
Spontaneous Calcium Bursts Organize the Apical Actin Cytoskeleton of Multiciliated Cells


Abstract
:1. Introduction
2. Results
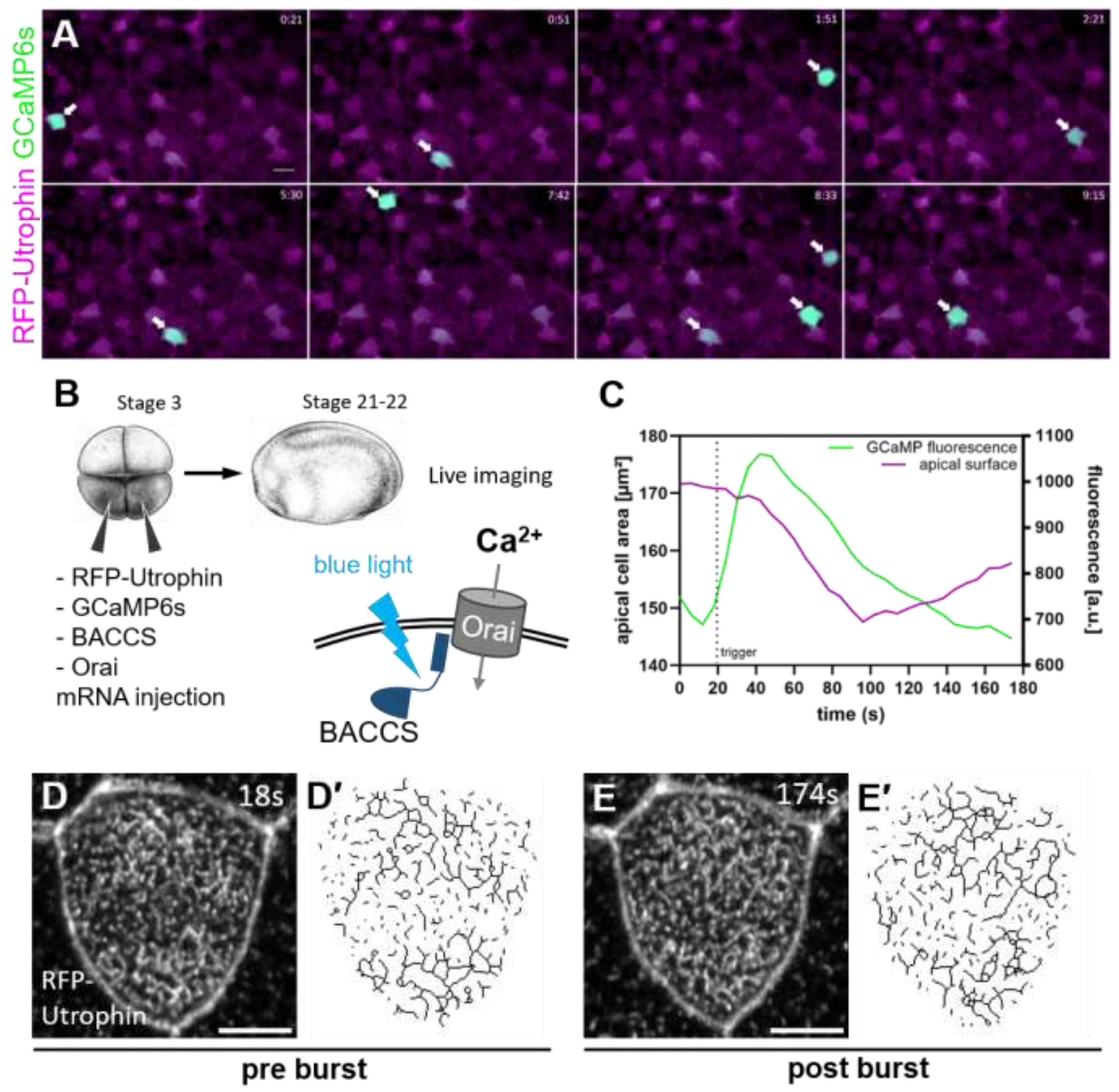
2.1. Calcium Activity in Multiciliated CELLS Drives Apical Actin Network Development
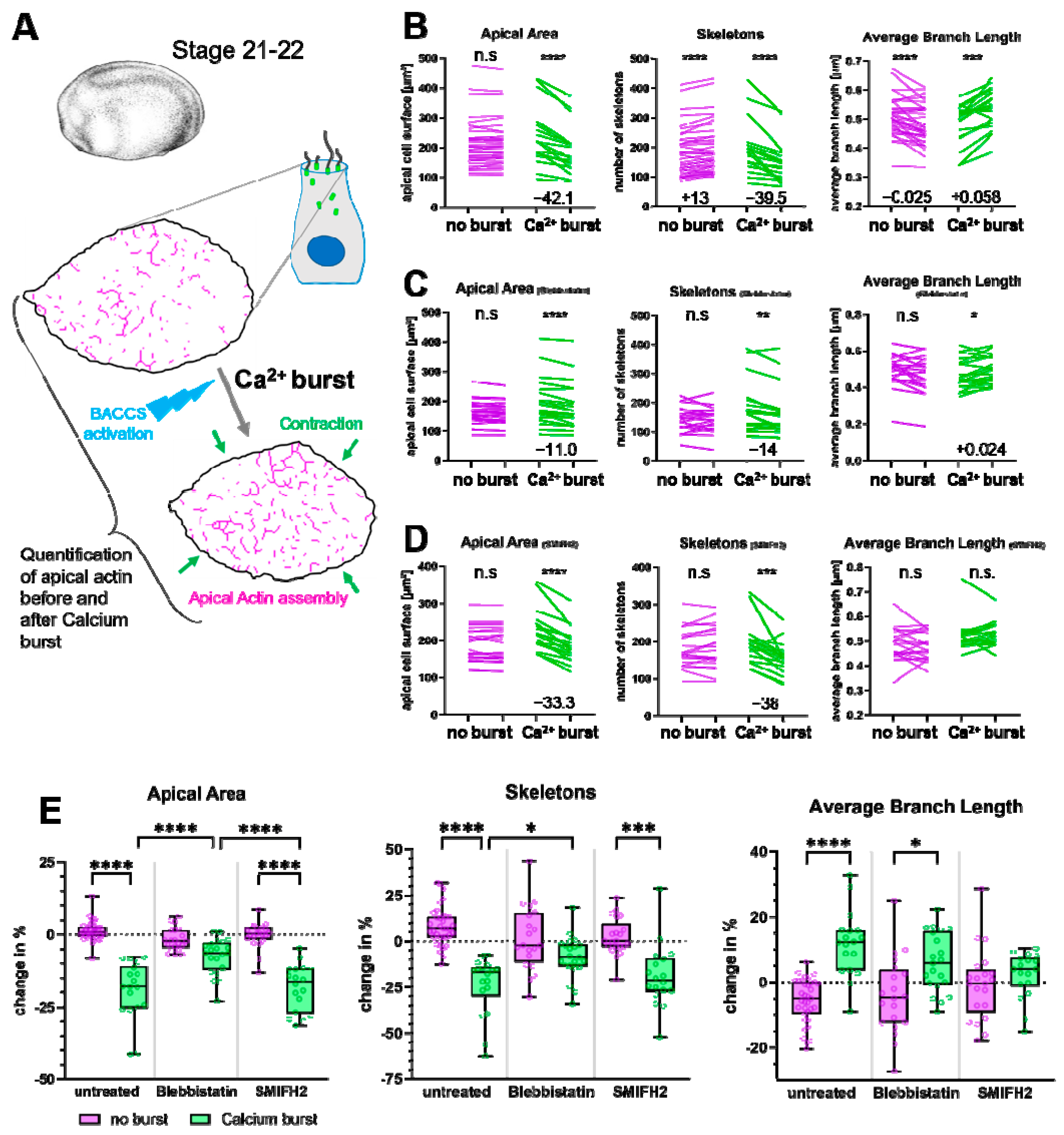
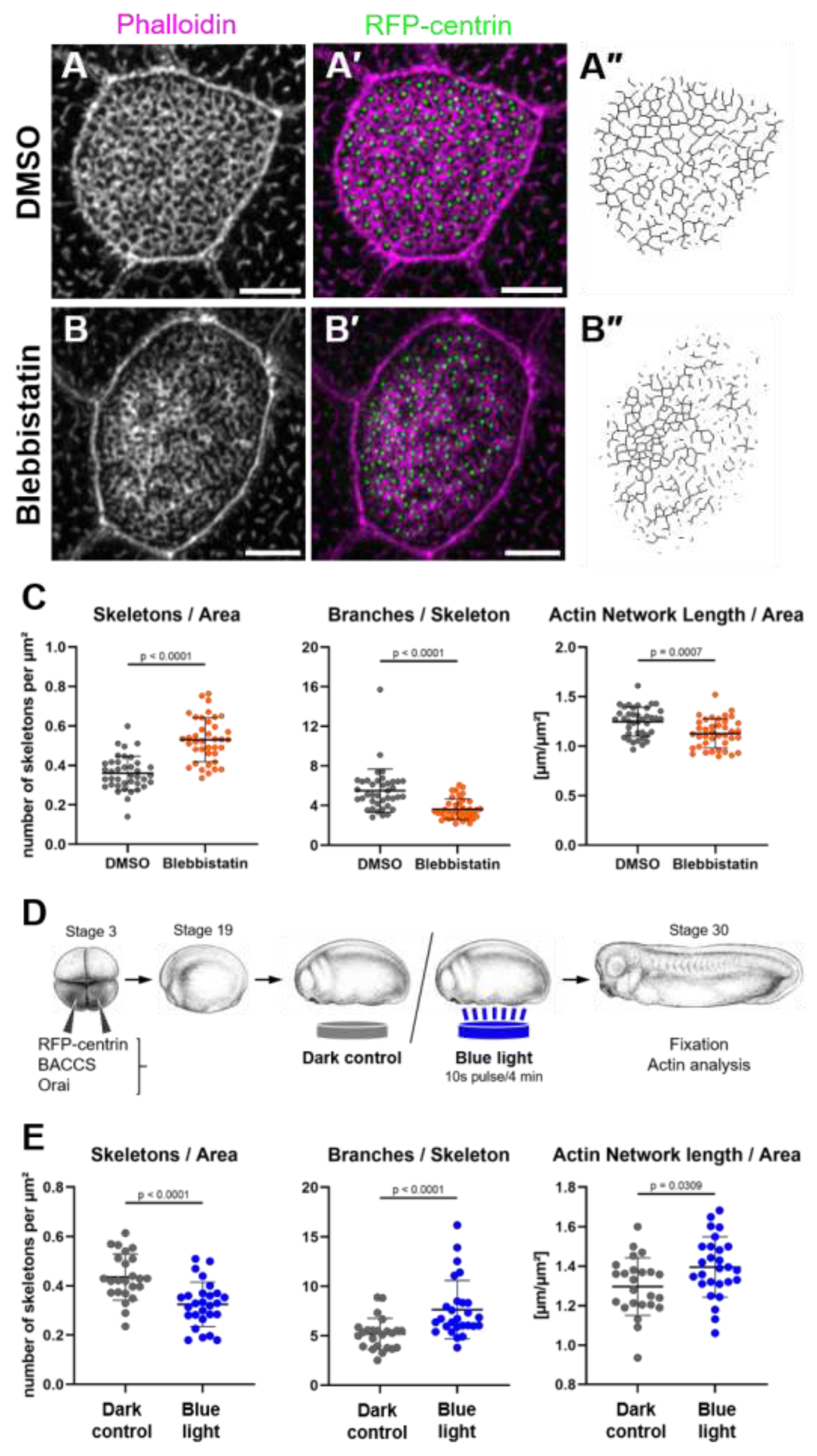
2.2. Non-Muscle Myosin II Promotes Coherence of the Apical Actin Network
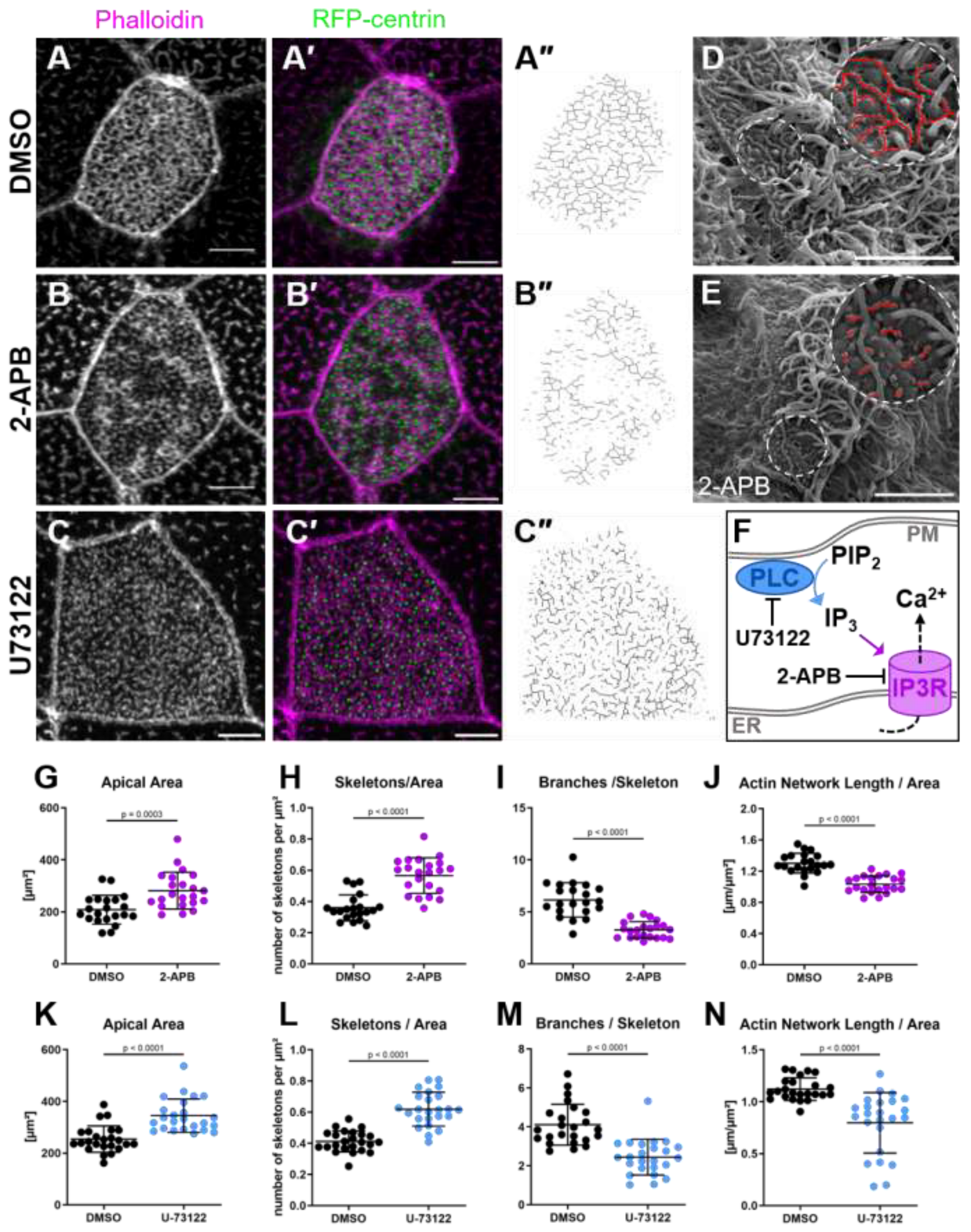
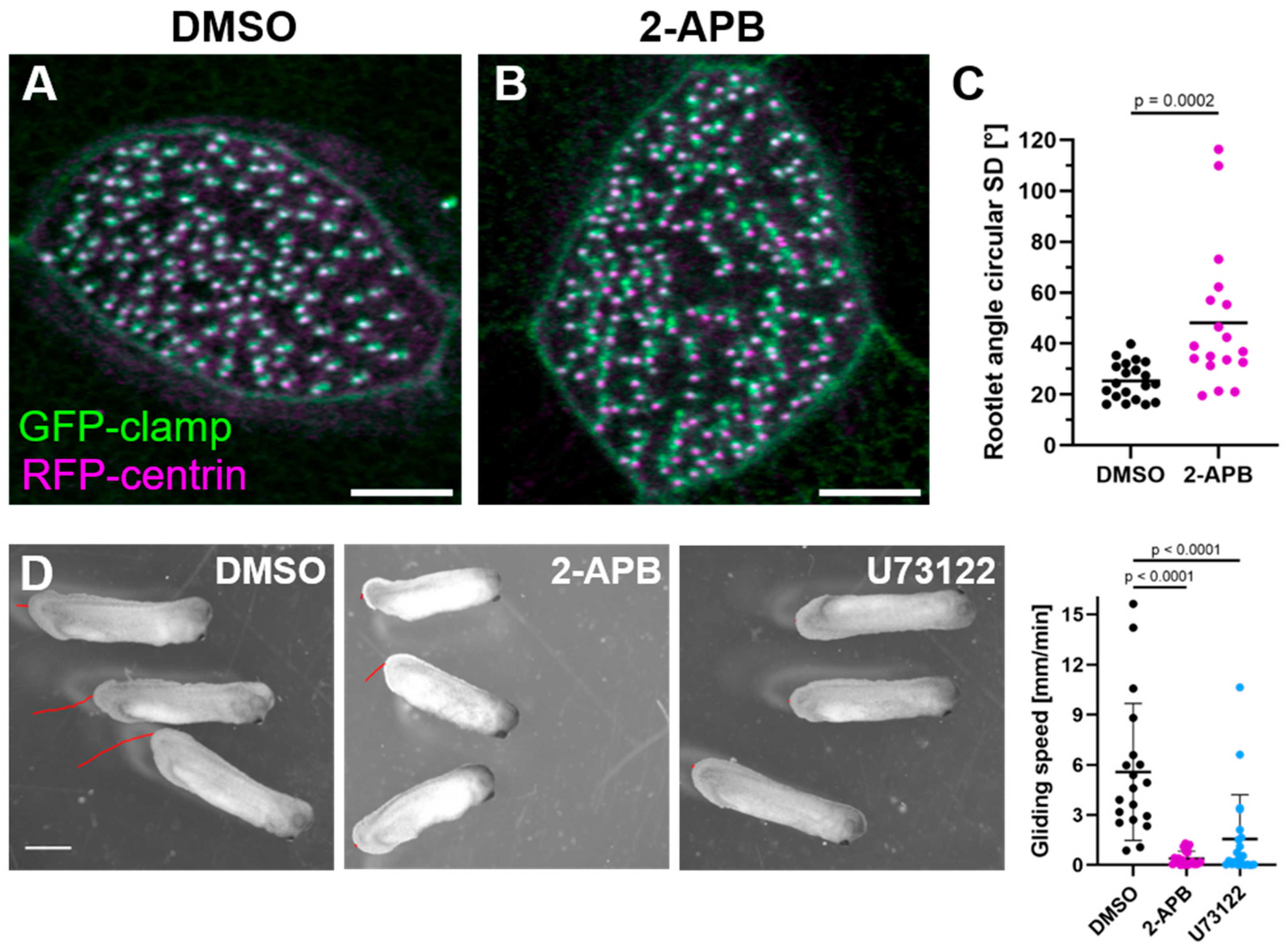
2.3. Inositol Trisphosphate/Calcium Signaling Is Required for Apical Actin Integrity
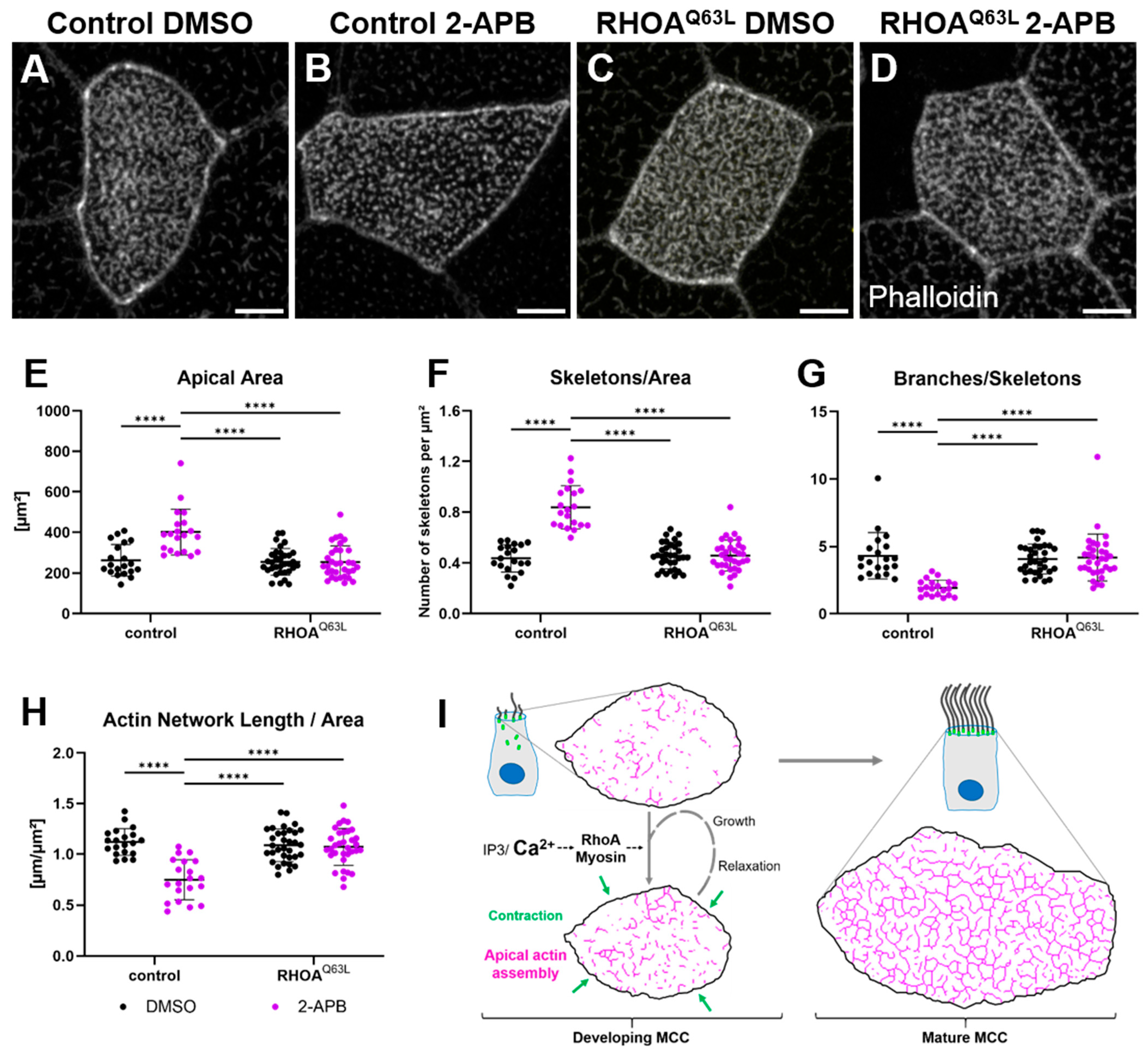
2.4. RhoA Acts Downstream of Calcium Controlling Apical Actin Development
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MCC | Multiciliated cell |
| Rho GTPase | Rho guanosine trisphosphase |
| PCP | Planar cell polarity |
| NMII | Non-muscle myosin II |
| MLCK | Myosin light chain kinase |
| BACCS | Blue light-activated calcium channel switch |
| STIM1 | Stromal interaction molecule 1 |
| SMIFH2 | Small molecule inhibitor of formin homology 2 domain |
| ER | Endoplasmic reticulum |
| IP3 | Inositol trisphosphate |
| 2-APB | 2-aminoetoxydiphenyl borate |
| SOCE | Store-operated calcium entry |
| IP3R | Inositol trisphosphate receptor |
| PLC | Phospholipase C |
| EMTB | Ensconsin microtubule-binding domain |
| rGBD | Rhotekin GTPase-binding domain |
| ROCK | Rho-associated coiled-coil-containing protein kinase |
| CRAC | Calcium release-activated calcium channel |
| ERM | Ezrin Radixin Moesin proteins |
| MMR | Marc’s modified Ringer’s medium |
| DMSO | Dimethyl sulfoxide |
| MOPS | 3-(N-morpholino)propanesulfonic acid |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid |
| PBST | Phosphate-buffered saline with 0.1% Tween-20 |
| FBS | Fetal bovine serum |
References
- Brooks, E.R.; Wallingford, J.B. Multiciliated Cells. Curr. Biol. 2014, 24, R973–R982. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Ventrella, R.; Mitchell, B.J. Chapter One—Building a Ciliated Epithelium: Transcriptional Regulation and Radial Intercalation of Multiciliated Cells. In Current Topics in Developmental Biology; Sokol, S.Y., Ed.; Amphibian Models of Development and Disease; Academic Press: New York, NY, USA, 2021; Volume 145, pp. 3–39. [Google Scholar]
- Mitchison, H.M.; Valente, E.M. Motile and Non-Motile Cilia in Human Pathology: From Function to Phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Walentek, P. Xenopus Epidermal and Endodermal Epithelia as Models for Mucociliary Epithelial Evolution, Disease, and Metaplasia. Genesis 2021, 59, e23406. [Google Scholar] [CrossRef]
- Klos Dehring, D.A.; Vladar, E.K.; Werner, M.E.; Mitchell, J.W.; Hwang, P.; Mitchell, B.J. Deuterosome-Mediated Centriole Biogenesis. Dev. Cell 2013, 27, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, I.; Stylianou, P.; Skourides, P.A. Making the Connection: Ciliary Adhesion Complexes Anchor Basal Bodies to the Actin Cytoskeleton. Dev. Cell 2014, 28, 70–80. [Google Scholar] [CrossRef]
- Epting, D.; Slanchev, K.; Boehlke, C.; Hoff, S.; Loges, N.T.; Yasunaga, T.; Indorf, L.; Nestel, S.; Lienkamp, S.S.; Omran, H.; et al. The Rac1 Regulator ELMO Controls Basal Body Migration and Docking in Multiciliated Cells through Interaction with Ezrin. Development 2015, 142, 174–184. [Google Scholar] [CrossRef]
- Pan, J.; You, Y.; Huang, T.; Brody, S.L. RhoA-Mediated Apical Actin Enrichment Is Required for Ciliogenesis and Promoted by Foxj1. J. Cell Sci. 2007, 120, 1868–1876. [Google Scholar] [CrossRef]
- Sedzinski, J.; Hannezo, E.; Tu, F.; Biro, M.; Wallingford, J.B. Emergence of an Apical Epithelial Cell Surface In Vivo. Dev. Cell 2016, 36, 24–35. [Google Scholar] [CrossRef]
- Sedzinski, J.; Hannezo, E.; Tu, F.; Biro, M.; Wallingford, J.B. RhoA Regulates Actin Network Dynamics during Apical Surface Emergence in Multiciliated Epithelial Cells. J. Cell Sci. 2017, 130, 420–428. [Google Scholar] [CrossRef]
- Lee, M.; Hwang, Y.-S.; Yoon, J.; Sun, J.; Harned, A.; Nagashima, K.; Daar, I.O. Developmentally Regulated GTP-Binding Protein 1 Modulates Ciliogenesis via an Interaction with Dishevelled. J. Cell Biol. 2019, 218, 2659–2676. [Google Scholar] [CrossRef]
- Park, T.J.; Mitchell, B.J.; Abitua, P.B.; Kintner, C.; Wallingford, J.B. Dishevelled Controls Apical Docking and Planar Polarization of Basal Bodies in Ciliated Epithelial Cells. Nat. Genet. 2008, 40, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, T.; Hoff, S.; Schell, C.; Helmstädter, M.; Kretz, O.; Kuechlin, S.; Yakulov, T.A.; Engel, C.; Müller, B.; Bensch, R.; et al. The Polarity Protein Inturned Links NPHP4 to Daam1 to Control the Subapical Actin Network in Multiciliated Cells. J. Cell Biol. 2015, 211, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, J.; Insinna, C.; Lu, Q.; Wang, Z.; Nagashima, K.; Stauffer, J.; Andresson, T.; Specht, S.; Perera, S.; et al. Male Infertility-Associated Ccdc108 Regulates Multiciliogenesis via the Intraflagellar Transport Machinery. EMBO Rep. 2022, 23, e52775. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Griffin, J.N.; Date, P.P.; Liem, K.F.; Khokha, M.K. WDR5 Stabilizes Actin Architecture to Promote Multiciliated Cell Formation. Dev. Cell 2018, 46, 595–610.e3. [Google Scholar] [CrossRef]
- Mahuzier, A.; Shihavuddin, A.; Fournier, C.; Lansade, P.; Faucourt, M.; Menezes, N.; Meunier, A.; Garfa-Traoré, M.; Carlier, M.-F.; Voituriez, R.; et al. Ependymal Cilia Beating Induces an Actin Network to Protect Centrioles against Shear Stress. Nat. Commun. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Werner, M.E.; Hwang, P.; Huisman, F.; Taborek, P.; Yu, C.C.; Mitchell, B.J. Actin and Microtubules Drive Differential Aspects of Planar Cell Polarity in Multiciliated Cells. J. Cell Biol. 2011, 195, 19–26. [Google Scholar] [CrossRef]
- Yasunaga, T.; Wiegel, J.; Bergen, M.D.; Helmstädter, M.; Epting, D.; Paolini, A.; Çiçek, Ö.; Radziwill, G.; Engel, C.; Brox, T.; et al. Microridge-like Structures Anchor Motile Cilia. Nat. Commun. 2022, 13, 2056. [Google Scholar] [CrossRef]
- Depasquale, J.A. Actin Microridges. Anat. Rec. 2018, 301, 2037–2050. [Google Scholar] [CrossRef]
- Lam, P.; Mangos, S.; Green, J.M.; Reiser, J.; Huttenlocher, A. In Vivo Imaging and Characterization of Actin Microridges. PLoS ONE 2015, 10, e0115639. [Google Scholar] [CrossRef]
- Pinto, C.S.; Khandekar, A.; Bhavna, R.; Kiesel, P.; Pigino, G.; Sonawane, M. Microridges Are Apical Epithelial Projections Formed of F-Actin Networks That Organize the Glycan Layer. Sci. Rep. 2019, 9, 12191. [Google Scholar] [CrossRef]
- van Loon, A.P.; Erofeev, I.S.; Maryshev, I.V.; Goryachev, A.B.; Sagasti, A. Cortical Contraction Drives the 3D Patterning of Epithelial Cell Surfaces. J. Cell Biol. 2020, 219, e201904144. [Google Scholar] [CrossRef] [PubMed]
- van Loon, A.P.; Erofeev, I.S.; Goryachev, A.B.; Sagasti, A. Stochastic Contraction of Myosin Minifilaments Drives Evolution of Microridge Protrusion Patterns in Epithelial Cells. Mol. Biol. Cell 2021, 32, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.; Cram, E.J.; Zaidel-Bar, R. Stretch-Induced Actomyosin Contraction in Epithelial Tubes: Mechanotransduction Pathways for Tubular Homeostasis. Semin. Cell Dev. Biol. 2017, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Salathe, M. Ciliary Beat Co-Ordination by Calcium. Biol. Cell 2011, 103, 159–169. [Google Scholar] [CrossRef]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive Fluorescent Proteins for Imaging Neuronal Activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef]
- Ishii, T.; Sato, K.; Kakumoto, T.; Miura, S.; Touhara, K.; Takeuchi, S.; Nakata, T. Light Generation of Intracellular Ca2+ Signals by a Genetically Encoded Protein BACCS. Nat. Commun. 2015, 6, 8021. [Google Scholar] [CrossRef]
- Zahn, N.; James-Zorn, C.; Ponferrada, V.G.; Adams, D.S.; Grzymkowski, J.; Buchholz, D.R.; Nascone-Yoder, N.M.; Horb, M.; Moody, S.A.; Vize, P.D.; et al. Normal Table of Xenopus Development: A New Graphical Resource. Dev. Camb. Engl. 2022, 149, dev200356. [Google Scholar] [CrossRef]
- Slusarski, D.C.; Pelegri, F. Calcium Signaling in Vertebrate Embryonic Patterning and Morphogenesis. Dev. Biol. 2007, 307, 1–13. [Google Scholar] [CrossRef]
- Seidl, C.; Da Silva, F.; Zhang, K.; Wohlgemuth, K.; Omran, H.; Niehrs, C. Mucociliary Wnt Signaling Promotes Cilia Biogenesis and Beating. Nat. Commun. 2023, 14, 1259. [Google Scholar] [CrossRef]
- Benink, H.A.; Bement, W.M. Concentric Zones of Active RhoA and Cdc42 around Single Cell Wounds. J. Cell Biol. 2005, 168, 429–439. [Google Scholar] [CrossRef]
- Wang, Y.; Sherrard, A.; Zhao, B.; Melak, M.; Trautwein, J.; Kleinschnitz, E.-M.; Tsopoulidis, N.; Fackler, O.T.; Schwan, C.; Grosse, R. GPCR-Induced Calcium Transients Trigger Nuclear Actin Assembly for Chromatin Dynamics. Nat. Commun. 2019, 10, 5271. [Google Scholar] [CrossRef] [PubMed]
- Kage, F.; Vicente-Manzanares, M.; McEwan, B.C.; Kettenbach, A.N.; Higgs, H.N. Myosin II Proteins Are Required for Organization of Calcium-Induced Actin Networks Upstream of Mitochondrial Division. Mol. Biol. Cell 2022, 33, ar63. [Google Scholar] [CrossRef]
- Wales, P.; Schuberth, C.E.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schäfer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-Mediated Actin Reset (CaAR) Mediates Acute Cell Adaptations. eLife 2016, 5, e19850. [Google Scholar] [CrossRef]
- Sadhu, L.; Tsopoulidis, N.; Hasanuzzaman, M.; Laketa, V.; Way, M.; Fackler, O.T. ARPC5 Isoforms and Their Regulation by Calcium-Calmodulin-N-WASP Drive Distinct Arp2/3-Dependent Actin Remodeling Events in CD4 T Cells. eLife 2023, 12, e82450. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, C.A.; Jankowska, K.I.; Burkhardt, J.K.; Lewis, R.S. Calcium Influx through CRAC Channels Controls Actin Organization and Dynamics at the Immune Synapse. eLife 2016, 5, e14850. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, J.; Shevchuk, A.I.; Frolenkov, G.I.; Diakonov, I.A.; Lab, M.J.; Kros, C.J.; Richardson, G.P.; Vodyanoy, I.; Edwards, C.R.W.; Klenerman, D.; et al. Dynamic Assembly of Surface Structures in Living Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5819–5822. [Google Scholar] [CrossRef]
- Higashi, T.; Stephenson, R.E.; Miller, A.L. Comprehensive Analysis of Formin Localization in Xenopus Epithelial Cells. Mol. Biol. Cell 2019, 30, 82–95. [Google Scholar] [CrossRef]
- Hou, W.; Izadi, M.; Nemitz, S.; Haag, N.; Kessels, M.M.; Qualmann, B. The Actin Nucleator Cobl Is Controlled by Calcium and Calmodulin. PLoS Biol. 2015, 13, e1002233. [Google Scholar] [CrossRef]
- Ravanelli, A.M.; Klingensmith, J. The Actin Nucleator Cordon-Bleu Is Required for Development of Motile Cilia in Zebrafish. Dev. Biol. 2011, 350, 101–111. [Google Scholar] [CrossRef]
- Nishimura, Y.; Shi, S.; Zhang, F.; Liu, R.; Takagi, Y.; Bershadsky, A.D.; Viasnoff, V.; Sellers, J.R. The Formin Inhibitor SMIFH2 Inhibits Members of the Myosin Superfamily. J. Cell Sci. 2021, 134, jcs253708. [Google Scholar] [CrossRef]
- Zhang, J.; Chandrasekaran, G.; Li, W.; Kim, D.-Y.; Jeong, I.Y.; Lee, S.-H.; Liang, T.; Bae, J.Y.; Choi, I.; Kang, H.; et al. Wnt-PLC-IP3-Connexin-Ca2+ Axis Maintains Ependymal Motile Cilia in Zebrafish Spinal Cord. Nat. Commun. 2020, 11, 1860. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, B.; Adamiok, A.; Mercey, O.; Revinski, D.R.; Zaragosi, L.-E.; Pasini, A.; Kodjabachian, L.; Barbry, P.; Marcet, B. miR-34/449 Control Apical Actin Network Formation during Multiciliogenesis through Small GTPase Pathways. Nat. Commun. 2015, 6, 8386. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.R.; Wallingford, J.B. The Small GTPase Rsg1 Is Important for the Cytoplasmic Localization and Axonemal Dynamics of Intraflagellar Transport Proteins. Cilia 2013, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Lombardo, A.; Sauvanet, C.; Viswanatha, R.; Awad, V.; Bonomo, L.E.-R.; McDermitt, D.; Bretscher, A. Effector-Mediated ERM Activation Locally Inhibits RhoA Activity to Shape the Apical Cell Domain. J. Cell Biol. 2021, 220, e202007146. [Google Scholar] [CrossRef]
- Lienkamp, S.; Ganner, A.; Boehlke, C.; Schmidt, T.; Arnold, S.J.; Schäfer, T.; Romaker, D.; Schuler, J.; Hoff, S.; Powelske, C.; et al. Inversin Relays Frizzled-8 Signals to Promote Proximal Pronephros Development. Proc. Natl. Acad. Sci. USA 2010, 107, 20388–20393. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Shcherbakova, D.M.; Alvelid, J.; Baloban, M.; Pennacchietti, F.; Shemetov, A.A.; Testa, I.; Verkhusha, V.V. A Set of Monomeric Near-Infrared Fluorescent Proteins for Multicolor Imaging across Scales. Nat. Commun. 2020, 11, 239. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Supplier | Stock Concentration | Final Concentration (DMSO conc.) |
|---|---|---|---|
| 2-Aminoethylborate (2-APB) | Tocris (Bio-Techne GmbH, Wiesbaden, Germany) | 20 mM | 15 µM (0.075%) |
| U73122 | Tocris (Bio-Techne GmbH, Wiesbaden, Germany) | 5 mM | 0.5 µM (0.01%) |
| Y27632 | Tocris (Bio-Techne GmbH, Wiesbaden, Germany) | 50 mM | 20 µM (0.04%) |
| (−)-Blebbistatin | Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany | 25 mM | 50 µM (0.2%) |
| para-amino-Blebbistatin | Cayman Chemical, Ann Arbor, MI, USA | 100 mM | 100 µM (0.1%) |
| SMIFH2 | Tocris (Bio-Techne GmbH, Wiesbaden, Germany) | 20 mM | 10 µM (0.05%) |
| YM 58483 | Tocris (Bio-Techne GmbH, Wiesbaden, Germany) | 20 mM | 40 µM (0.2%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiegel, J.; Helmstädter, M.; Walz, G.; Bergen, M.D. Spontaneous Calcium Bursts Organize the Apical Actin Cytoskeleton of Multiciliated Cells. Int. J. Mol. Sci. 2025, 26, 2507. https://doi.org/10.3390/ijms26062507
Wiegel J, Helmstädter M, Walz G, Bergen MD. Spontaneous Calcium Bursts Organize the Apical Actin Cytoskeleton of Multiciliated Cells. International Journal of Molecular Sciences. 2025; 26(6):2507. https://doi.org/10.3390/ijms26062507
Chicago/Turabian StyleWiegel, Johannes, Martin Helmstädter, Gerd Walz, and Max D. Bergen. 2025. "Spontaneous Calcium Bursts Organize the Apical Actin Cytoskeleton of Multiciliated Cells" International Journal of Molecular Sciences 26, no. 6: 2507. https://doi.org/10.3390/ijms26062507
APA StyleWiegel, J., Helmstädter, M., Walz, G., & Bergen, M. D. (2025). Spontaneous Calcium Bursts Organize the Apical Actin Cytoskeleton of Multiciliated Cells. International Journal of Molecular Sciences, 26(6), 2507. https://doi.org/10.3390/ijms26062507