
Unusual Partners: γδ-TCR-Based T Cell Therapy in Combination with Oncolytic Virus Treatment for Diffuse Midline Gliomas

 ,
,  , ,
, ,  , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
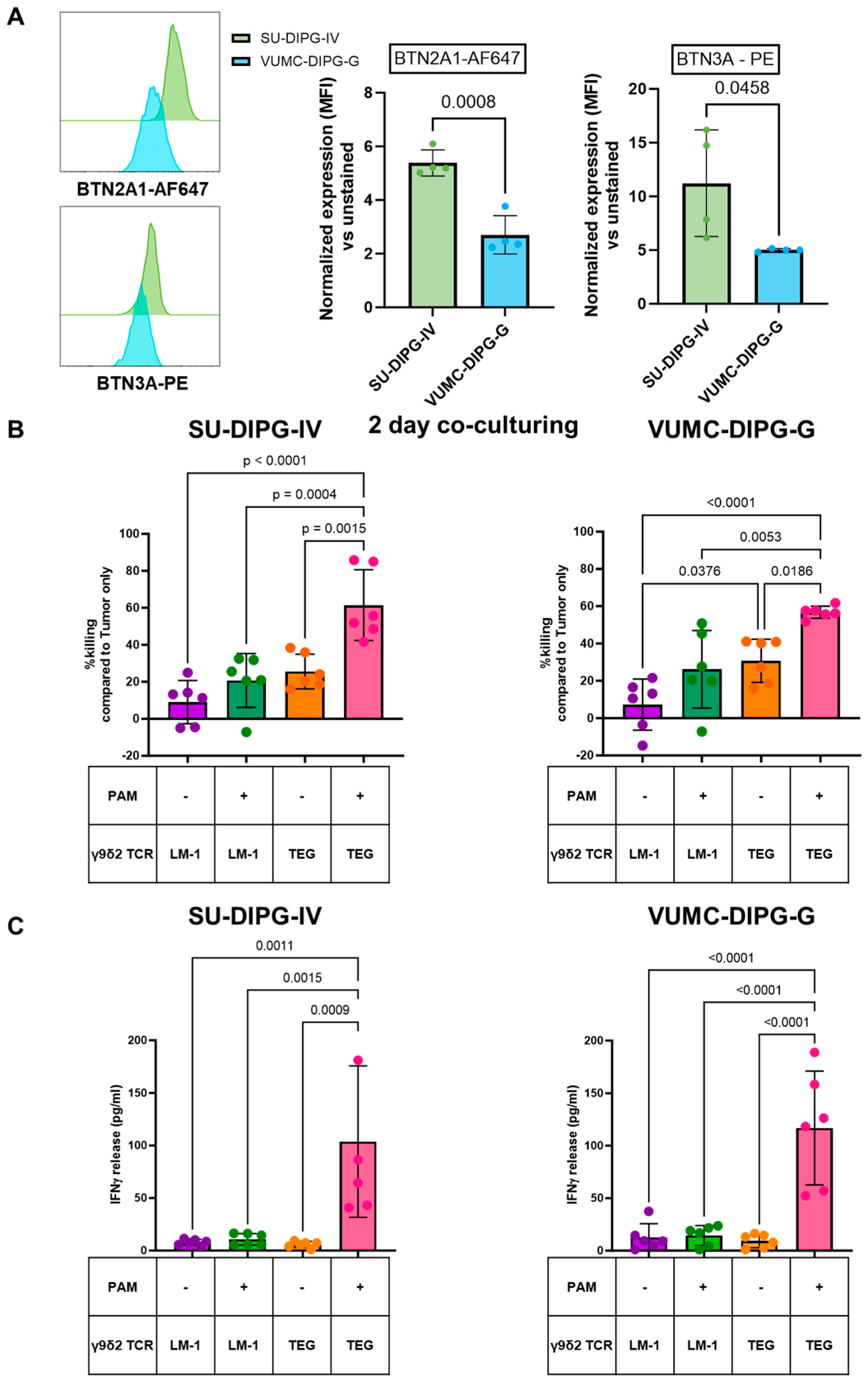
2.1. TEGs Recognize Pediatric DMGs Through Their γ9δ2TCR
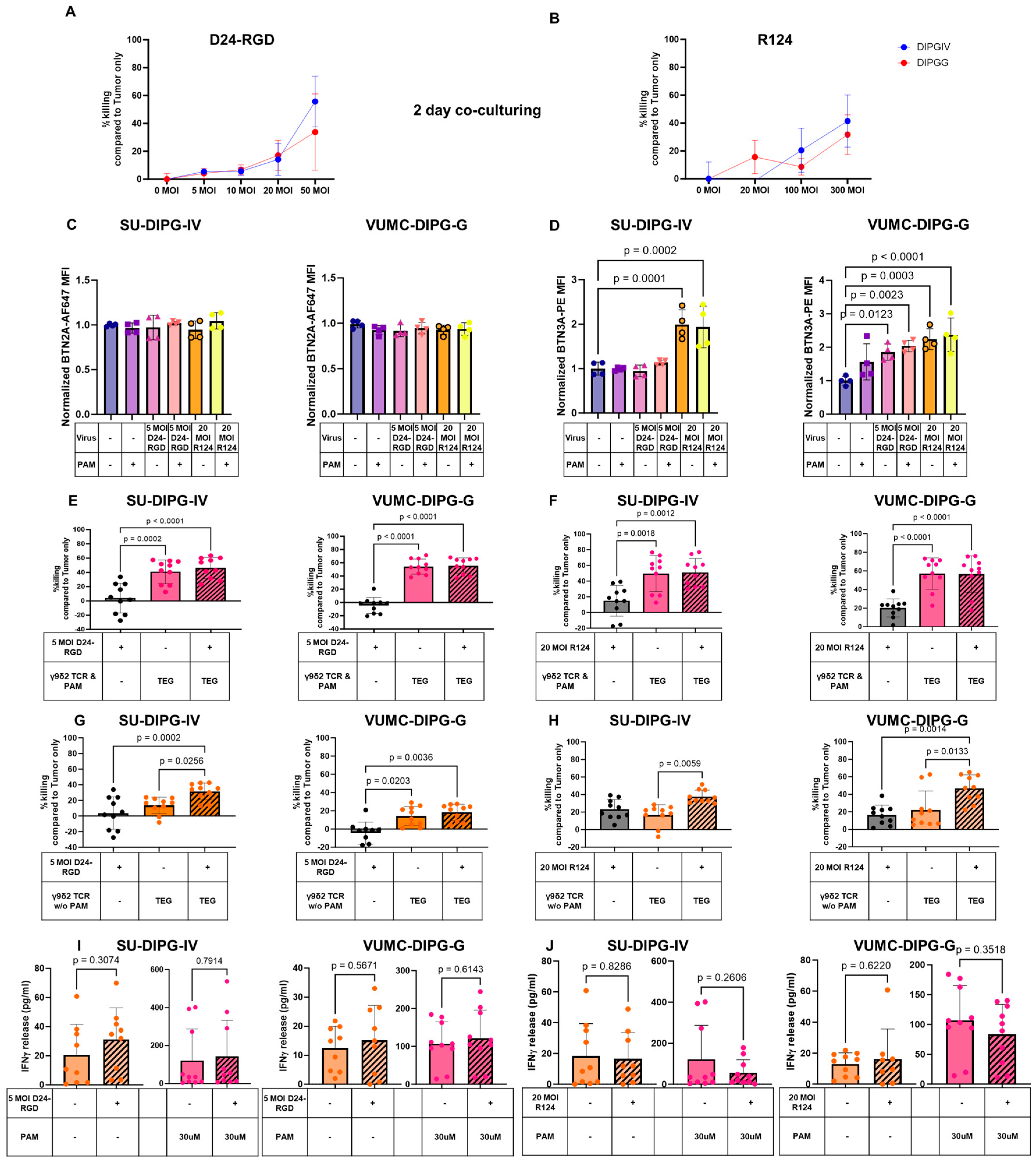
2.2. OVs Kill DMGs and Enhance BTN3A but Not BTN2A1 Expression
2.3. Additive Effect of the Combination of OVs and TEGs to Enhanced DMG Killing
3. Discussion
4. Materials and Methods
4.1. DMG Cell Cultures
4.2. T Cell Transduction to Express a γδ TCR (TEGs and LM1s)
4.3. Co-Culture Functional Assays
4.4. IFN-γ Release Assay
4.5. BTN2A1 and BTN3A Staining
4.6. Plots and Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Pachocki, C.J.; Hol, E.M. Current perspectives on diffuse midline glioma and a different role for the immune microenvironment compared to glioblastoma. J. Neuroinflammation 2022, 19, 276. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.C.; Staudt, D.E.; Douglas, A.M.; Monje, M.; Vitanza, N.A.; Dun, M.D. CAR T cell therapies for diffuse midline glioma. Trends Cancer 2023, 9, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, K.A.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef]
- Yuan, L.; Ma, X.; Yang, Y.; Qu, Y.; Li, X.; Zhu, X.; Ma, W.; Duan, J.; Xue, J.; Yang, H.; et al. Phosphoantigens glue butyrophilin 3A1 and 2A1 to activate Vγ9Vδ2 T cells. Nature 2023, 621, 840–848. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Scheper, W.; Vyborova, A.; Gu, S.; Rychnavska, Z.; Schiffler, M.; Cleven, A.; Chéneau, C.; van Noorden, M.; Peigné, M.-C.; et al. RhoB Mediates Phosphoantigen Recognition by Vgamma9Vdelta2 T Cell Receptor. Cell Rep. 2016, 15, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Vyborova, A.; Janssen, A.; Gatti, L.; Karaiskaki, F.; Yonika, A.; van Dooremalen, S.; Sanders, J.; Beringer, X.D.; Straetemans, T.; Sebestyen, Z.; et al. γ9δ2 T-Cell Expansion and Phenotypic Profile Are Reflected in the CDR3δ Repertoire of Healthy Adults. Front. Immunol. 2022, 13, 915366. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, M.R.; Vedova, S.; Freimer, J.W.; Sahu, A.D.; Ramesh, A.; Arce, M.M.; Meringa, D.A.; Ota, M.; Chen, A.P.; Hanspers, K.; et al. CRISPR screens decode cancer cell pathways that trigger gammadelta T cell detection. Nature 2023, 621, 188–195. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef]
- Hernández-López, P.; van Diest, E.; Brazda, P.; Heijhuurs, S.; Meringa, A.; van Heyningen, L.H.; Riillo, C.; Schwenzel, C.; Zintchenko, M.; Johanna, I.; et al. Dual targeting of cancer metabolome and stress antigens affects transcriptomic heterogeneity and efficacy of engineered T cells. Nat. Immunol. 2024, 25, 88–101. [Google Scholar] [CrossRef]
- de Witte, M.; Scheepstra, J.; Weertman, N.; Daudeij, A.; van der Wagen, L.; Oostvogels, R.; de Haar, C.; Prins, J.-H.; Dohmen, W.; Bertels-Wilmer, C.; et al. First in Human Clinical Responses and Persistence Data on TEG001: A Next Generation of Engineered Aβ T Cells Targeting AML and MM with a High Affinity γ9δ2TCR. Blood 2022, 140 (Suppl. S1), 12737–12739. [Google Scholar] [CrossRef]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.-T.; Chan, G.C.-F.; Yang, Y.; et al. Targeted Activation of Human Vγ9Vδ2-T Cells Controls Epstein-Barr Virus-Induced B Cell Lymphoproliferative Disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef]
- Janssen, A.; an Diest, E.; Vyborova, A.; Schrier, L.; Bruns, A.; Sebestyen, Z.; Straetemans, T.; de Witte, M.; Kuball, J. The Role of gammadelta T Cells as a Line of Defense in Viral Infections after Allogeneic Stem Cell Transplantation: Opportunities and Challenges. Viruses 2022, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, X.; Jin, Z.; Xu, L.; Chen, S.; Yang, L.; Grabarczyk, P.; Przybylski, K.G.; Schmidt, C.; Li, Y. Specific Gamma Delta T Cells for Cellular Immunotherapy of EBV-Associated Diseases after Allo-HSCT By T-Cell Re-ceptor Gene Modification. Blood 2014, 124, 5811. [Google Scholar] [CrossRef]
- Pérez-Larraya, J.G.; Garcia-Moure, M.; Labiano, S.; Patiño-García, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. New Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Vazaios, K.; van Berkum, R.E.; Calkoen, F.G.; van der Lugt, J.; Hulleman, E. OV Modulators of the Paediatric Brain TIME: Current Status, Combination Strategies, Limitations and Future Directions. Int. J. Mol. Sci. 2024, 25, 5007. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef]
- Garcia-Moure, M.; Gonzalez-Huarriz, M.; Labiano, S.; Guruceaga, E.; Bandres, E.; Zalacain, M.; Marrodan, L.; de Andrea, C.; Villalba, M.; Martinez-Velez, N.; et al. Delta-24-RGD, an Oncolytic Adenovirus, Increases Survival and Promotes Proinflammatory Immune Landscape Remodeling in Models of AT/RT and CNS-PNET. Clin. Cancer Res. 2021, 27, 1807–1820. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar]
- Shmulevitz, M.; Marcato, P.; Lee, P.W.K. Activated Ras signaling significantly enhances reovirus replication and spread. Cancer Gene Ther. 2010, 17, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Van den Hengel, S.K.; Balvers, R.K.; Dautzenberg, I.J.C.; van den Wollenberg, D.J.M.; Kloezeman, J.J.; Lamfers, M.L.; Sillivis-Smit, P.A.E.; Hoeben, R.C. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer Gene Ther. 2013, 20, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Vazaios, K.; Stavrakaki, Ε.; Vogelezang, L.B.; Ju, J.; Waranecki, P.; Metselaar, D.S.; Meel, M.H.; Kemp, V.; Hoogen, B.G.v.D.; Hoeben, R.C.; et al. The heterogeneous sensitivity of pediatric brain tumors to different oncolytic viruses is predicted by unique gene expression profiles. Mol. Ther. Oncol. 2024, 32, 200804. [Google Scholar] [CrossRef]
- Wang, S.S.; Bandopadhayay, P.; Jenkins, M.R. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol. 2019, 40, 748–761. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Cleven, A.; Meringa, A.D.; Brazda, P.; Fasci, D.; Koorman, T.; Aarts, T.; Johanna, I.; Beringer, X.D.; Hernandez-Lopez, P.; Heijhuurs, S.; et al. Sensitivity to Vγ9Vδ2TCR T Cells is Imprinted After Single Mutations During Early Oncogenesis. bioRxiv 2024. bioRxiv:2024.11.19.624272. [Google Scholar]
- Cimini, E.; Agrati, C. γδ T Cells in Emerging Viral Infection: An Overview. Viruses 2022, 14, 1166. [Google Scholar] [CrossRef] [PubMed]
- Dybas, J.M.; Lum, K.K.; Kulej, K.; Reyes, E.D.; Lauman, R.; Charman, M.; Purman, C.E.; Steinbock, R.T.; Grams, N.; Price, A.M.; et al. Adenovirus Remodeling of the Host Proteome and Host Factors Associated with Viral Genomes. mSystems 2021, 6, e0046821. [Google Scholar] [CrossRef]
- González-Morales, A.; Zabaleta, A.; Guruceaga, E.; Alonso, M.M.; García-Moure, M.; Fernández-Irigoyen, J.; Santamaría, E. Spatial and temporal proteome dynamics of glioma cells during oncolytic adenovirus Delta-24-RGD infection. Oncotarget 2018, 9, 31045–31065. [Google Scholar] [CrossRef]
- Wada, A.; Fukui, K.; Sawai, Y.; Imanaka, K.; Kiso, S.; Tamura, S.; Shimomura, I.; Hayashi, N. Pamidronate induced anti-proliferative, apoptotic, and anti-migratory effects in hepatocellular carcinoma. J. Hepatol. 2006, 44, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Hirasawa, K.; Yang, A.-D.; Shields, M.A.; Lee, P.W.K. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wu, W.-L.; Liu, Y.; Xiang, Z.; Liu, M.; Chan, K.-H.; Lau, S.-Y.; Lam, K.-T.; To, K.K.-W.; Chan, J.F.-W.; et al. The Therapeutic Effect of Pamidronate on Lethal Avian Influenza A H7N9 Virus Infected Humanized Mice. PLoS ONE 2015, 10, e0135999. [Google Scholar] [CrossRef]
- Acott, P.D.; Wong, J.A.; Crocker, J.F.S.; Lang, B.; O’regan, P.; Renton, K.W. Pamidronate distribution in pediatric renal and rheumatologic patients. Eur. J. Clin. Pharmacol. 2006, 62, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hoggarth, C.R.; Bennett, R.; Daley-Yates, P.T. The pharmacokinetics and distribution of pamidronate for a range of doses in the mouse. Calcif. Tissue Int. 1991, 49, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Volovat, S.R.; Scripcariu, D.V.; Vasilache, I.A.; Stolniceanu, C.R.; Volovat, C.; Augustin, I.G.; Volovat, C.C.; Ostafe, M.-R.; Andreea-Voichița, S.-G.; Bejusca-Vieriu, T.; et al. Oncolytic Virotherapy: A New Paradigm in Cancer Immunotherapy. Int. J. Mol. Sci. 2024, 25, 1180. [Google Scholar] [CrossRef]
- Bhatt, D.K.; Chammas, R.; Daemen, T. Resistance Mechanisms Influencing Oncolytic Virotherapy, a Systematic Analysis. Vaccines 2021, 9, 1166. [Google Scholar] [CrossRef] [PubMed]
- Stavrakaki, E.; Bossche, W.B.v.D.; Vogelezang, L.B.; Teodosio, C.; Mustafa, D.M.; van Dongen, J.J.; Dirven, C.M.; Balvers, R.K.; Lamfers, M.L. An autologous ex vivo model for exploring patient-specific responses to viro-immunotherapy in glioblastoma. Cell Rep. Methods 2024, 4, 100716. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Xella, A.; Woodall, R.T.; Cassady, K.A.; Branciamore, S.; Brown, E.C.; Rockne, C.R. CAR T-Cell and Oncolytic VIRUS Dynamics and Determinants of Combination Therapy Success for Glioblastoma. bioRxiv 2025. [Google Scholar] [CrossRef]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The Efficacy of Oncolytic Adenovirus Is Mediated by T-cell Responses against Virus and Tumor in Syrian Hamster Model. Clin. Cancer Res. 2017, 23, 239–249. [Google Scholar] [CrossRef]
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.X.; Daniel, P.; Bradshaw, G.; Shi, H.; Loi, M.; Chew, N.; Parackal, S.; Tsui, V.; Liang, Y.; Koptyra, M.; et al. Generation and multi-dimensional profiling of a childhood cancer cell line atlas defines new therapeutic opportunities. Cancer Cell 2023, 41, 660–677.e7. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Vyborova, A.; Beringer, D.X.; Fasci, D.; Karaiskaki, F.; van Diest, E.; Kramer, L.; de Haas, A.; Sanders, J.; Janssen, A.; Straetemans, T.; et al. γ9δ2T cell diversity and the receptor interface with tumor cells. J. Clin. Investig. 2020, 130, 4637–4651. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vazaios, K.; Hernández López, P.; Aarts-Riemens, T.; Daudeij, A.; Kemp, V.; Hoeben, R.C.; Straetemans, T.; Hulleman, E.; Calkoen, F.G.; van der Lugt, J.; et al. Unusual Partners: γδ-TCR-Based T Cell Therapy in Combination with Oncolytic Virus Treatment for Diffuse Midline Gliomas. Int. J. Mol. Sci. 2025, 26, 2167. https://doi.org/10.3390/ijms26052167
Vazaios K, Hernández López P, Aarts-Riemens T, Daudeij A, Kemp V, Hoeben RC, Straetemans T, Hulleman E, Calkoen FG, van der Lugt J, et al. Unusual Partners: γδ-TCR-Based T Cell Therapy in Combination with Oncolytic Virus Treatment for Diffuse Midline Gliomas. International Journal of Molecular Sciences. 2025; 26(5):2167. https://doi.org/10.3390/ijms26052167
Chicago/Turabian StyleVazaios, Konstantinos, Patricia Hernández López, Tineke Aarts-Riemens, Annet Daudeij, Vera Kemp, Rob C. Hoeben, Trudy Straetemans, Esther Hulleman, Friso G. Calkoen, Jasper van der Lugt, and et al. 2025. "Unusual Partners: γδ-TCR-Based T Cell Therapy in Combination with Oncolytic Virus Treatment for Diffuse Midline Gliomas" International Journal of Molecular Sciences 26, no. 5: 2167. https://doi.org/10.3390/ijms26052167
APA StyleVazaios, K., Hernández López, P., Aarts-Riemens, T., Daudeij, A., Kemp, V., Hoeben, R. C., Straetemans, T., Hulleman, E., Calkoen, F. G., van der Lugt, J., & Kuball, J. (2025). Unusual Partners: γδ-TCR-Based T Cell Therapy in Combination with Oncolytic Virus Treatment for Diffuse Midline Gliomas. International Journal of Molecular Sciences, 26(5), 2167. https://doi.org/10.3390/ijms26052167