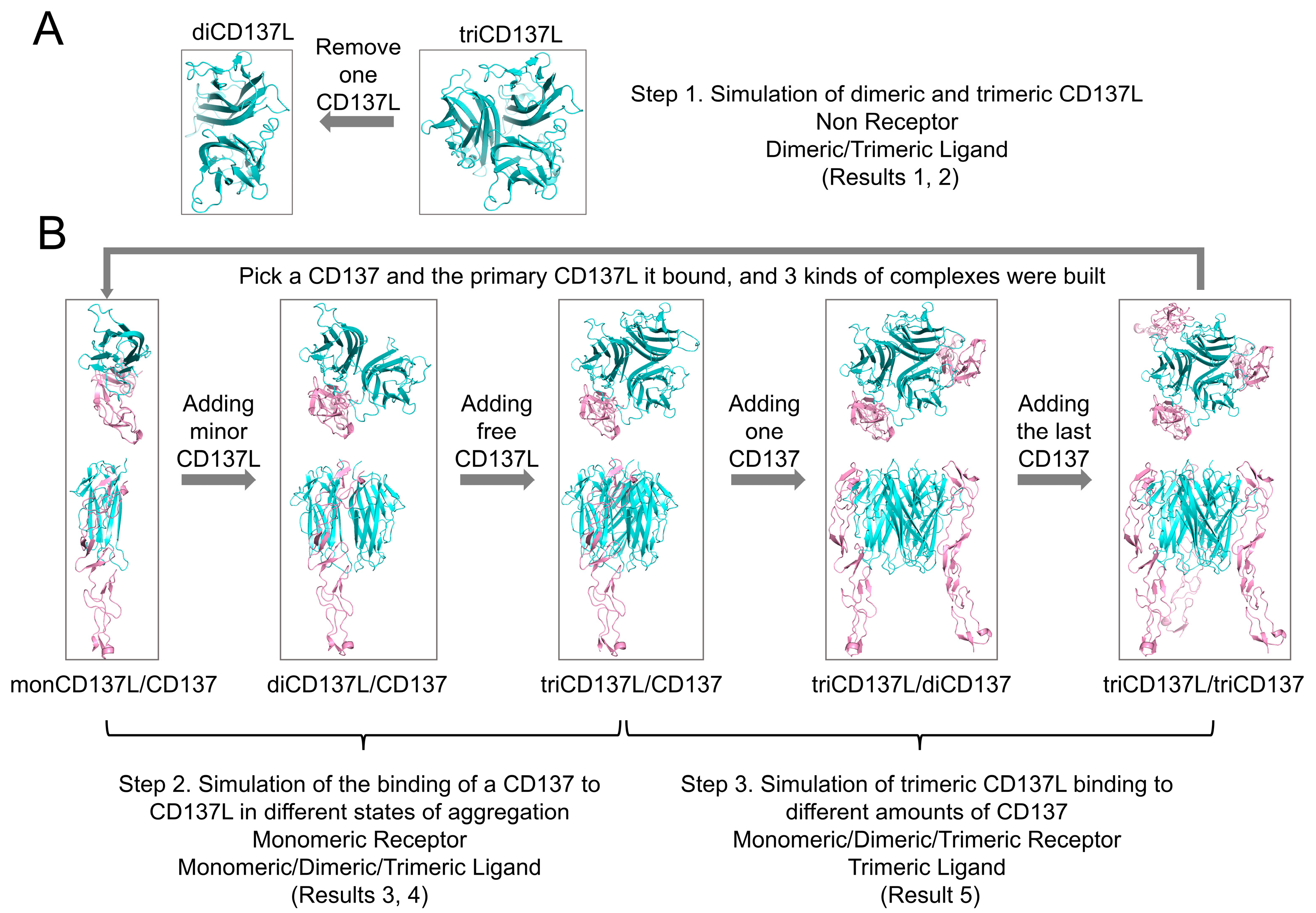
4. Materials and Methods
4.1. System Setup
4.2. MD Simulations
4.3. Data Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF family of ligands and receptors: Communication modules in the immune system and beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Dadas, O.; Ertay, A.; Cragg, M.S. Delivering co-stimulatory tumor necrosis factor receptor agonism for cancer immunotherapy: Past, current and future perspectives. Front. Immunol. 2023, 14, 1147467. [Google Scholar] [CrossRef]
- Croft, M.; Salek-Ardakani, S.; Ware, C.F. Targeting the TNF and TNFR superfamilies in autoimmune disease and cancer. Nat. Rev. Drug Discov. 2024, 23, 939–961. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- Eissner, G.; Kirchner, S.; Lindner, H.; Kolch, W.; Janosch, P.; Grell, M.; Scheurich, P.; Andreesen, R.; Holler, E. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J. Immunol. 2000, 164, 6193–6198. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, D.; Shi, X.; Liang, H.F.; Pang, Y.; Qin, N.L.; Chen, H.; Wang, J.; Yin, B.J.; Jiang, X.D.; et al. Transmembrane TNF-α mediates “forward” and “reverse” signaling, inducing cell death or survival via the NF-κB pathway in Raji Burkitt lymphoma cells. J. Leukoc. Biol. 2008, 84, 789–797. [Google Scholar] [CrossRef]
- Lückerath, K.; Kirkin, V.; Melzer, I.M.; Thalheimer, F.B.; Siele, D.; Milani, W.; Adler, T.; Aguilar-Pimentel, A.; Horsch, M.; Michel, G.; et al. Immune modulation by Fas ligand reverse signaling: Lymphocyte proliferation is attenuated by the intracellular Fas ligand domain. Blood 2011, 117, 519–529. [Google Scholar] [CrossRef]
- Muller, J.; Baeyens, A.; Dustin, M.L. Tumor necrosis factor receptor superfamily in T Cell priming and effector function. Adv. Immunol. 2018, 140, 21–57. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 2009, 9, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, B.; Wu, M.H.; Abdullah, N.S.; Rajendran, S.; Ishak, N.D.; Nickles, E.; Harfuddin, Z.; Schwarz, H. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology 2016, 5, e1113367. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.Y.; Schwarz, H. CD137/CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J. Autoimmun. 2020, 112, 102499. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Immunotherapy of cancer with 4-1BB. Mol. Cancer Ther. 2012, 11, 1062–1070. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhou, Y.B.; Schwarz, H. CD137L-DCs, potent immune-stimulators-History, characteristics, and perspectives. Front. Immunol. 2019, 10, 2216. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Choi, B.K.; Lee, D.G.; Kim, Y.H.; Kim, C.H.; Lee, S.J.; Kwon, B.S. 4-1BB signaling activates the T cell factor 1 effector/β-catenin pathway with delayed kinetics via ERK signaling and delayed PI3K/AKT activation to promote the proliferation of CD8 T cells. PLoS ONE 2013, 8, e6967. [Google Scholar] [CrossRef]
- Cannons, J.L.; Choi, Y.; Watts, T.H. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J. Immunol. 2000, 165, 6193–6204. [Google Scholar] [CrossRef]
- Soellner, L.; Shaqireen Kwajah, M.M.; Wu, J.T.; Schwarz, H. Signal transduction mechanisms of CD137 ligand in human monocytes. Cell Signal 2007, 19, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Schwarz, H. The progress of investigating the CD137-CD137L axis as a potential target for systemic lupus erythematosus. Cells 2019, 8, 1044. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Bang, B.R.; Lu, J.W.; Eun, S.Y.; Otsuka, M.; Croft, M.; Tobias, P.; Han, J.H.; Takeuchi, O.; Akira, S.; et al. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci. Signal 2013, 6, e2004431. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, A.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2019, 4, e000733. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Nuti, M. CD137+ T-cells: Protagonists of the immunotherapy revolution. Cancers 2021, 13, 456. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolaños, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef]
- Shen, X.L.; Zhang, R.S.; Nie, X.J.; Yang, Y.H.; Hua, Y.; Lue, P. 4-1BB targeting immunotherapy: Mechanism, antibodies, and chimeric antigen receptor T. Cancer Biother. Radiopharm. 2023, 38, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Z.; Luo, P.T. Targeting CD137 (4-1BB) towards improved safety and efficacy for cancer immunotherapy. Front. Immunol. 2023, 14, 1208788. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an integrated safety analysis of Urelumab, an agonist anti-CD137 monoclonal antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef]
- Dubrot, J.; Milheiro, F.; Alfaro, C.; Palazón, A.; Martinez-Forero, I.; Perez-Gracia, J.L.; Morales-Kastresana, A.; Romero-Trevejo, J.L.; Ochoa, M.C.; Hervás-Stubbs, S.; et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol. Immun. 2010, 59, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Khushalani, N.I.; Ott, P.A.; Ferris, R.L.; Cascone, T.; Schadendorf, D.; Le, D.T.; Sharma, M.R.; Barlesi, F.; Sharfman, W.; Luke, J.J.; et al. Final results of urelumab, an anti-CD137 agonist monoclonal antibody, in combination with cetuximab or nivolumab in patients with advanced solid tumors. J. Immunother. Cancer 2024, 12, e007364. [Google Scholar] [CrossRef]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I study of single-agent Utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.M.; Kimberlin, C.R.; Roe-Zurz, Z.; Zhang, P.; Xu, A.; Liao-Chan, S.; Sen, D.; Nager, A.R.; Oakdale, N.S.; Brown, C.; et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat. Commun. 2018, 9, 71436. [Google Scholar] [CrossRef]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 activated by extracellular adenosine triphosphate is tumor selective and broadly effective without systemic immune activation. Cancer Discov. 2021, 11, 158–175. [Google Scholar] [CrossRef]
- Liu, G.Z.; Du, F.; She, X.H.; Zhu, Y.; Tolcher, A.W.; Luo, P. A safe and potent agonist ADG106 targeting a unique epitope of CD137 with novel mechanism of actions. Cancer Res. 2020, 80, 4538. [Google Scholar] [CrossRef]
- Liu, L.; Chen, F.H.; Li, S.; Yang, T.; Chen, S.Z.; Zhou, Y.; Lin, Z.J.; Zeng, G.D.; Feng, P.J.; Shu, H.B.; et al. Human/mouse CD137 agonist, JNU-0921, effectively shrinks tumors through enhancing the cytotoxicity of CD8+ T cells in cis and in trans. Sci. Adv. 2024, 10, eadp8647. [Google Scholar] [CrossRef]
- O’Connell, J.; Porter, J.; Kroeplien, B.; Norman, T.; Rapecki, S.; Davis, R.; McMillan, D.; Arakaki, T.; Burgin, A.; Fox, D.; et al. Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer. Nat. Commun. 2019, 10, 13616. [Google Scholar] [CrossRef] [PubMed]
- van Schie, K.A.; Ooijevaar-de Heer, P.; Dijk, L.; Kruithof, S.; Wolbink, G.; Rispens, T. Therapeutic TNF inhibitors can differentially stabilize trimeric TNF by inhibiting monomer exchange. Sci. Rep. 2016, 6, 32747. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tan, S.G.; Zhang, C.; Chai, Y.; He, M.N.; Zhang, C.W.H.; Wang, Q.H.; Tong, Z.; Liu, K.F.; Lei, Y.F.; et al. Limited cross-linking of 4-1BB by 4-1BB ligand and the agonist monoclonal antibody utomilumab. Cell Rep. 2018, 25, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Gilbreth, R.N.; Oganesyan, V.Y.; Amdouni, H.; Novarra, S.; Grinberg, L.; Barnes, A.; Baca, M. Crystal structure of the human 4-1BB/4-1BBL complex. J. Biol. Chem. 2018, 293, 9880–9891. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Lau, P.; Ghumman, B.; DeBenedette, M.A.; Yagita, H.; Okumura, K.; Watts, T.H. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J. Immunol. 2001, 167, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.M.; Perez-Chacon, G.; Carr-Baena, P.; Martinez-Forero, I.; Azpilikueta, A.; Otano, I.; Melero, I. CD137 (4-1BB) signalosome: Complexity is a matter of TRAFs. Front. Immunol. 2018, 9, 2618. [Google Scholar] [CrossRef]
- Zapata, J.M.; Lefebvre, S.; Reed, J.C. Targeting TRAFs for therapeutic intervention. Adv. Exp. Med. Biol. 2007, 597, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. CD137 as an attractive T cell co-stimulatory target in the TNFRSF for immuno-oncology drug development. Cancers 2021, 13, 2288. [Google Scholar] [CrossRef]
- Somekh, I.; Thian, M.; Medgyesi, D.; Gülez, N.; Magg, T.; Duque, A.G.; Stauber, T.; Lev, A.; Genel, F.; Unal, E.; et al. CD137 deficiency causes immune dysregulation with predisposition to lymphomagenesis. Blood 2019, 134, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.Y.; Jia, K.Y.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.Y.; Zhou, C.C. CD137, an attractive candidate for the immunotherapy of lung cancer. Cancer Sci. 2020, 111, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.P.; Reynoso, G.V.; Andhey, P.S.; Swain, A.; Turner, J.S.; Boon, A.C.M.; Krammer, F.; Ellebedy, A.H.; Zanini, F.; Artyomov, M.; et al. An agonistic anti-CD137 antibody disrupts lymphoid follicle structure and T-cell-dependent antibody responses. Cell Rep. Med. 2020, 1, 100035. [Google Scholar] [CrossRef]
- Wyzgol, A.; Müller, N.; Fick, A.; Munkel, S.; Grigoleit, G.U.; Pfizenmaier, K.; Wajant, H. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J. Immunol. 2009, 183, 1851–1861. [Google Scholar] [CrossRef]
- Blevitt, J.M.; Hack, M.D.; Herman, K.L.; Jackson, P.F.; Krawczuk, P.J.; Lebsack, A.D.; Liu, A.X.; Mirzadegan, T.; Nelen, M.I.; Patrick, A.N.; et al. Structural basis of small-molecule aggregate induced inhibition of a protein protein interaction. J. Med. Chem. 2017, 60, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Rabu, C.; Quéméner, A.; Jacques, Y.; Echasserieau, K.; Vusio, P.; Lang, F. Production of recombinant human trimeric CD137L (4-1BBL): Cross-linking is essential to its T cell co-stimulation activity. J. Biol. Chem. 2005, 280, 41472–41481. [Google Scholar] [CrossRef]
- Won, E.Y.; Cha, K.; Byun, J.S.; Kim, D.U.; Shin, S.; Ahn, B.; Kim, Y.H.; Rice, A.J.; Walz, T.; Kwon, B.S.; et al. The structure of the trimer of human 4-1BB ligand is unique among members of the tumor necrosis factor superfamily. J. Biol. Chem. 2010, 285, 9202–9210. [Google Scholar] [CrossRef]
- Yi, L.; Yan, Z.; Jia, H.; Wang, X.; Zhao, Y.; Zhang, H. CD137-CRDI is not necessary in the role of contacting its natural ligand. Immunol. Cell Biol. 2016, 95, 24–32. [Google Scholar] [CrossRef]
- Kucka, K.; Wajant, H. Receptor oligomerization and its relevance for signaling by receptors of the tumor necrosis factor receptor superfamily. Front. Cell. Dev. Biol. 2021, 8, 615141. [Google Scholar] [CrossRef]
- Votapka, L.W.; Stokely, A.M.; Ojha, A.A.; Amaro, R.E. SEEKR2: Versatile multiscale milestoning utilizing the OpenMM molecular dynamics engine. J. Chem. Inf. Model. 2022, 62, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.A.; Votapka, L.W.; Amaro, R.E. QMrebind: Incorporating quantum mechanical force field reparameterization at the ligand binding site for improved drug-target kinetics through milestoning simulations. Chem. Sci. 2023, 14, 13159–13175. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.A.; Srivastava, A.; Votapka, L.W.; Amaro, R.E. Selectivity and ranking of tight-binding JAK-STAT inhibitors using Markovian milestoning with voronoi tessellations. J. Chem. Inf. Model. 2023, 63, 2469–2482. [Google Scholar] [CrossRef] [PubMed]
- Votapka, L.W.; Ojha, A.A.; Asada, N.; Amaro, R.E. Prediction of threonine-tyrosine kinase receptor-ligand unbinding kinetics with multiscale milestoning and metadynamics. J. Phys. Chem. Lett. 2024, 15, 10473–10478. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Jagger, B.R.; Amaro, R.E. Ranking of ligand binding kinetics using a weighted ensemble approach and comparison with a multiscale milestoning approach. J. Chem. Inf. Model. 2020, 60, 5340–5352. [Google Scholar] [CrossRef]
- Jagger, B.R.; Ojha, A.A.; Amaro, R.E. Predicting ligand binding kinetics using a Markovian milestoning with Voronoi tessellations multiscale approach. J. Chem. Theory Comput. 2020, 16, 5348–5357. [Google Scholar] [CrossRef]
- Van Wart, A.T.; Durrant, J.; Votapka, L.; Amaro, R.E. Weighted Implementation of Suboptimal Paths (WISP): An optimized algorithm and tool for dynamical network analysis. J. Chem. Theory Comput. 2014, 10, 511–517. [Google Scholar] [CrossRef]
- Ojha, A.A.; Thakur, S.; Ahn, S.H.; Amaro, R.E. DeepWEST: Deep learning of kinetic models with the weighted ensemble simulation toolkit for enhanced sampling. J. Chem. Theory Comput. 2023, 19, 1342–1359. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, S.; Cavalli, A. Thermodynamics and kinetics of drug-target binding by molecular simulation. Chem. Rev. 2020, 120, 12788–12833. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Fei, P.; Zhang, T.; Yao, D.; Gao, Y.; Liu, J.; Chen, H.; Lu, Q.; Mudianto, T.; et al. Mechanical activation of spike fosters SARS-CoV-2 viral infection. Cell Res. 2021, 31, 1047–1060. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, M.; Ogino, S.; Umemoto, R.; Nishida, N.; Shimada, I. Mechanical force effect on the two-state equilibrium of the hyaluronan-binding domain of CD44 in cell rolling. Proc. Natl. Acad. Sci. USA 2015, 112, 6991–6996. [Google Scholar] [CrossRef]
- Liu, F.F.; Wu, Q.; Han, W.; Laster, K.; Hu, Y.M.; Ma, F.Y.; Chen, H.Y.; Tian, X.L.; Qiao, Y.; Liu, H.; et al. Targeting integrin αvβ3 with indomethacin inhibits patient-derived xenograft tumour growth and recurrence in oesophageal squamous cell carcinoma. Clin. Transl. Med. 2021, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Mackoy, T.; Kale, B.; Papka, M.E.; Wheeler, R.A. viewSq, a Visual Molecular Dynamics (VMD) module for calculating, analyzing, and visualizing X-ray and neutron structure factors from atomistic simulations (vol 264, 107881, 2021). Comput. Phys. Commun. 2022, 276, 108358. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 014475. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.Y.; Wu, J.H.; Fang, Y. Moderate constraint facilitates association and force-dependent dissociation of HA-CD44 complex. Int. J. Mol. Sci. 2023, 24, 2243. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, Z.Y.; Fang, Y.; Wu, J.H. Prediction of catch-slip bond transition of kindlin2/β3 integrin via steered molecular dynamics simulation. J. Chem. Inf. Model. 2020, 60, 5132–5141. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Dis. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wu, J.; Fang, Y.; Li, Q. MD Simulation Reveals a Trimerization-Enhanced Interaction of CD137L with CD137. Int. J. Mol. Sci. 2025, 26, 1903. https://doi.org/10.3390/ijms26051903
Wang H, Wu J, Fang Y, Li Q. MD Simulation Reveals a Trimerization-Enhanced Interaction of CD137L with CD137. International Journal of Molecular Sciences. 2025; 26(5):1903. https://doi.org/10.3390/ijms26051903
Chicago/Turabian StyleWang, Hefeng, Jianhua Wu, Ying Fang, and Quhuan Li. 2025. "MD Simulation Reveals a Trimerization-Enhanced Interaction of CD137L with CD137" International Journal of Molecular Sciences 26, no. 5: 1903. https://doi.org/10.3390/ijms26051903
APA StyleWang, H., Wu, J., Fang, Y., & Li, Q. (2025). MD Simulation Reveals a Trimerization-Enhanced Interaction of CD137L with CD137. International Journal of Molecular Sciences, 26(5), 1903. https://doi.org/10.3390/ijms26051903