

Bacterial Communities and Resistance and Virulence Genes in Hospital and Community Wastewater: Metagenomic Analysis

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DNA Extraction and Sequencing
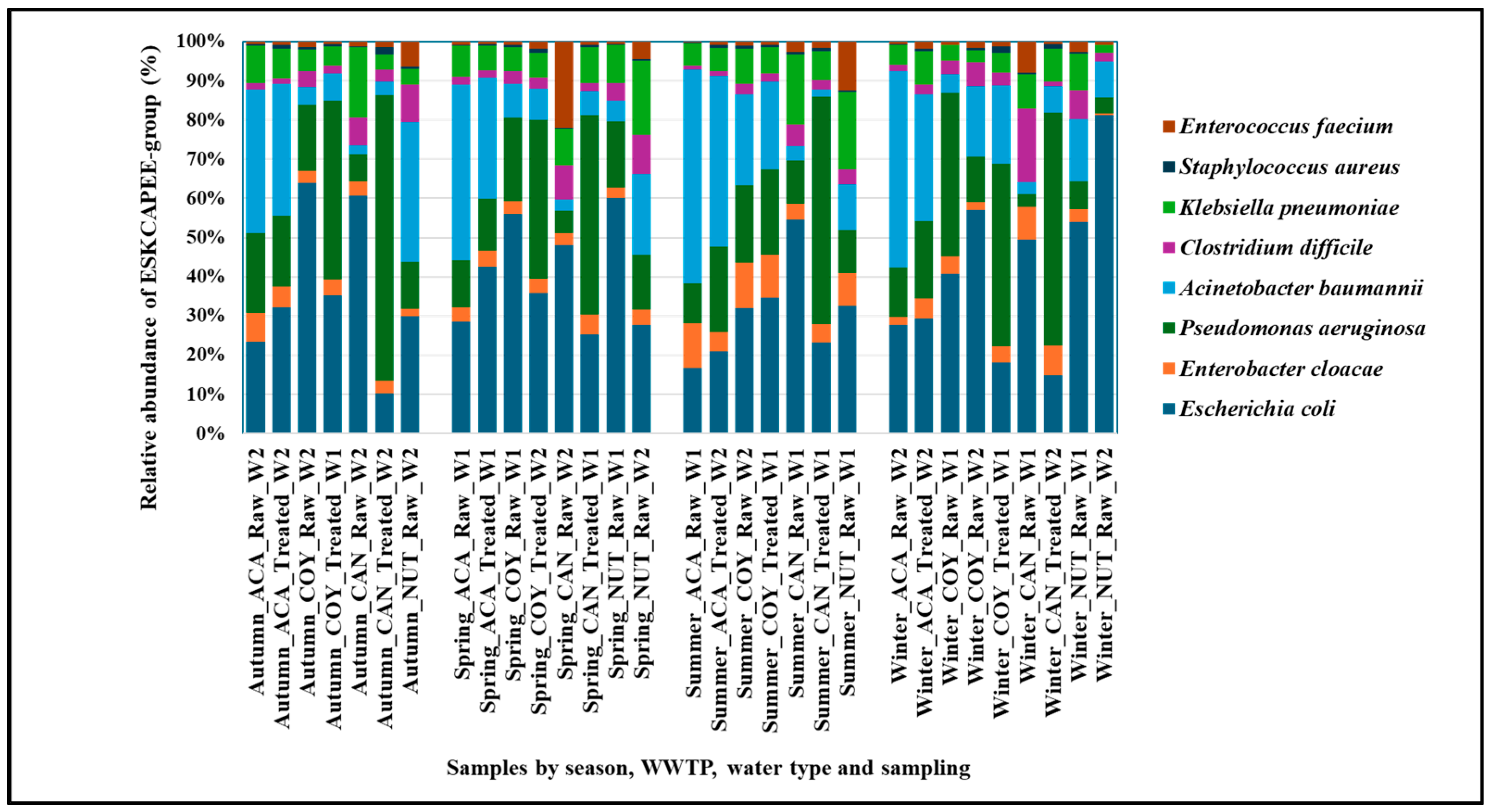
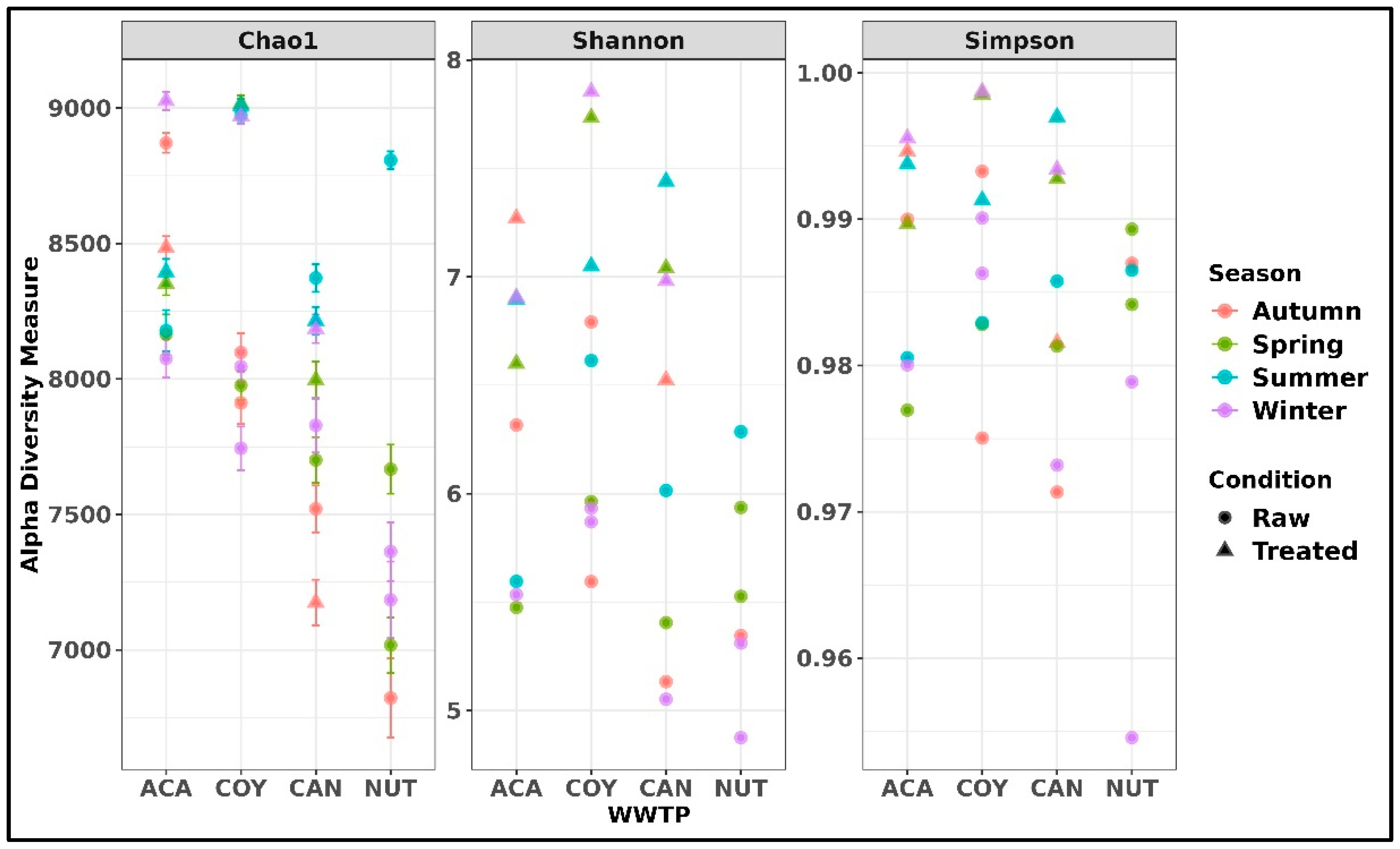
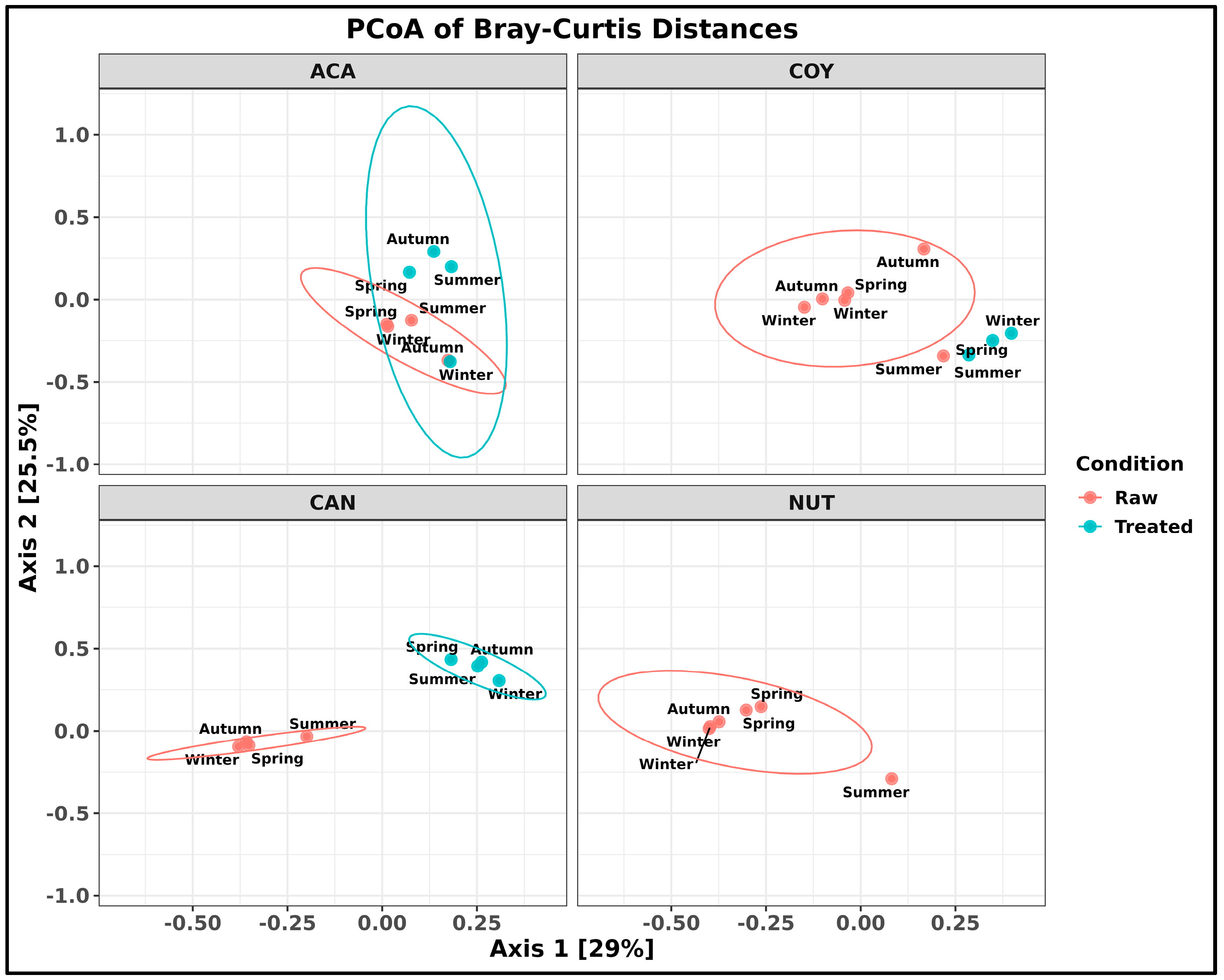
2.2. Taxonomic Composition of Bacteria in Community and Hospital Wastewater
2.3. Analysis of Antimicrobial Resistance Genes
2.4. Analysis of Virulence Factor Genes
3. Discussion
3.1. Taxonomic Composition of Bacteria in Community and Hospital Wastewater
3.2. Analysis of Antimicrobial Resistance Genes
3.3. Analysis of Virulence Factor Genes
4. Materials and Methods
4.1. Study Design and Sample Collection
4.2. Wastewater Treatment Plant Characteristics
4.3. DNA Extraction
4.4. Sequencing and Bioinformatics Analysis
4.5. Analysis of Antimicrobial Resistance Genes and Virulence Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Antimicrobial Resistance Surveillance System (GLASS) Report: Early Implementation 2020, 1st ed.; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-000558-7. [Google Scholar]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis (accessed on 7 January 2025).
- Sinclair, R.G.; Choi, C.Y.; Riley, M.R.; Gerba, C.P. Pathogen Surveillance Through Monitoring of Sewer Systems. In Advances in Applied Microbiology; Academic Press Inc.: Cambridge, MA, USA, 2008; Volume 65, pp. 249–269. ISBN 9780123744296. [Google Scholar]
- Daughton, C.G. Wastewater Surveillance for Population-Wide COVID-19: The Present and Future. Sci. Total Environ. 2020, 736, 139631. [Google Scholar] [CrossRef] [PubMed]
- Manaia, C.M.; Rocha, J.; Scaccia, N.; Marano, R.; Radu, E.; Biancullo, F.; Cerqueira, F.; Fortunato, G.; Iakovides, I.C.; Zammit, I.; et al. Antibiotic Resistance in Wastewater Treatment Plants: Tackling the Black Box. Environ. Int. 2018, 115, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Warnes, S.L.; Highmore, C.J.; Keevil, C.W. Horizontal Transfer of Antibiotic Resistance Genes on Abiotic Touch Surfaces: Implications for Public Health. MBio 2012, 3, 1010–1128. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, F.M.; Woolhouse, M.E.J. Using Sewage for Surveillance of Antimicrobial Resistance. Science 2020, 367, 630–632. [Google Scholar] [CrossRef]
- Miłobedzka, A.; Ferreira, C.; Vaz-Moreira, I.; Calderón-Franco, D.; Gorecki, A.; Purkrtova, S.; Bartacek, J.; Dziewit, L.; Singleton, C.M.; Nielsen, P.H.; et al. Monitoring Antibiotic Resistance Genes in Wastewater Environments: The Challenges of Filling a Gap in the One-Health Cycle. J. Hazard. Mater. 2022, 424, 127407. [Google Scholar] [CrossRef]
- Pärnänen, K.M.M.; Narciso-da-Rocha, C.; Kneis, D.; Berendonk, T.U.; Cacace, D.; Do, T.T.; Elpers, C.; Fatta-Kassinos, D.; Henriques, I.; Jaeger, T.; et al. Antibiotic Resistance in European Wastewater Treatment Plants Mirrors the Pattern of Clinical Antibiotic Resistance Prevalence. Sci. Adv. 2019, 5, eaau9124. [Google Scholar] [CrossRef]
- Raza, S.; Shin, H.; Hur, H.G.; Unno, T. Higher Abundance of Core Antimicrobial Resistant Genes in Effluent from Wastewater Treatment Plants. Water Res. 2022, 208, 117882. [Google Scholar] [CrossRef]
- Mao, G.; Liang, J.; Wang, Q.; Zhao, C.; Bai, Y.; Liu, R.; Liu, H.; Qu, J. Epilithic Biofilm as a Reservoir for Functional Virulence Factors in Wastewater-Dominant Rivers after WWTP Upgrade. J. Environ. Sci. 2021, 101, 27–35. [Google Scholar] [CrossRef]
- Galarde-López, M.; Velazquez-Meza, M.E.; Godoy-Lozano, E.E.; Carrillo-Quiroz, B.A.; Cornejo-Juárez, P.; Sassoé-González, A.; Ponce-de-León, A.; Saturno-Hernández, P.; Alpuche-Aranda, C.M. Presence and Persistence of ESKAPEE Bacteria before and after Hospital Wastewater Treatment. Microorganisms 2024, 12, 1231. [Google Scholar] [CrossRef]
- Numberger, D.; Ganzert, L.; Zoccarato, L.; Mühldorfer, K.; Sauer, S.; Grossart, H.P.; Greenwood, A.D. Characterization of Bacterial Communities in Wastewater with Enhanced Taxonomic Resolution by Full-Length 16S RRNA Sequencing. Sci. Rep. 2019, 9, 9673. [Google Scholar] [CrossRef]
- Lu, X.M.; Lu, P.Z. Characterization of Bacterial Communities in Sediments Receiving Various Wastewater Effluents with High-Throughput Sequencing Analysis. Microb. Ecol. 2014, 67, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Poopedi, E.; Singh, T.; Gomba, A. Potential Exposure to Respiratory and Enteric Bacterial Pathogens among Wastewater Treatment Plant Workers, South Africa. Int. J. Environ. Res. Public Health 2023, 20, 4338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Peng, Y.; Chan, C.L.; On, H.; Wai, H.K.F.; Shekhawat, S.S.; Gupta, A.B.; Varshney, A.K.; Chuanchuen, R.; Zhou, X.; et al. Metagenomic Survey Reveals More Diverse and Abundant Antibiotic Resistance Genes in Municipal Wastewater Than Hospital Wastewater. Front. Microbiol. 2021, 12, 712843. [Google Scholar] [CrossRef] [PubMed]
- Lepper, H.C.; Perry, M.R.; Wee, B.A.; Wills, D.; Nielsen, H.; Otani, S.; Simon, M.; Aarestrup, F.M.; Woolhouse, M.E.J.; van Bunnik, B.A.D. Distinctive Hospital and Community Resistomes in Scottish Urban Wastewater: Metagenomics of a Paired Wastewater Sampling Design. Sci. Total Environ. 2023, 902, 165978. [Google Scholar] [CrossRef]
- Fresia, P.; Antelo, V.; Salazar, C.; Giménez, M.; D’Alessandro, B.; Afshinnekoo, E.; Mason, C.; Gonnet, G.H.; Iraola, G. Urban Metagenomics Uncover Antibiotic Resistance Reservoirs in Coastal Beach and Sewage Waters. Microbiome 2019, 7, 35. [Google Scholar] [CrossRef]
- Berglund, F.; Ebmeyer, S.; Kristiansson, E.; Larsson, D.G.J. Evidence for Wastewaters as Environments Where Mobile Antibiotic Resistance Genes Emerge. Commun. Biol. 2023, 6, 321. [Google Scholar] [CrossRef]
- Lin, Q.; Xavier, B.B.; Alako, B.T.F.; Mitchell, A.L.; Rajakani, S.G.; Glupczynski, Y.; Finn, R.D.; Cochrane, G.; Malhotra-Kumar, S. Screening of Global Microbiomes Implies Ecological Boundaries Impacting the Distribution and Dissemination of Clinically Relevant Antimicrobial Resistance Genes. Commun. Biol. 2022, 5, 1217. [Google Scholar] [CrossRef]
- He, Y.; Li, K.X.; Wang, J.W.; Wang, W.; Fan, P.C.; Chen, H.H.; Wang, J.J. Microbial Community Structure of Wastewater Treatment Plants in Different Seasons. Environ. Sci. 2021, 42, 1488–1495. [Google Scholar] [CrossRef]
- Kang, X.H.; Leng, Y.; Macdonald, O.M.; Zeng, X.Y.; Li, S.W. The Seasonal Changes of Core Bacterial Community Decide Sewage Purification in Sub-Plateau Municipal Sewage Treatment Plants. Bioprocess Biosyst. Eng. 2020, 43, 1609–1617. [Google Scholar] [CrossRef]
- Sanderson, H.; Ortega-Polo, R.; Zaheer, R.; Goji, N.; Amoako, K.K.; Brown, R.S.; Majury, A.; Liss, S.N.; McAllister, T.A. Comparative Genomics of Multidrug-Resistant Enterococcus spp. Isolated from Wastewater Treatment Plants. BMC Microbiol. 2020, 20. [Google Scholar] [CrossRef]
- Moradigaravand, D.; Gouliouris, T.; Ludden, C.; Reuter, S.; Jamrozy, D.; Blane, B.; Naydenova, P.; Judge, K.; Aliyu, S.H.; Hadjirin, N.F.; et al. Genomic Survey of Clostridium difficile Reservoirs in the East of England Implicates Environmental Contamination of Wastewater Treatment Plants by Clinical Lineages. Microb. Genom. 2018, 4, e000162. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Meza, M.E.; Galarde-López, M.; Cornejo-Juárez, P.; Carrillo-Quiroz, B.A.; Velázquez-Acosta, C.; Bobadilla-del-Valle, M.; Ponce-de-León, A.; Alpuche-Aranda, C.M. Multidrug-Resistant Staphylococcus sp. and Enterococcus sp. in Municipal and Hospital Wastewater: A Longitudinal Study. Microorganisms 2024, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Hubeny, J.; Korzeniewska, E.; Ciesielski, S.; Płaza, G.; Harnisz, M. The Resistome of ESKAPEE Pathogens in Untreated and Treated Wastewater: A Polish Case Study. Biomolecules 2022, 12, 1160. [Google Scholar] [CrossRef] [PubMed]
- Dijkshoorn, L.; van Aken, E.; Shunburne, L.; van der Reijden, T.J.K.; Bernards, A.T.; Nemec, A.; Towner, K.J. Prevalence of Acinetobacter baumannii and Other Acinetobacter spp. in Faecal Samples from Non-Hospitalised Individuals. Clin. Microbiol. Infect. 2005, 11, 329–332. [Google Scholar] [CrossRef]
- Ma, X.; Dong, X.; Cai, J.; Fu, C.; Yang, J.; Liu, Y.; Zhang, Y.; Wan, T.; Lin, S.; Lou, Y.; et al. Metagenomic Analysis Reveals Changes in Bacterial Communities and Antibiotic Resistance Genes in an Eye Specialty Hospital and a General Hospital Before and After Wastewater Treatment. Front. Microbiol. 2022, 13, 848167. [Google Scholar] [CrossRef]
- Gholami, S.; Tabatabaei, M.; Sohrabi, N. Comparison of Biofilm Formation and Antibiotic Resistance Pattern of Pseudomonas aeruginosa in Human and Environmental Isolates. Microb. Pathog. 2017, 109, 94–98. [Google Scholar] [CrossRef]
- Auguet, O.; Pijuan, M.; Borrego, C.M.; Rodriguez-Mozaz, S.; Triadó-Margarit, X.; Della Giustina, S.V.; Gutierrez, O. Sewers as Potential Reservoirs of Antibiotic Resistance. Sci. Total Environ. 2017, 605–606, 1047–1054. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D.G.J. Fecal Pollution Can Explain Antibiotic Resistance Gene Abundances in Anthropogenically Impacted Environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Pilmis, B.; Le Monnier, A.; Zahar, J.R. Gut Microbiota, Antibiotic Therapy and Antimicrobial Resistance: A Narrative Review. Microorganisms 2020, 8, 269. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Zou, S.; Fang, H.H.P.; Zhang, T. Fate of Antibiotic Resistance Genes in Sewage Treatment Plant Revealed by Metagenomic Approach. Water Res. 2014, 62, 97–106. [Google Scholar] [CrossRef]
- Gupta, S.K.; Shin, H.; Han, D.; Hur, H.G.; Unno, T. Metagenomic Analysis Reveals the Prevalence and Persistence of Antibiotic- and Heavy Metal-Resistance Genes in Wastewater Treatment Plant. J. Microbiol. 2018, 56, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowski, R.; Linke, B.; Krahn, I.; Gartemann, K.H.; Gützkow, T.; Eichler, W.; Pühler, A.; Schlüter, A. Detection of 140 Clinically Relevant Antibiotic-Resistance Genes in the Plasmid Metagenome of Wastewater Treatment Plant Bacteria Showing Reduced Susceptibility to Selected Antibiotics. Microbiology 2009, 155, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.M.; Baker-Austin, C.; Verner-Jeffreys, D.W.; Ryan, J.J.; Micallef, C.; Maskell, D.J.; Pearce, G.P. Overexpression of Antibiotic Resistance Genes in Hospital Effluents over Time. J. Antimicrob. Chemother. 2017, 72, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, J.; Zhao, Z.; Cao, Y.; Li, B. Hospital Wastewater as a Reservoir for Antibiotic Resistance Genes: A Meta-Analysis. Front. Public Health 2020, 8, 679. [Google Scholar] [CrossRef] [PubMed]
- Heir, E.; Sundheim, G.; Holck, A.L. The Staphylococcus QacH Gene Product: A New Member of the SMR Family Encoding Multidrug Resistance. FEMS Microbiol. Lett. 1998, 163, 49–56. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, Y.; Li, Y.; Zhang, K.; Liu, L.; Wang, H.; Tian, J.; Ying, H.; Shi, L.; Yu, T. Characterization and Horizontal Transfer of QacH-Associated Class 1 Integrons in Escherichia coli Isolated from Retail Meats. Int. J. Food Microbiol. 2017, 258, 12–17. [Google Scholar] [CrossRef]
- Correa, J.E.; De Paulis, A.; Predari, S.; Sordelli, D.O.; Jeric, P.E. First Report of QacG, QacH and QacJ Genes in Staphylococcus haemolyticus Human Clinical Isolates. J. Antimicrob. Chemother. 2008, 62, 956–960. [Google Scholar] [CrossRef]
- Bischoff, M.; Bauer, J.; Preikschat, P.; Schwaiger, K.; Mölle, G.; Hölzel, C. First Detection of the Antiseptic Resistance Gene QacA/B in Enterococcus faecalis. Microb. Drug Resist. 2012, 18, 7–12. [Google Scholar] [CrossRef]
- Hefzy, E.M.; Radwan, T.E.E.; Hozayen, B.M.M.; Mahmoud, E.E.; Khalil, M.A.F. Antiseptics and Mupirocin Resistance in Clinical, Environmental, and Colonizing Coagulase Negative Staphylococcus Isolates. Antimicrob. Resist. Infect. Control 2023, 12, 110. [Google Scholar] [CrossRef]
- Xia, R.; Ren, Y.; Guo, X.; Xu, H. Molecular Diversity of Class 2 Integrons in Antibiotic-Resistant Gram-Negative Bacteria Found in Wastewater Environments in China. Ecotoxicology 2013, 22, 402–414. [Google Scholar] [CrossRef]
- Morgado, S.M.; Fonseca, É.L.; Vicente, A.C.P. Prevalence and Characterization of an Integrative and Conjugative Element Carrying Tet(X) Gene in Elizabethkingia meningoseptica. J. Glob. Antimicrob. Resist. 2024, 38, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.N.; Li, M. Comparative Resistomics Analysis of Multidrug-Resistant Chryseobacteria. Environ. Microbiol. Rep. 2024, 16, e13288. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Ribeiro, J.; Rocha, J.; Manaia, C.M.; Silva, A.; Pereira, J.E.; Maltez, L.; Capelo, J.L.; Igrejas, G.; Poeta, P. High Frequency of the EMRSA-15 Clone (ST22-MRSA-IV) in Hospital Wastewater. Microorganisms 2022, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.E.Z.; Zaheer, R.; Poulin-Laprade, D.; Scott, A.; Rehman, M.A.; Diarra, M.; Topp, E.; Van Domselaar, G.; Zovoilis, A.; McAllister, T.A. Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum. Microorganisms 2023, 11, 727. [Google Scholar] [CrossRef]
- Ben Said, L.; Klibi, N.; Lozano, C.; Dziri, R.; Ben Slama, K.; Boudabous, A.; Torres, C. Diversity of Enterococcal Species and Characterization of High-Level Aminoglycoside Resistant Enterococci of Samples of Wastewater and Surface Water in Tunisia. Sci. Total Environ. 2015, 530–531, 11–17. [Google Scholar] [CrossRef]
- Surleac, M.; Barbu, I.C.; Paraschiv, S.; Popa, L.I.; Gheorghe, I.; Marutescu, L.; Popa, M.; Sarbu, I.; Talapan, D.; Nita, M.; et al. Whole Genome Sequencing Snapshot of Multidrug Resistant Klebsiella Pneumoniae Strains from Hospitals and Receiving Wastewater Treatment Plants in Southern Romania. PLoS ONE 2020, 15, e0228079. [Google Scholar] [CrossRef]
- Moura, A.; Pereira, C.; Henriques, I.; Correia, A. Novel Gene Cassettes and Integrons in Antibiotic-Resistant Bacteria Isolated from Urban Wastewaters. Res. Microbiol. 2012, 163, 92–100. [Google Scholar] [CrossRef]
- Pallares-Vega, R.; Blaak, H.; van der Plaats, R.; de Roda Husman, A.M.; Hernandez Leal, L.; van Loosdrecht, M.C.M.; Weissbrodt, D.G.; Schmitt, H. Determinants of Presence and Removal of Antibiotic Resistance Genes during WWTP Treatment: A Cross-Sectional Study. Water Res. 2019, 161, 319–328. [Google Scholar] [CrossRef]
- Rafraf, I.D.; Lekunberri, I.; Sànchez-Melsió, A.; Aouni, M.; Borrego, C.M.; Balcázar, J.L. Abundance of Antibiotic Resistance Genes in Five Municipal Wastewater Treatment Plants in the Monastir Governorate, Tunisia. Environ. Pollut. 2016, 219, 353–358. [Google Scholar] [CrossRef]
- Sekizuka, T.; Itokawa, K.; Tanaka, R.; Hashino, M.; Yatsu, K.; Kuroda, M. Metagenomic Analysis of Urban Wastewater Treatment Plant Effluents in Tokyo. Infect. Drug Resist. 2022, 15, 4763–4777. [Google Scholar] [CrossRef]
- Liang, Z.; Yao, J.; Ma, H.; Peng, W.; Xia, X.; Chen, Y. A Sludge Bulking Wastewater Treatment Plant with an Oxidation Ditch-Denitrification Filter in a Cold Region: Bacterial Community Composition and Antibiotic Resistance Genes. Environ. Sci. Pollut. Res. 2023, 30, 33767–33779. [Google Scholar] [CrossRef] [PubMed]
- Donchev, D.; Ivanov, I.N.; Stoikov, I.; Ivanova, M. Metagenomic Investigation of the Short-Term Temporal and Spatial Dynamics of the Bacterial Microbiome and the Resistome Downstream of a Wastewater Treatment Plant in the Iskar River in Bulgaria. Microorganisms 2024, 12, 1250. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Ramudzulu, M.; Munk, P.; Avot, B.J.P.; Esterhuyse, K.C.M.; van Blerk, N.; Kwenda, S.; Sekwadi, P. Metagenomics Analysis of Sewage for Surveillance of Antimicrobial Resistance in South Africa. PLoS ONE 2024, 19, e0309409. [Google Scholar] [CrossRef] [PubMed]
- Agramont, J.; Gutiérrez-Cortez, S.; Joffré, E.; Sjöling, Å.; Toledo, C.C. Fecal Pollution Drives Antibiotic Resistance and Class 1 Integron Abundance in Aquatic Environments of the Bolivian Andes Impacted by Mining and Wastewater. Microorganisms 2020, 8, 1122. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.; Lourenço, A.B.; Monteiro, S.; Santos, R.; Cunha, M.V. Metagenomic Profiling of Raw Wastewater in Portugal Highlights Microbiota and Resistome Signatures of Public Health Interest beyond the Usual Suspects. Sci. Total Environ. 2024, 946, 174272. [Google Scholar] [CrossRef]
- WHO. The 2019 WHO AWaRe Classification of Antibiotics for Evaluation and Monitoring of Use. Available online: https://www.who.int/publications/i/item/WHOEMPIAU2019.11 (accessed on 18 July 2024).
- Xin, R.; Zhang, K.; Wu, N.; Zhang, Y.; Niu, Z. The Pollution Level of the BlaOXA-58 Carbapenemase Gene in Coastal Water and Its Host Bacteria Characteristics. Environ. Pollut. 2019, 244, 66–71. [Google Scholar] [CrossRef]
- Desmet, S.; Nepal, S.; van Dijl, J.M.; Van Ranst, M.; Chlebowicz, M.A.; Rossen, J.W.; Van Houdt, J.K.J.; Maes, P.; Lagrou, K.; Bathoorn, E. Antibiotic Resistance Plasmids Cointegrated into a Megaplasmid Harboring the Bla OXA-427 Carbapenemase Gene. Antimicrob. Agents Chemother. 2018, 62, 1010–1128. [Google Scholar] [CrossRef]
- Begmatov, S.; Beletsky, A.V.; Dorofeev, A.G.; Pimenov, N.V.; Mardanov, A.V.; Ravin, N.V. Metagenomic Insights into the Wastewater Resistome before and after Purification at Large-scale Wastewater Treatment Plants in the Moscow City. Sci. Rep. 2024, 14, 6349. [Google Scholar] [CrossRef]
- Woegerbauer, M.; Zeinzinger, J.; Springer, B.; Hufnagl, P.; Indra, A.; Korschineck, I.; Hofrichter, J.; Kopacka, I.; Fuchs, R.; Steinwider, J.; et al. Prevalence of the Aminoglycoside Phosphotransferase Genes Aph(39)-IIIa and Aph(39)-IIa in Escherichia coli, Enterococcus faecalis, Enterococcus faecium, Pseudomonas aeruginosa, Salmonella enterica Subsp. enterica and Staphylococcus aureus Isolates in Aust. J. Med. Microbiol. 2014, 63, 210–217. [Google Scholar] [CrossRef]
- Salgueiro, V.; Manageiro, V.; Rosado, T.; Bandarra, N.M.; Botelho, M.J.; Dias, E.; Caniça, M. Snapshot of Resistome, Virulome and Mobilome in Aquaculture. Sci. Total Environ. 2023, 905, 166351. [Google Scholar] [CrossRef]
- Zhang, G.; Leclercq, S.O.; Tian, J.; Wang, C.; Yahara, K.; Ai, G.; Liu, S.; Feng, J. A New Subclass of Intrinsic Aminoglycoside Nucleotidyltransferases, ANT(3″)-II, Is Horizontally Transferred among Acinetobacter spp. by Homologous Recombination. PLoS Genet. 2017, 13, e1006602. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Sha, Y.; Chen, Q.; Lin, N.; Zhao, J.; Zhang, Y.; Ji, Y.; Jiang, W.; Zhang, X.; et al. Identification and Characterization of a Novel 6′-N-Aminoglycoside Acetyltransferase AAC(6′)-Va from a Clinical Isolate of Aeromonas hydrophila. Front. Microbiol. 2023, 14, 1229593. [Google Scholar] [CrossRef]
- Asad, A.; Jahan, I.; Munni, M.A.; Begum, R.; Mukta, M.A.; Saif, K.; Faruque, S.N.; Hayat, S.; Islam, Z. Multidrug-Resistant Conjugative Plasmid Carrying MphA Confers Increased Antimicrobial Resistance in Shigella. Sci. Rep. 2024, 14, 6947. [Google Scholar] [CrossRef]
- Chen, Q.; Lu, W.; Zhou, D.; Zheng, G.; Liu, H.; Qian, C.; Zhou, W.; Lu, J.; Ni, L.; Bao, Q.; et al. Characterization of Two Macrolide Resistance-Related Genes in Multidrug-Resistant Pseudomonas aeruginosa Isolates. Pol. J. Microbiol. 2020, 69, 349–356. [Google Scholar] [CrossRef]
- Xiang, Y.; Wu, F.; Chai, Y.; Xu, X.; Yang, L.; Tian, S.; Zhang, H.; Li, Y.; Yang, C.; Liu, H.; et al. A New Plasmid Carrying MphA Causes Prevalence of Azithromycin Resistance in Enterotoxigenic Escherichia coli Serogroup O6. BMC Microbiol. 2020, 20, 247. [Google Scholar] [CrossRef]
- Marutescu, L.G.; Popa, M.; Gheorghe-Barbu, I.; Barbu, I.C.; Rodríguez-Molina, D.; Berglund, F.; Blaak, H.; Flach, C.F.; Kemper, M.A.; Spießberger, B.; et al. Wastewater Treatment Plants, an “Escape Gate” for ESCAPE Pathogens. Front. Microbiol. 2023, 14, 1193907. [Google Scholar] [CrossRef]
- Ovejero, C.M.; Delgado-Blas, J.F.; Calero-Caceres, W.; Muniesa, M.; Gonzalez-Zorn, B. Spread of Mcr-1-Carrying Enterobacteriaceae in Sewage Water from Spain. J. Antimicrob. Chemother. 2017, 72, 1050–1053. [Google Scholar] [CrossRef]
- Bréchet, C.; Plantin, J.; Sauget, M.; Thouverez, M.; Talon, D.; Cholley, P.; Guyeux, C.; Hocquet, D.; Bertrand, X. Wastewater Treatment Plants Release Large Amounts of Extended-Spectrum β-Lactamase-Producing Escherichia coli into the Environment. Clin. Infect. Dis. 2014, 58, 1658–1665. [Google Scholar] [CrossRef]
- Hembach, N.; Schmid, F.; Alexander, J.; Hiller, C.; Rogall, E.T.; Schwartz, T. Occurrence of the mcr-1 Colistin Resistance Gene and Other Clinically Relevant Antibiotic Resistance Genes in Microbial Populations at Different Municipal Wastewater Treatment Plants in Germany. Front. Microbiol. 2017, 8, 267477. [Google Scholar] [CrossRef]
- Chukamnerd, A.; Pomwised, R.; Jeenkeawpiam, K.; Sakunrang, C.; Chusri, S.; Surachat, K. Genomic Insights into BlaNDM-Carrying Carbapenem-Resistant Klebsiella pneumoniae Clinical Isolates from a University Hospital in Thailand. Microbiol. Res. 2022, 263, 127136. [Google Scholar] [CrossRef]
- Bönemann, G.; Stiens, M.; Pühler, A.; Schlüter, A. Mobilizable IncQ-Related Plasmid Carrying a New Quinolone Resistance Gene, QnrS2, Isolated from the Bacterial Community of a Wastewater Treatment Plant. Antimicrob. Agents Chemother. 2006, 50, 3075–3080. [Google Scholar] [CrossRef]
- Loftie-Eaton, W.; Rawlings, D.E. Diversity, Biology and Evolution of IncQ-Family Plasmids. Plasmid 2012, 67, 15–34. [Google Scholar] [CrossRef]
- Piotrowska, M.; Dziewit, L.; Ostrowski, R.; Chmielowska, C.; Popowska, M. Molecular Characterization and Comparative Genomics of Incq-3 Plasmids Conferring Resistance to Various Antibiotics Isolated from a Wastewater Treatment Plant in Warsaw (Poland). Antibiotics 2020, 9, 613. [Google Scholar] [CrossRef]
- Galarde-López, M.; Velazquez-Meza, M.E.; Bobadilla-del-Valle, M.; Carrillo-Quiroz, B.A.; Cornejo-Juárez, P.; Ponce-de-León, A.; Sassoé-González, A.; Alpuche-Aranda, C.M. Surveillance of Antimicrobial Resistance in Hospital Wastewater: Identification of Carbapenemase-Producing Klebsiella spp. Antibiotics 2022, 11, 288. [Google Scholar] [CrossRef]
- Galarde-López, M.; Velazquez-Meza, M.E.; Bobadilla-del-Valle, M.; Cornejo-Juárez, P.; Carrillo-Quiroz, B.A.; Ponce-de-León, A.; Sassoé-González, A.; Saturno-Hernández, P.; Alpuche-Aranda, C.M. Antimicrobial Resistance Patterns and Clonal Distribution of E. coli, Enterobacter spp. and Acinetobacter spp. Strains Isolated from Two Hospital Wastewater Plants. Antibiotics 2022, 11, 601. [Google Scholar] [CrossRef]
- Chung, M.; De Lencastre, H.; Matthews, P.; Tomasz, A.; Adamsson, I.; Aires de Sousa, M.; Camou, T.; Cocuzza, C.; Corso, A.; Couto, I.; et al. Molecular Typing of Methicillin-Resistant Staphylococcus aureus by Pulsed-Field Gel Electrophoresis: Comparison of Results Obtained in a Multilaboratory Effort Using Identical Protocols and MRSA Strains. Microb. Drug Resist. 2009, 6, 189–198. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics-FastQC a Quality Control Tool for High Throughput Sequence Data 2010. 2023. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 July 2024).
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA—A practical iterative de bruijn graph de novo assembler. In Proceedings of the Research in Computational Molecular Biology: 14th Annual International Conference, RECOMB 2010, Lisbon, Portugal, 25–28 April 2010; Volume 6044 LNBI, pp. 426–440. [Google Scholar]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial Strain-Level Population Structure and Genetic Diversity from Metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome Analysis Using the Kraken Software Suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Oksanen, A.J.; Blanchet, F.G.; Kindt, R.; Legen, P.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H. Community Ecology Package. r-project.org 2022. Available online: https://github.com/vegandevs/vegan (accessed on 18 July 2024).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N.; Sommerfield, P.J.; Warwick, R.M. Change in Marine Communities—Statistical Analysis, 3rd ed.; Plymouth Marine Laboratory: Plymouth, UK, 2014. [Google Scholar]
- Valero-Mora, P.M. Ggplot2: Elegant Graphics for Data Analysis. J. Stat. Softw. 2010, 35, 180–185. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015; Available online: https://www.rstudio.com/ (accessed on 18 July 2024).
- Seemann, T. Abricate. Github. 2023. Available online: https://github.com/tseemann/abricate (accessed on 18 July 2024).
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In Silico Serotyping of E. coli from Short Read Data Identifies Limited Novel o-Loci but Extensive Diversity of O:H Serotype Combinations within and between Pathogenic Lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velazquez-Meza, M.E.; Galarde-López, M.; Cornejo-Juárez, P.; Bobadilla-del-Valle, M.; Godoy-Lozano, E.; Aguilar-Vera, E.; Carrillo-Quiroz, B.A.; Ponce de León-Garduño, A.; Velazquez Acosta, C.; Alpuche-Aranda, C.M. Bacterial Communities and Resistance and Virulence Genes in Hospital and Community Wastewater: Metagenomic Analysis. Int. J. Mol. Sci. 2025, 26, 2051. https://doi.org/10.3390/ijms26052051
Velazquez-Meza ME, Galarde-López M, Cornejo-Juárez P, Bobadilla-del-Valle M, Godoy-Lozano E, Aguilar-Vera E, Carrillo-Quiroz BA, Ponce de León-Garduño A, Velazquez Acosta C, Alpuche-Aranda CM. Bacterial Communities and Resistance and Virulence Genes in Hospital and Community Wastewater: Metagenomic Analysis. International Journal of Molecular Sciences. 2025; 26(5):2051. https://doi.org/10.3390/ijms26052051
Chicago/Turabian StyleVelazquez-Meza, Maria Elena, Miguel Galarde-López, Patricia Cornejo-Juárez, Miriam Bobadilla-del-Valle, Ernestina Godoy-Lozano, Edgar Aguilar-Vera, Berta Alicia Carrillo-Quiroz, Alfredo Ponce de León-Garduño, Consuelo Velazquez Acosta, and Celia Mercedes Alpuche-Aranda. 2025. "Bacterial Communities and Resistance and Virulence Genes in Hospital and Community Wastewater: Metagenomic Analysis" International Journal of Molecular Sciences 26, no. 5: 2051. https://doi.org/10.3390/ijms26052051
APA StyleVelazquez-Meza, M. E., Galarde-López, M., Cornejo-Juárez, P., Bobadilla-del-Valle, M., Godoy-Lozano, E., Aguilar-Vera, E., Carrillo-Quiroz, B. A., Ponce de León-Garduño, A., Velazquez Acosta, C., & Alpuche-Aranda, C. M. (2025). Bacterial Communities and Resistance and Virulence Genes in Hospital and Community Wastewater: Metagenomic Analysis. International Journal of Molecular Sciences, 26(5), 2051. https://doi.org/10.3390/ijms26052051