
Integrated Transcriptome and Metabolome Analysis Elucidates the Defense Mechanisms of Pumpkin Against Gummy Stem Blight



Abstract
:1. Introduction
2. Results
2.1. Transcriptome Analyses
2.1.1. Sequencing Data and Quality Analysis
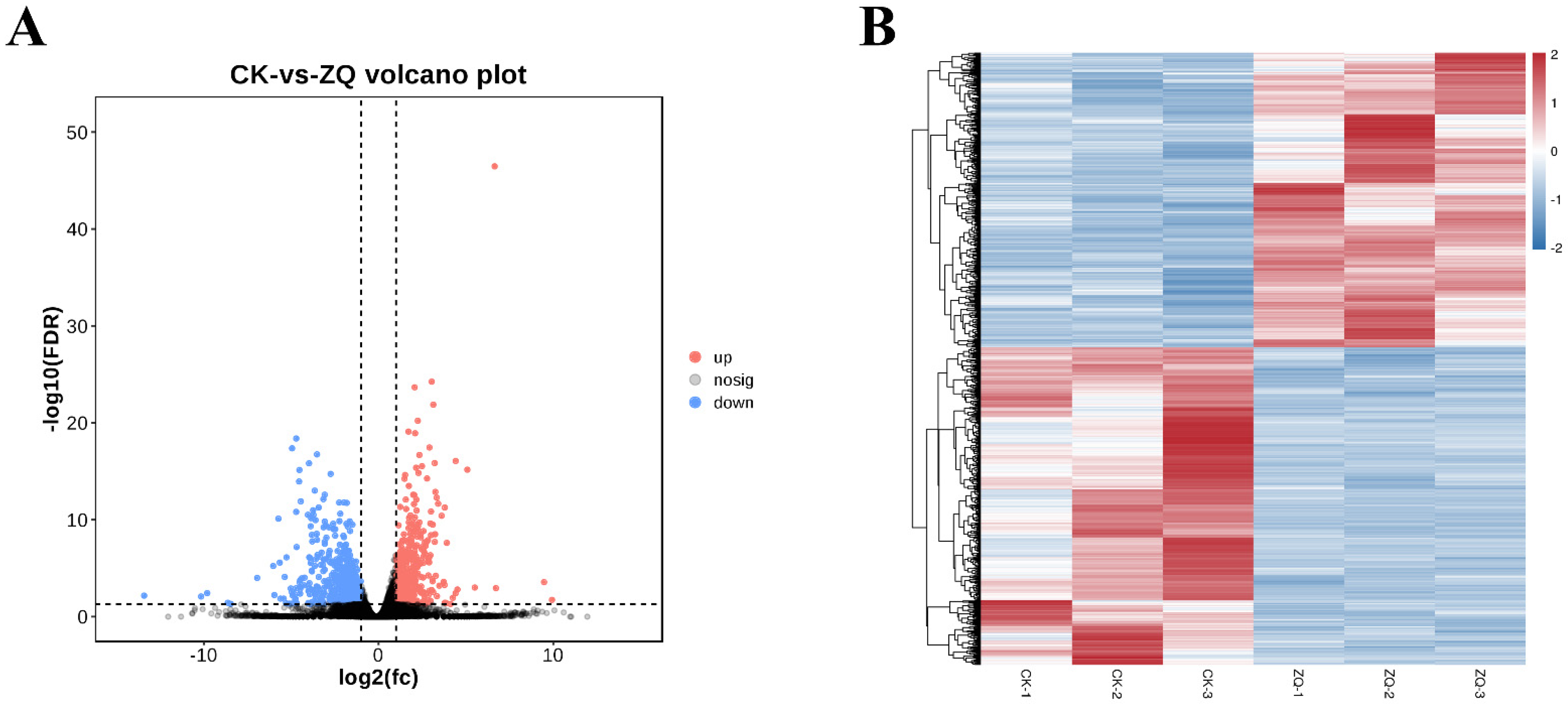
2.1.2. Identification and Functional Enrichment Analysis of DEGs
2.1.3. Novel Gene Mining and Functional Annotation
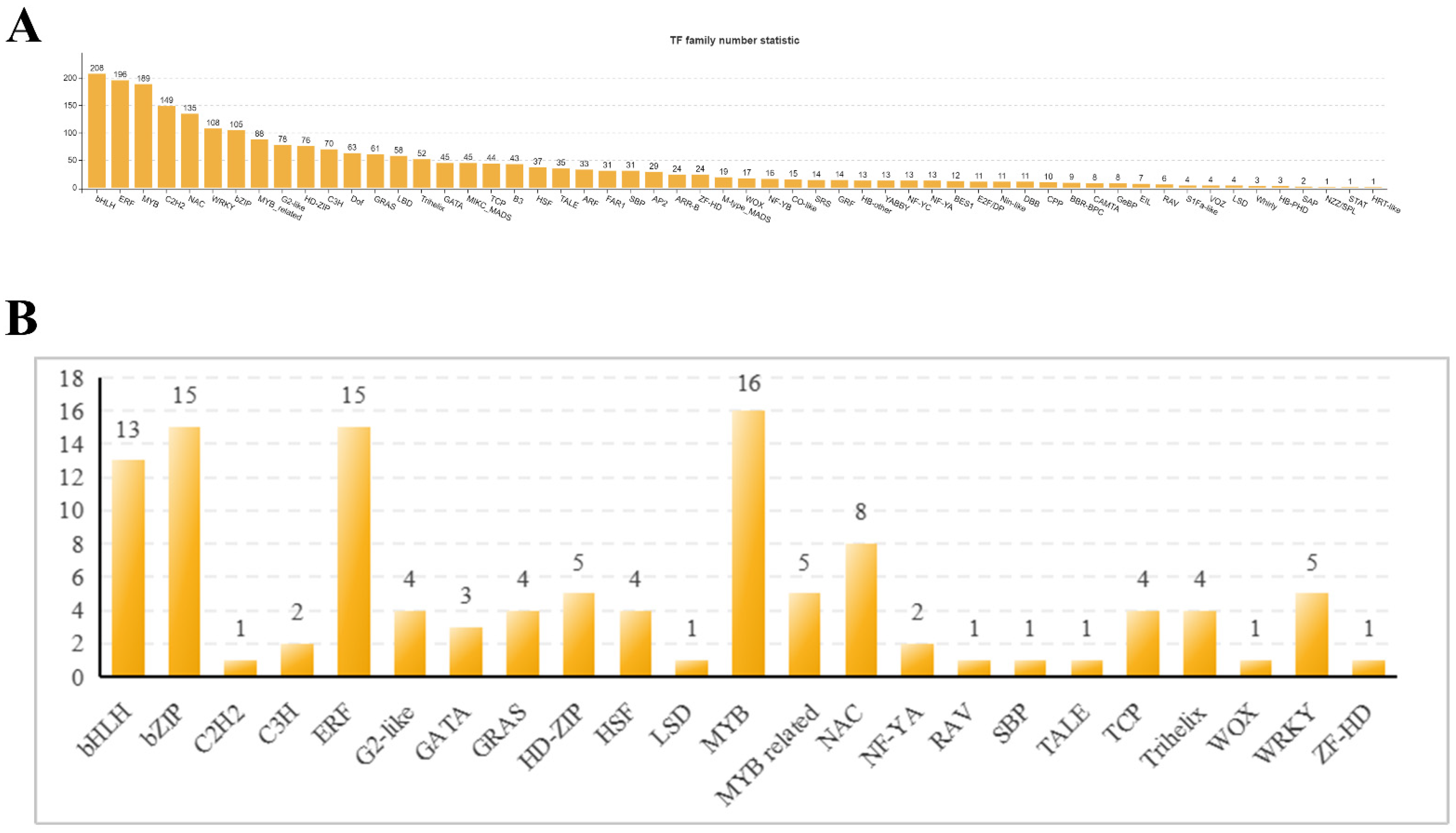
2.1.4. Transcription Factor Analysis
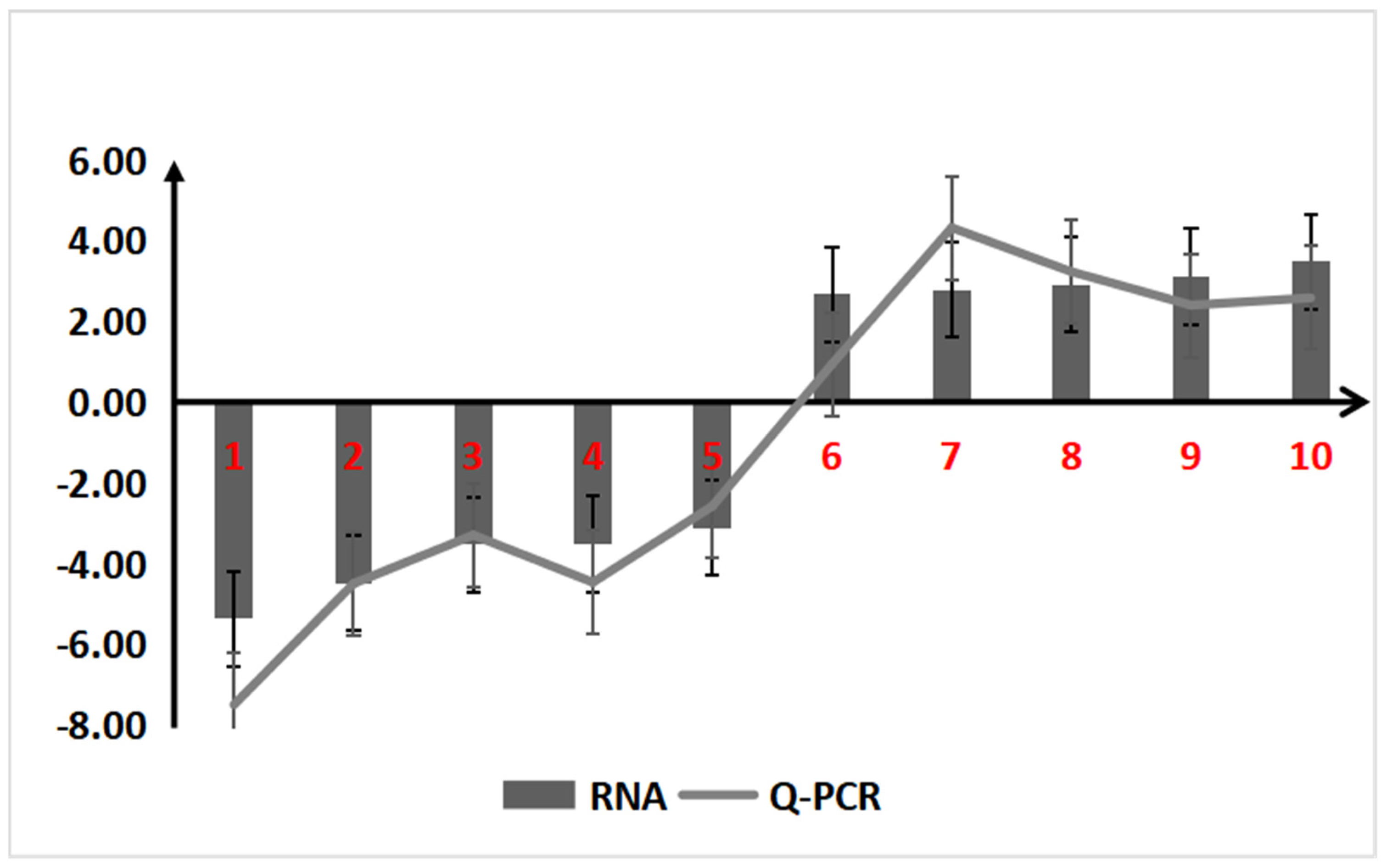
2.1.5. qRT-PCR Validation
2.2. Metabolome Analyses
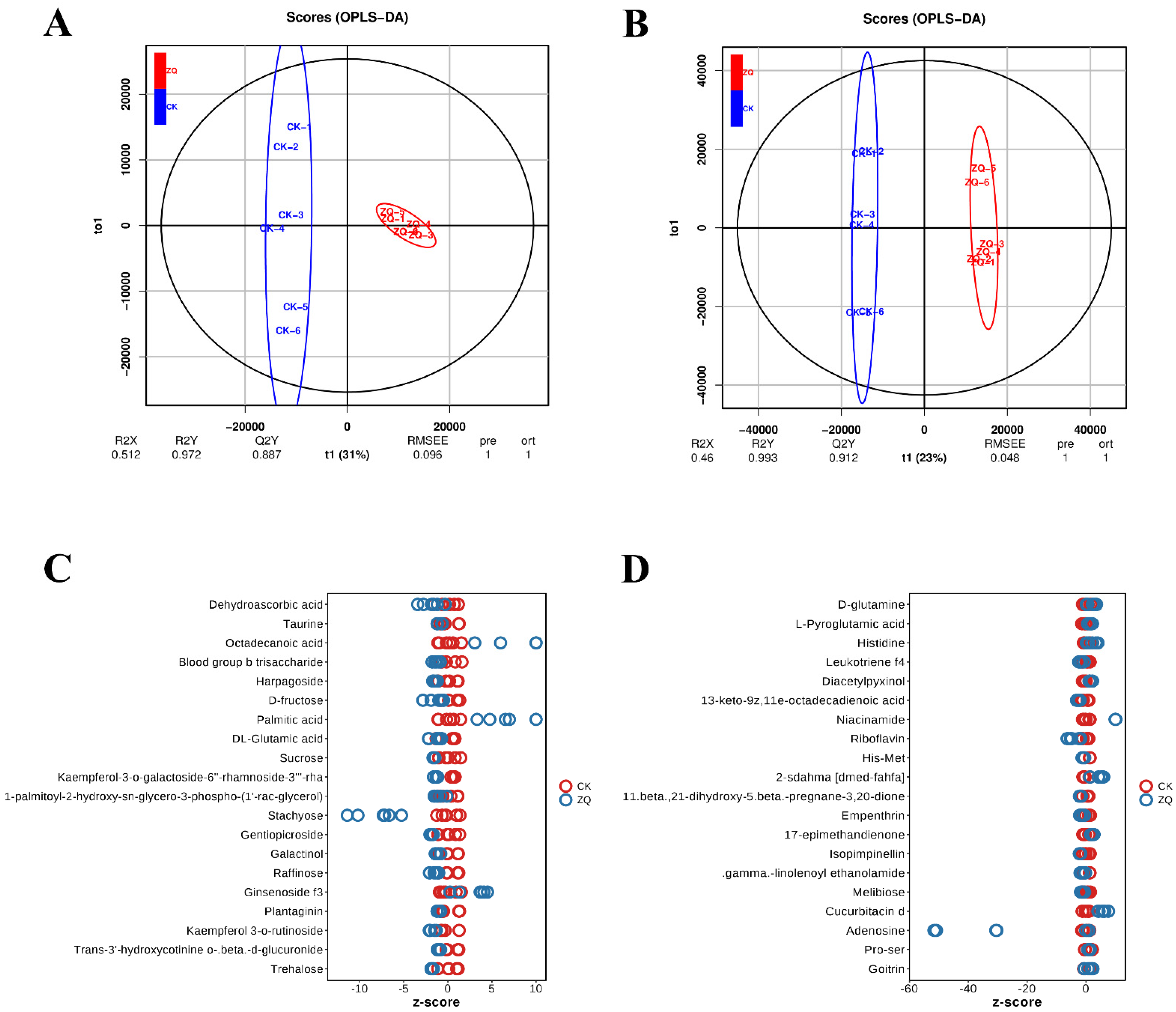
2.2.1. Reliability Analysis of Metabolomics Data
2.2.2. Identification and Functional Enrichment Analysis of DAMs
2.3. Integration of Transcriptome and Metabolome Profiles
3. Discussion
3.1. Transcriptomic Analysis
3.2. Transcription Factors Analysis
3.3. Metabolome Analysis
3.4. Metabolome and Transcriptome Analysis
3.5. Functional Validation of Novel Genes in Plant Defense Mechanisms
4. Materials and Methods
4.1. Plant Materials and Artificial Inoculation
4.2. Transcriptome Analysis
4.2.1. Sampling and Experimental Design
4.2.2. Library Construction and RNA-seq and Quality Control
4.2.3. Differential Expression, Gene Annotation and Novel Gene
4.2.4. Quantitative Real-Time PCR (qRT-PCR) Validation
4.3. Metabolome Analysis
4.3.1. Metabolite Extraction
4.3.2. LC-MS/MS Analysis
4.3.3. Metabolite Detection
4.4. Metabolome and Transcriptome Association Analysis
4.4.1. Pathway Model
4.4.2. O2PLS Model
4.4.3. Pearson Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paris, H.S.; Brown, R.N. The genes of pumpkin and squash. HortScience 2005, 40, 1620–1630. [Google Scholar] [CrossRef]
- Vaghefi, N.; Hay, F.S.; Pethybridge, S.J.; Ford, R.; Taylor, P.W. Development of a multiplex PCR diagnostic assay for the detection of Stagonosporopsis species associated with ray blight of Asteraceae. Eur. J. Plant Pathol. 2016, 146, 581–595. [Google Scholar] [CrossRef]
- Li, H.-X.; Gottilla, T.M.; Brewer, M.T. Organization and evolution of mating-type genes in three Stagonosporopsis species causing gummy stem blight of cucurbits and leaf spot and dry rot of papaya. Fungal Biol. 2017, 121, 849–857. [Google Scholar] [CrossRef]
- Keinath, A.P. From native plants in central Europe to cultivated crops worldwide: The emergence of Didymella bryoniae as a cucurbit pathogen. HortScience 2011, 4, 532–535. [Google Scholar] [CrossRef]
- Tsutsumi, C.Y.; Silva, N. Screening of melon populations for resistance to Didymella bryonia. Braz. Arch. Biol. Technol. 2004, 47, 171–177. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Fu, C.; Wang, L.; Liu, H.; Cheng, Y.; Li, S.; Deng, Q.; Wang, S.; Zhu, J.; et al. Comparative transcriptome analyses of gene expression changes triggered by Rhizoctonia solani AG1-IA infection in resistant and susceptible rice varieties. Front. Plant Sci. 2017, 8, 1422. [Google Scholar] [CrossRef]
- Yang, L.R.; Xie, L.H.; Xue, B.G. Comparative transcriptome profiling of the early infection of wheat roots by Gaeumannomycesgraminis var. tritici. PLoS ONE 2015, 10, e0120691. [Google Scholar]
- Hassan, M.Z.; Robin, A.H.K.; Rahim, M.A.; Natarajan, S.; Kim, H.T.; Park, J.I.; Nou, I.S. Screening of melon genotypes identifies gummy stem blight resistance associated with Gsb1 resistant loci. J. Plant Biotechnol. 2018, 45, 217–227. [Google Scholar] [CrossRef]
- Strange, R.N.; Scott, P.R. Plant disease: A threat to global food security. Annu. Rev. Phytopathol. 2005, 43, 83–116. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, G.; Yu, Q.; Gao, T.; Zhu, Q.; Wang, R.; Huang, S.; Han, Z.; Cervone, F.; Yin, H.; et al. A plant mechanism of hijacking pathogen virulence factors to trigger innate immunity. Science 2024, 383, 732–739. [Google Scholar] [CrossRef]
- Nga, N.T.T.; de Neergaard, E.; Jørgensen, H.J.L. Infection biology of Stagonosporopsis cucurbitacearum in watermelon and defence responses in the host. Agriculture 2024, 14, 380. [Google Scholar] [CrossRef]
- Hassan, M.Z.; Rahim, M.A.; Natarajan, S.; Robin, A.H.K.; Kim, H.T.; Park, J.I.; Nou, I.S. Gummy stem blight resistance in melon: Inheritance pattern and development of molecular markers. Int. J. Mol. Sci. 2018, 19, 2914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kyle, M.; Anagnostou, K.; Zitter, T.A. Screening melon (Cucumis melo) for resistance to gummy stem blight in the greenhouse and field. HortScience 1997, 32, 117–121. [Google Scholar] [CrossRef]
- Wolukau, J.N.; Zhou, X.H.; Li, Y.; Zhang, Y.B.; Chen, J.F. Resistance to gummy stem blight in melon (Cucumis melo L.) germplasm and inheritance of resistance from plant introductions 157076, 420145, and 323498. HortScience 2007, 42, 215–221. [Google Scholar] [CrossRef]
- Zhao, Q.; Gong, Z.; Jiang, S.; Li, Z.; Zhang, L.; Wang, L.; Yang, X.; Li, M.; Li, Z.; Chi, L.; et al. Gene mining to discover pumpkin response genes to gummy stem blight infection caused by Stagonosporopsis cucurbitacearum. J. Plant Interact. 2022, 17, 1–8. [Google Scholar] [CrossRef]
- Bhadauria, V.; Popescu, L.; Zhao, W.S.; Peng, Y.L. Fungal transcriptomics. Microbiol. Res. 2007, 162, 285–298. [Google Scholar] [CrossRef]
- Feussner, I.; Polle, A. What the transcriptome does not tell—Proteomics and metabolomics are closer to the plants’ patho-phenotype. Curr. Opin. Plant Biol. 2015, 26, 26–31. [Google Scholar] [CrossRef]
- Hu, X.; Puri, K.D.; Gurung, S.; Klosterman, S.J.; Wallis, C.M.; Britton, M.; Durbin-Johnson, B.; Phinney, B.; Salemi, M.; Short, D.P.; et al. Proteome and metabolome analyses reveal differential responses in tomato-Verticillium dahliae-interactions. J. Proteom. 2019, 207, 103449. [Google Scholar] [CrossRef]
- Panwar, V.; Bakkeren, G. Editorial: Plant genomics and pathogenomics: From technology to application in improving crop disease resistance. Front. Plant Sci. 2024, 15, 1349113. [Google Scholar] [CrossRef]
- Bhadauria, V.; Zhao, W. The Molecular Genetics and Genomics of Plant–Pathogen Interactions. Int. J. Mol. Sci. 2024, 25, 3970. [Google Scholar] [CrossRef]
- Seblani, R.; Keinath, A.P.; Munkvold, G. Gummy stem blight: One disease, three pathogens. Mol. Plant Pathol. 2023, 24, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Tan, G.F.; Ma, Y.Q.; Meng, P.H.; Zhang, J. Cucurbitaceous Vegetables’ Gummy Stem Blight Research. Agronomy 2022, 12, 1283. [Google Scholar] [CrossRef]
- Boughalleb, N.; Mahjoub, M.E.; Abad-Campos, P.; Pérez-Sierra, A.; Armengol, J. First report of gummy stem blight caused by Didymellabryoniae on grafted watermelon in Tunisia. Plant Dis. 2007, 91, 468. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Mathur, S.B.; Neergaard, P. Detection and location of seed-borne inoculum of Didymellabryoniae and its transmission in seedlings of cucumber and pumpkin. J. Phytopathol. 1984, 109, 301–308. [Google Scholar] [CrossRef]
- Grube, M.; Fürnkranz, M.; Zitzenbacher, S.; Huss, H.; Berg, G. Emerging multi-pathogen disease caused by Didymella bryoniae and pathogenic bacteria on Styrian oil pumpkin. Eur. J. Plant Pathol. 2011, 131, 539–548. [Google Scholar] [CrossRef]
- Cho, M.; Cheoul, P.; Hyo, G.; Kim, H.; Tae, K. Spore production and inoculation methods for resistance screening to gummy stem blight (Mycosphaerellamelonis) in muskmelon seedlings. Hortic. Environ. Biotechnol. 1997, 38, 364–367. [Google Scholar]
- Afroz, M.; Rahman, M.A.; Nahar, M.S.; Yasmin, L.; Alam, S.M.K. Selection of resistant variety, effective organic amendment and fungicide against gummy stem blight (Didymella bryoniae) of bottle gourd. Diss. Saarc J. Agric. 2012, 10, 1–8. [Google Scholar]
- Thinggaard, K. Attack of Didymellabryoniae on roots of cucumber. J. Phytopathol. 1987, 120, 372–375. [Google Scholar] [CrossRef]
- Café-Filho, A.C.; Santos, G.; Laranjeira, F.F. Temporal and spatial dynamics of watermelon gummstem blight epidemics. Eur. J. Plant Pathol. 2010, 128, 473–482. [Google Scholar] [CrossRef]
- Brewer, M.T.; Rath, M.; Li, H.X. Genetic diversity and population structure of cucurbit gummy stem blight fungi based on microsatellite markers. Phytopathology 2015, 105, 815–824. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, J.; Zhang, L.; Xu, L.; Yan, C.; Gong, Z. Identification and characterization of Cucurbita gummy stem blight fungi in Northeast China. J. Phytopathol. 2018, 166, 305–313. [Google Scholar] [CrossRef]
- Ishida, K.; Yokoyama, R. Reconsidering the function of the xyloglucan endotransglucosylase /hydrolase family. J. Plant Res. 2022, 135, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, L.; Li, D.; Qi, D.; Fang, F.; Luo, Y.; Zhang, H.; Zhang, S. Genome-wide characterization of the xyloglucan endotransglucosylase/hydrolase family genes and their response to plant hormone in sugar beet. Plant Physiol. Biochem. 2024, 206, 108239. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.D.; Felix MD, R.; Patanita, M.; Materatski, P.; Albuquerque, A.; Ribeiro, J.A.; Varanda, C. Defense strategies: The role of transcription factors in tomato–pathogen interaction. Biology 2022, 11, 235. [Google Scholar] [CrossRef]
- Bakshi, M.; Oelmüller, R. Wrky transcription factors jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Wang, H.; Guo, Z. WRKY transcription factors: Evolution, binding, and action. Phytopathol. Res. 2019, 1, 13. [Google Scholar] [CrossRef]
- Fang, Y.; You, J.; Xie, K.; Xie, W.; Xiong, L. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol. Genet. Genom. 2008, 280, 547–563. [Google Scholar] [CrossRef]
- Jin, J.F.; Wang, Z.Q.; He, Q.Y.; Wang, J.Y.; Li, P.F.; Xu, J.M.; Zheng, S.J.; Fan, W.; Yang, J.L. Genome-wide identification and expression analysis of the NAC transcription factor family in tomato (Solanum lycopersicum) during aluminum stress. BMC Genom. 2020, 21, 288. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Chao, J.; Zhang, Z.; Wang, W.; Guo, Y. NAC Family Transcription factors in tobacco and their potential role in regulating leaf senescence. Front. Plant Sci. 2018, 9, 1900. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Bartholomew, E.; Black, K.; Dong, M.; Zhang, Y.; Yang, S.; Cai, Y.; Xue, S.; Weng, Y.; et al. Comprehensive analysis of NAC transcription factors and their expression during fruit spine development in cucumber (Cucumis sativus L.). Hortic. Res. 2018, 5, 31. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Akbudak, M.A.; Filiz, E.; Kontbay, K. DREB2 (dehydration-responsive element-binding protein 2) type transcription factor in sorghum (Sorghum bicolor): Genome-wide identification, characterization and expression profiles under cadmium and salt stresses. 3 Biotech 2018, 8, 426. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Guo, Z.H.; Hao, P.P.; Wang, G.M.; Jin, Z.M.; Zhang, S.L. Multiple regulatory roles of AP2/ERF transcription factor in angiosperm. Bot. Stud. 2017, 58, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Song, J.; Liu, S.; Chen, C.; Zhang, S.; Wang, J.; Xiao, Y.; Cao, B.; Lei, J.; Zhu, Z. Genome-wide identification of the capsicum bHLH transcription factor family: Discovery of a candidate regulator involved in the regulation of species-specific bioactive metabolites. BMC Plant Biol. 2021, 21, 262. [Google Scholar] [CrossRef]
- Bai, G.; Yang, D.H.; Chao, P.; Yao, H.; Fei, M.; Zhang, Y.; Chen, X.; Xiao, B.; Li, F.; Wang, Z.Y.; et al. Genome-wide identification and expression analysis of NtbHLH gene family in tobacco (Nicotiana tabacum L.) and the role of NtbHLH86 in drought adaptation. Plant Divers. 2021, 43, 510–522. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, P.; Kong, N.; Lu, R.; Pei, Y.; Huang, C.; Ma, H.; Chen, Q. Genome-wide identification and characterization of the potato bHLH transcription factor family. Genes 2018, 9, 54. [Google Scholar] [CrossRef]
- Sun, H.; Fan, H.J.; Ling, H.Q. Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genom. 2015, 16, 9. [Google Scholar] [CrossRef]
- Seo, E.; Choi, D. Functional studies of transcription factors involved in plant defenses in the genomics era. Brief. Funct. Genom. 2015, 14, 260–267. [Google Scholar] [CrossRef]
- van Ver, M.C.; Gatz, C.; Linthorst, H.J.M. Chapter 10 Transcriptional regulation of plant defense responses. Adv. Bot. Res. 2009, 51, 397–438. [Google Scholar]
- Wei, K.; Chen, J.; Wang, Y.; Chen, Y.; Chen, S.; Lin, Y.; Pan, S.; Zhong, X.; Xie, D. Genome-wide analysis of bZIP-encoding genes in maize. DNA Res. 2012, 19, 463–476. [Google Scholar] [CrossRef]
- Kesarwani, M.; Yoo, J.; Dong, X. Genetic interactions of TGA transcription factors in the regulation of pathogenesis-related genes and disease resistance in Arabidopsis. Plant Physiol. 2007, 144, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Amorim, L.; Santos, R.; Neto, J.; Guida-Santos, M.; Crovella, S.; Benko-Iseppon, A. Transcription factors involved in plant resistance to pathogens. Curr. Protein Pept. Sci. 2016, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ma, R.; Chen, X.L. Advances of metabolomics in fungal pathogen–plant interactions. Metabolites 2019, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, T.; Harimoto, Y.; Akimitsu, K.; Ohtani, K.; Kodama, M.; Akagi, Y.; Egusa, M.; Yamamoto, M.; Otani, H. Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 2013, 37, 44–66. [Google Scholar] [CrossRef]
- Inayavekhin, N.; Saghatelian, A. Untargeted Metabolomics. In Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; Chapter 30; pp. 1–24. [Google Scholar]
- Castro-Moretti, F.R.; Gentzel, I.N.; Mackey, D.; Alonso, A.P. Metabolomics as an emerging tool for the study of plant–pathogen interactions. Metabolites 2020, 10, 52. [Google Scholar] [CrossRef]
- Allwood, J.W.; Ellis, D.I.; Goodacre, R. Metabolomic technologies and their application to the study of plants and plant–host interactions. Physiol. Plant. 2008, 132, 117–135. [Google Scholar] [CrossRef]
- Slisz, A.M.; Breksa, A.P.; Mishchuk, D.O.; McCollum, G.; Slupsky, C.M. Metabolomic analysis of citrus infection by Candidatus Liberibacter reveals insight into pathogenicity. J. Proteome Res. 2012, 11, 4223–4230. [Google Scholar] [CrossRef]
- Lowe RG, T.; Lord, M.; Rybak, K.; Trengove, R.D.; Oliver, R.P.; Solomon, P.S. A metabolomic approach to dissecting osmotic stress in the wheat pathogen Stagonospora nodorum. Fungal Genet. Biol. 2008, 45, 1479–1486. [Google Scholar] [CrossRef]
- Parker, D.; Beckmann, M.; Zubair, H.; Enot, D.P.; Caracuel-Rios, Z.; Overy, D.P.; Snowdon, S.; Talbot, N.J.; Draper, J. Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J. 2009, 59, 723–737. [Google Scholar] [CrossRef]
- Wang, S.; Wang, S.; Li, M.; Su; Sun, Z.; Ma, H. Combined transcriptome and metabolome analysis of Nerium indicum L. elaborates the key pathways that are activated in response to witches’ broom disease. BMC Plant Biol. 2022, 22, 291. [Google Scholar] [CrossRef]
- Duan, W.; Peng, L.; Jiang, J.; Zhang, H.; Tong, G. Combined transcriptome and metabolome analysis of strawberry fruits in response to powdery mildew infection. Agron. J. 2022, 114, 1027–1039. [Google Scholar] [CrossRef]
- Deutsch, J.C. Dehydroascorbic acid. J. Chromatogr. A 2000, 881, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, X.; Wang, L.; Liu, H.; Zhao, Y.; Yi, K.; Cui, G.; Yin, X. Transcriptome profiling unveils the mechanism of phenylpropane biosynthesis in rhizome development of Caucasian clover. PLoS ONE 2021, 16, e0254669. [Google Scholar] [CrossRef]
- Amjad, M.; Wang, Y.; Han, S.; Haider, M.Z.; Sami, A.; Batool, A.; Shafiq, M.; Ali, Q.; Dong, J.; Sabir, I.A.; et al. Genome wide identification of phenylalanine ammonia-lyase (PAL) gene family in Cucumis sativus (cucumber) against abiotic stress. BMC Genom. Data 2024, 25, 76. [Google Scholar] [CrossRef]
- Chai, P.; Cui, M.; Zhao, Q.; Chen, L.; Guo, T.; Guo, J.; Wu, C.; Du, P.; Liu, H.; Xu, J.; et al. Genome-Wide Characterization of the Phenylalanine Ammonia-Lyase Gene Family and Their Potential Roles in Response to Aspergillus flavus L. Infection in Cultivated Peanut (Arachis hypogaea L.). Genes 2024, 15, 265. [Google Scholar] [CrossRef]
- Hoffmann, L.; Besseau, S.; Geoffroy, P.; Ritzenthaler, C.; Meyer, D.; Lapierre, C.; Pollet, B.; Legrand, M. Silencing of Hydroxycinnamoyl-Coenzyme A Shikimate/Quinate Hydroxycinnamoyl transferase Affects Phenylpropanoid Biosynthesis. Plant Cell 2004, 16, 1446–1465. [Google Scholar] [CrossRef]
- Xu, R.; Wang, Y.; Li, X. CRISPR-Cas9-mediated genome editing in plants: A review. Trends Plant Sci. 2021, 26, 123–135. [Google Scholar]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2019, 444, 323–329. [Google Scholar] [CrossRef]
- Huang, R.; Liu, J.; Wang, L. Silencing of necrosis-related gene (NRL) in watermelon reduces susceptibility to Gummy stem blight. Mol. Plant Pathol. 2023, 24, 123–135. [Google Scholar]
- Wang, L.; Liu, J.; Huang, R. Genome comparison of Cucumis sativus and Cucumis melo reveals conserved defense-related gene families. Plant Genome 2022, 15, e20120. [Google Scholar]
- Li, Z.; Yu, Q.; Zhang, W. OrthoMCL software for ortholog identification and gene family analysis in plant genomes. Bioinformatics 2021, 37, 1200–1206. [Google Scholar]
- Chen, Y.; Zhang, H.; Li, X. Comparative genomics of watermelon and melon reveals conserved signaling pathways associated with Gummy stem blight resistance. Plant Dis. 2023, 107, 456–465. [Google Scholar]
- Smith, A.; Brown, T.; Johnson, K. Limitations of experimental designs in studying plant pathogen interactions. PLoS ONE 2021, 16, e0251443. [Google Scholar]
- Li, X.; Chen, Y.; Zhang, H. Metabolomic profiling reveals cucurbitacins as critical secondary metabolites in Gummy stem blight resistance. J. Agric. Food Chem. 2023, 71, 4567–4578. [Google Scholar]
- Kumar, P.; Sharma, R. Marker-assisted breeding for disease resistance in crops. Annu. Rev. Plant Biol. 2023, 74, 123–150. [Google Scholar]
- Wang, H.; Li, X.; Zhang, Y. Overexpression of a fungal resistance gene in watermelon enhances resistance to Didymella bryoniae. Transgenic Res. 2023, 32, 78–89. [Google Scholar]
- Zhao, Q.; Wu, J.; Zhang, L.; Yan, C.; Jiang, S.; Li, Z.; Sun, D.; Lai, Y.; Gong, Z. Genome-scale analyses and characteristics of putative pathogenicity genes of Stagonosporopsis cucurbitacearum, a pumpkin gummy stem blight fungus. Sci. Rep. 2020, 10, 8065. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Harel, A.; Bercovich, S.; Yarden, O. Calcineurin is required for sclerotial development and pathogenicity of Sclerotinia sclerotiorum in an oxalic acid-independent manner. Mol. Plant Microbe Interact. 2006, 19, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17, S11. [Google Scholar] [CrossRef]
- Csardi, G. The igraph software package for complex network research. Inter. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | mRNAs | Proteins |
|---|---|---|
| All Genes | 26,596 | 8146 |
| Diff Genes | 1176 | 370 |
| Up Genes | 566 | 70 |
| Down Genes | 610 | 300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Zhang, L.; Han, W.; Wang, Z.; Wu, J. Integrated Transcriptome and Metabolome Analysis Elucidates the Defense Mechanisms of Pumpkin Against Gummy Stem Blight. Int. J. Mol. Sci. 2025, 26, 2586. https://doi.org/10.3390/ijms26062586
Zhao Q, Zhang L, Han W, Wang Z, Wu J. Integrated Transcriptome and Metabolome Analysis Elucidates the Defense Mechanisms of Pumpkin Against Gummy Stem Blight. International Journal of Molecular Sciences. 2025; 26(6):2586. https://doi.org/10.3390/ijms26062586
Chicago/Turabian StyleZhao, Qian, Liyan Zhang, Weibo Han, Ziyu Wang, and Jianzhong Wu. 2025. "Integrated Transcriptome and Metabolome Analysis Elucidates the Defense Mechanisms of Pumpkin Against Gummy Stem Blight" International Journal of Molecular Sciences 26, no. 6: 2586. https://doi.org/10.3390/ijms26062586
APA StyleZhao, Q., Zhang, L., Han, W., Wang, Z., & Wu, J. (2025). Integrated Transcriptome and Metabolome Analysis Elucidates the Defense Mechanisms of Pumpkin Against Gummy Stem Blight. International Journal of Molecular Sciences, 26(6), 2586. https://doi.org/10.3390/ijms26062586